

Abstract
The expression of ABO(H) blood group antigens causes deletion of cells that generate self anti-blood group antibodies, but this deletion limits adaptive immunity toward pathogens bearing cognate blood group antigens. To explore potential defense mechanisms against these pathogens, given such limitations in adaptive immunity, we screened for innate proteins that could recognize human blood group antigens. Here we report that two innate immune lectins, galectins-4 and -8, which are expressed in the intestinal tract, recognize and kill human blood group antigen-expressing E. coli, while failing to alter viability of other E. coli strains or other gram-negative or gram-positive organisms both in vitro and in vivo. Killing by both galectins-4 and -8 resides within their C-terminal domains, occurs rapidly and independently of complement, and is accompanied by disruption of membrane integrity. These results demonstrate that innate defense lectins can provide immunity against pathogens that display blood group self-antigens on their surface.
Recent studies suggest that blood group antigen diversity may provide a mechanism of pathogen evasion whereby distinct ABO(H) antigen structures may reduce pathogen attachment and therefore infectivity1. Because ABO(H) antigens are composed of carbohydrate structures that only differ by distinct monosaccharides on the terminal structures of glycans2, potential factors responsible for providing innate immunity toward pathogens expressing blood group antigens must recognize carbohydrates. A growing list of glycan-binding proteins, including galectins and C-type lectins, recognize carbohydrate determinants on pathogens and participate in innate immune responses3-5. Importantly, previous studies suggest that several galectins may recognize blood group antigens6 along with various other carbohydrate ligands. We analyzed publicly available data from the screening of nearly 100 different lectins from the Consortium for Functional Glycomics (www.functionalglycomics.org/), many of which are mammalian lectins with documented immunological activity, including members of the galectin family. Some of the most specific interactions observed among the lectins tested following screening of over 300 structurally diverse glycans was seen by members of the galectin family. Human galectin-3 (Gal-3), galectin-4 (Gal-4), and galectin-8 (Gal-8), which recognize multiple glycan structures at higher concentrations6,7, displayed novel specificity for human blood group A and B antigens at submicromolar concentrations and did not bind to blood group O(H) at these concentrations, while human galectin-1 (Gal-1), a related galectin family member, did not recognize blood group antigens (Fig. 1a–d). This specificity was not as striking in our previous studies concerning members of this protein family, where we tested binding at high concentrations when saturation of binding detection showed that the lectins recognized multiple carbohydrate ligands along with blood group antigens6,7.
Figure 1. Gal-3, Gal-4, and Gal-8 recognize blood group B positive E. coli.
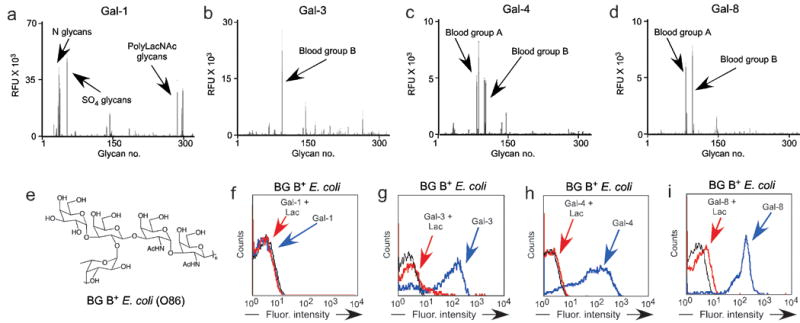
(a–d) Glycan microarray data obtained following incubation with (a) 0.2 μM Gal-1, (b) 0.2 μM Gal-3, (c) 0.5 μM Gal-4, and (d) 0.02 μM Gal-8. RFU = relative fluorescence units represented on the y-axis. Error bars = +/- 1 SEM. See Supplementary Table 1 for complete list of glycans represented on the x-axis. (e) Structure of E. coli O86 O antigen. (f-i) Flow cytometric analysis following incubation of E. coli O86 with (f) Gal-1, (g) Gal-3, (h) Gal-4, and (i) Gal-8 all tested at ~0.1 μM with or without inclusion of 20 mM lactose (Lac) where indicated.
Bacteria generate a wide variety of glycan-based antigenic structures, many of which can possess blood group antigen activity8,9. The best characterized of these, E. coli O86, cross-reacts with human anti-blood group B antibodies and induces significant blood group B antibodies in previously unexposed individuals10. Importantly, while blood group A or O individuals produce antibodies that kill E. coli O86, blood group B individuals do not generate antibodies capable of altering E. coli O86 viability10,11, providing a specific example of the immunological limitation in adaptive immunity toward a blood group antigen-bearing pathogen. The ability of human Gal-3, Gal-4, and Gal-8 to specifically recognize blood group A and B antigens suggests that they may be uniquely poised to provide innate immunity toward blood group-bearing pathogens regardless of the blood group antigen status of an individual. However, although E. coli O86 generates an identical blood group B epitope (Fig. 1e) to that of humans12, the context of this epitope may differ from the common human presentations found on the glycan microarray. We examined, therefore, whether Gal-3, Gal-4, and Gal-8 recognize E. coli O86. Consistent with their ability to specifically recognize blood group A and B antigens on the glycan microarray, human Gal-3, Gal-4, and Gal-8, but not Gal-1, bound to E. coli O86, hereafter referred to as blood group B positive E. coli (BG B+ E. coli) (Fig. 1f–i). Binding of all galectins to bacteria was inhibited by lactose, an inhibitor of galectin-carbohydrate interactions, indicating that galectin binding was toward glycan determinants on the surface of BG B+ E. coli.
Previous studies demonstrated high levels of galectin expression in the intestinal mucosa where they may serve as pathogen recognition proteins13,14, suggesting Gal-3, Gal-4, and Gal-8 may facilitate innate immunity toward BG B+ pathogens. While previous studies have shown that several innate immune lectins can directly affect pathogen viability3,15,16, including potential roles for galectins in pathogen adhesion, recognition, and killing14, there is no direct evidence as to whether galectins can alter prokaryote viability. Thus, we asked whether Gal-3, Gal-4, and Gal-8 might confer intrinsic immunity by directly killing BG B+ E. coli. Incubation with both Gal-4 and Gal-8 caused direct killing of BG B+ E. coli, whereas Gal-3, which also binds BG B+ E. coli did not affect viability, and Gal-1, which does not bind BG B+ E. coli, had no effect (Fig. 2a). Similar to Gal-4 and Gal-8 binding to BG B+ E. coli, lactose completely inhibited both Gal-4 and Gal-8-induced death, while sucrose, a disaccharide unable to inhibit galectin-carbohydrate interactions, failed to alter killing of BG B+ E. coli (Fig. 2b–c). Gal-4 and Gal-8 displayed similarly potent concentration-dependent killing of BG B+ E. coli, with an LD50 of ∼0.1 μM (Fig. 2d), concentrations similar to those observed in vivo17 and used to evaluate glycan binding specificity on the glycan microarray. In addition, effects of Gal-8 treatment appeared to be rapid, since treated BG B+ E. coli lost all motility compared to untreated BG B+ E. coli nearly immediately following the addition of Gal-8 (Fig. 2e, Supplementary video 1). BG B+ E. coli positively stained for propidium iodide (PI) following 30 min incubation with Gal-8 (Fig. 2f), and displayed significant disruption of membrane morphology (Fig. 2g,h). These results show that Gal-8 kills BG B+ E. coli through directly altering membrane integrity. Comparable alterations were observed following incubation with Gal-4 (data not shown). Taken together, these results demonstrate that both human Gal-4 and Gal-8 directly kill BG B+ E. coli through recognition of bacterial surface carbohydrates via a mechanism that drastically alters membrane integrity and bacterial motility. Killing of BG B+ E. coli by human Gal-4 and Gal-8 did not require complement, demonstrating that these lectins fundamentally differ from other innate immune lectins, such as mannan binding proteins (MBP), which do not directly alter viability, but activate complement following pathogen recognition16.
Figure 2. Gal-4 and Gal-8 kill blood group B positive E. coli.
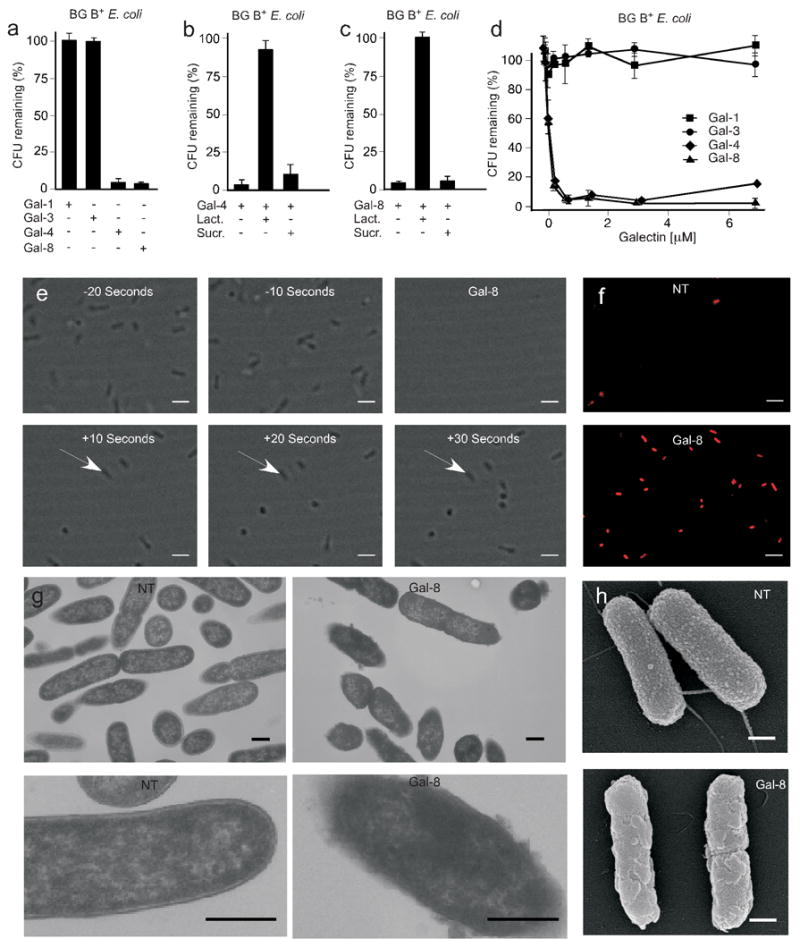
E. coli O86 (BG B+ E. coli) were mixed with (a) 5 μM Gal-1, Gal-3, Gal-4, or Gal-8, (b) 5 μM Gal-4 with or without 20 mM lactose (Lact.) or 20 mM sucrose (Sucr.), (c) 5 μM Gal-8 with or without 20 mM lactose (Lact.) or 20 mM sucrose (Sucr.), or (d) the indicated concentrations of Gal-1, Gal-3, Gal-4, or Gal-8. Viable bacteria were quantified by dilution plating, n=3, 1 representative experiment in duplicate over 2 dilutions shown (a–c), error bars=SD. (e) Still-frame images from real-time video microscopy demonstrating bacterial mobility at 10-s intervals before and after addition of 5 μM Gal-8 as indicated (see Supplementary video 1). Arrows indicate one group of immobilized bacteria. Scale bars = 100 μm. (f) E. coli O86 (BG B+ E. coli) were grown to mid-log phase followed by addition of 5 μM Gal-8. Untreated and Gal-8 treated bacteria were stained with propidium iodide (red) and visualized by fluorescence microscopy. Scale bars = 100 μm. (g) Transmission electron microscopy images of E. coli O86 (BG B+ E. coli) following addition of PBS (NT) or 5 μM Gal-8. Lower panels show close up view of single bacterium. Scale bars = 500 nm. (h) Scanning electron microscopy images of E. coli O86 (BG B+ E. coli) followed by addition of PBS (NT) or Gal-8. Scale bars = 500 nm.
Unlike Gal-1 and Gal-3, which contain a single carbohydrate recognition domain (CRD), Gal-4 and Gal-8 have two distinct CRDs18, suggesting that these galectins may utilize one domain for target recognition and the other domain for killing the target once bound, similar to many prokaryotic AB toxins19. To distinguish these possibilities, we mutated each CRD of Gal-8, as done previously6,7 in the context of the whole protein, to determine which domain recognizes BG B+ E. coli. Inactivation of the C-terminal CRD (Arg233→His) (Gal-8R233H) eliminated recognition of blood group antigens on both the glycan microarray and BG B+ E. coli, while the analogous mutation in the N-terminal CRD (Arg69→His) (Gal-8R69H) did not alter blood group antigen recognition in either context (data not shown). Importantly, Gal-8R69H, but not Gal-8R233H, killed BG B+ E. coli (Fig. 3a), which demonstrated that Gal-8-mediated killing requires carbohydrate recognition only by the blood group binding C-terminal domain of Gal-8. To determine whether the N-terminal domain may be required for Gal-8 killing independent of glycan recognition, we expressed the individual domains of Gal-8. While the N-terminal domain (Gal-8N) failed to bind to blood group antigens on either the glycan microarray or BG B+ E. coli (Fig. 3b, data not shown), the C-terminal domain alone of Gal-8 (Gal-8C) independently recognized blood group antigens and killed BG B+ E. coli (Fig. 3b,c). These results show that recognition and killing of BG B+ E. coli by Gal-8 resides entirely within its blood group binding domain. By contrast, both domains of Gal-4 displayed specific recognition toward BG B+ E. coli (Fig. 3d). Thus, we asked whether Gal-4N and Gal-4C might independently kill BG B+ E. coli. However, similar to Gal-3, Gal-4N displayed significant recognition of BG B+ E. coli, yet failed to alter BG B+ E. coli viability. By contrast, Gal-4C had substantial killing activity toward BG B+ E. coli (Fig. 3e). Interestingly, Gal-4C and Gal-8C domains display phylogenetic similarities not shared by Gal-3 and Gal-4N domains20, which suggests a conserved mechanism shared between these two protein domains.
Figure 3. Gal-4 and Gal-8 kill BG B+ E. coli solely through the C-terminal domain.
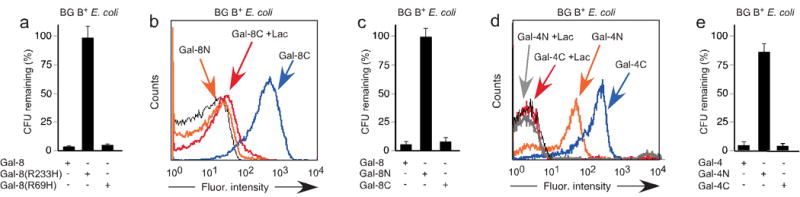
(a) 5 μM Gal-8, Gal-8R233H, or Gal-8R69H were added to mid-log phase E. coli O86 (BG B+ E. coli). Viable bacteria were quantified by dilution plating, n=3, 1 representative experiment in duplicate over 2 dilutions shown, error bars=SD. (b) Flow cytometric analysis following incubation of E. coli O86 (BG B+ E. coli) with Gal-8N or Gal-8C at ~0.1 μM with or without inclusion of 20 mM lactose (Lac) where indicated. (c) 5 μM Gal-8, Gal-8N, or Gal-8C were added to mid-log phase E. coli O86 (BG B+ E. coli). Viable bacteria were quantified by dilution plating, n=3, 1 representative experiment in duplicate over 2 dilutions shown, error bars=SD. (d) Flow cytometric analysis following incubation of E. coli O86 (BG B+ E. coli) with Gal-4N or Gal-4C at ~0.1 μM with or without inclusion of 20 mM lactose (Lac) where indicated. (e) 5 μM Gal-4, Gal-4N, or Gal-4C were added to mid-log phase E. coli O86 (BG B+ E. coli). Viable bacteria were quantified by dilution plating, n=3, 1 representative experiment in duplicate over 2 dilutions shown, error bars=SD.
The ability of the blood group binding domain of Gal-4 and Gal-8 to independently kill BG B+ E. coli (Fig. 3a,c,e) suggested that Gal-4 and Gal-8 might specifically kill BG B+ E. coli. To test this, we examined whether Gal-4 and Gal-8 recognize strains of E. coli that fail to express the blood group B antigen. Although both Gal-4 and Gal-8 recognize BG B+ E. coli, they did not significantly bind to or affect the viability of BG B− E. coli (Fig. 4a–c, data not shown). In addition, Gal-4 and Gal-8 did not recognize nor kill gram-negative BG B− species K. pneumoniae and P. aeruginosa, and neither bound nor altered the viability of gram-positive S. aureus (Fig. 4d–g, data not shown). We next sought to determine whether Gal-8 specifically kills BG B+ E. coli in a mixed population of BG B+ and BG B− bacteria. To accomplish this, we incubated GFP+ BG B− P. aeruginosa with Gal-8 to determine whether Gal-8 altered GFP expression or viability. Importantly, Gal-8 failed to alter GFP expression (Fig. 4h) or viability (data not shown), allowing discrimination of GFP+ P. aeruginosa and BG B+ E. coli within a mixed population. To examine whether Gal-8 specifically kills BG B+ E. coli, we incubated different ratios of BG B+ E. coli to GFP+ P. aeruginosa with or without Gal-8. Importantly, even at a ratio of 4:1 BG B+ E. coli to GFP+ P. aeruginosa, Gal-8 selectively eliminated the GFP− BG B+ E. coli (Fig. 4i,j). Furthermore, defined mutations that prevent synthesis of the blood group antigen formation on BG B+ E. coli (WaaL−) prevented recognition and killing by Gal-4 and Gal-8, while mutations that allow formation of at least one repeat of the blood group antigen (Wzy−) failed to substantially alter sensitivity to Gal-4 or Gal-8 (Fig. 5a–d), further illustrating the specificity of Gal-4 and Gal-8 for the blood group B antigen21. Importantly, lactose, but not sucrose, prevented Gal-4 and Gal-8 killing (data not shown). However, although both Gal-4 and Gal-8 recognized BG B+ human erythrocytes, neither affected the membrane integrity of these cells (data not shown), which demonstrated that the killing activity exhibited by Gal-4 and Gal-8 not only displays antigen specificity but also uniquely targets prokaryotes. Furthermore, Gal-4 and Gal-8-induced killing of BG B+ E. coli did not represent a simple agglutination-associated reduction in CFU counts, as Gal-4 and Gal-8 bound to BG B+ E. coli at 4 °C but did not alter viability (data not shown). In addition, human anti-BG B antibodies and Gal-3 recognized and agglutinated BG B+ E. coli at higher concentrations, yet failed to affect CFU counts of BG B+ E. coli following incubation (data not shown).
Figure 4. Gal-4 and Gal-8 specifically kill blood group B positive E. coli.
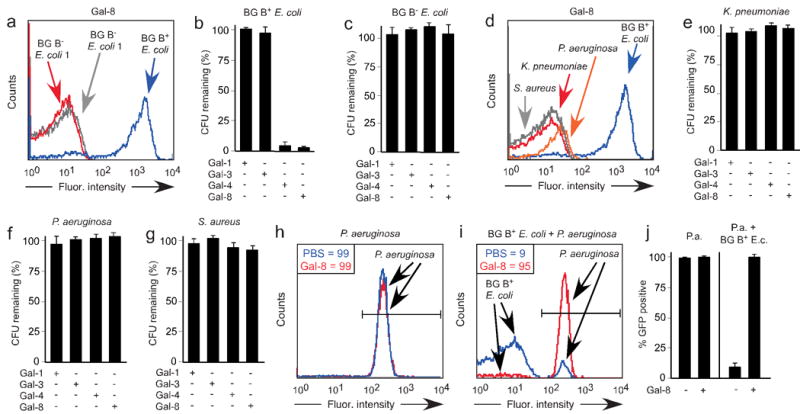
(a) Flow cytometric analysis of galectin binding after incubation of BG B+ E. coli and two different BG B− E. coli reference strains obtained from a clinical laboratory with ∼0.1 μM Gal-8. (b–c) Incubation of (b) BG B+ E. coli or (c) BG B− E. coli strain 1 with 5 μM Gal-1, Gal-3, Gal-4, or Gal-8 as indicated. Viable bacteria were quantified by dilution plating, n=3, representative experiment in duplicate over 2 dilutions shown, error bars=SD. (d) Flow cytometric analysis following incubation of BG B+ E. coli, K. pneumoniae, P. aeruginosa, and S. aureus with ∼0.1 μM Gal-8. (e–g) Incubation of (e) K. pneumoniae, (f) P. aeruginosa, or (g) S. aureus with 5 μM Gal-1, Gal-3, Gal-4, or Gal-8 as indicated. Viable bacteria were quantified by dilution plating, n=3, representative experiment in duplicate over 2 dilutions shown, error bars=SD. (h–i) Incubation with or without 5 μM Gal-8 with (h) GFP+ P. aeruginosa alone or (i) GFP+ P. aeruginosa mixed with BG B+ E. coli followed by determination of percent GFP+ P. aeruginosa by flow cytometric analysis in a mixing experiment. Gated values of GFP+ bacteria treated with PBS (blue) or Gal-8 (red) are shown. (j) Quantification of percent GFP+ bacteria utilizing flow cytometric analysis obtained following incubation of Gal-8 with either GFP+ P. aeruginosa alone (P.a.) or GFP+ P. aeruginosa mixed with BG B+ E. coli (P.a. + BG B+ E.c.).
Figure 5. Gal-4 and Gal-8 specifically recognize blood group B antigen on blood group B positive E. coli.
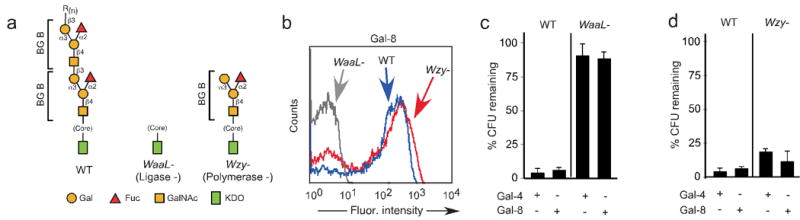
(a) Schematic of O antigen structures on wild type (WT) BG B+ E. coli and mutants of BG B+ E. coli WaaL− (Ligase-) and Wzy− (Polymerase-) lacking a complete O antigen. (b) Flow cytometric analysis following incubation of BG B+ E. coli and mutants WaaL− and Wzy− with ∼0.1 μM Gal-8. (c–d) Incubation of (c) WT and WaaL− mutant BG B+ E. coli or (d) WT and Wzy− mutant BG B+ E. coli with 5 μM Gal-4 or Gal-8 as indicated. Viable bacteria were quantified by dilution plating, n=3, representative experiment in duplicate over 2 dilutions shown, error bars=SD.
While these results show that Gal-4 and Gal-8 kill BG B+ E. coli in vitro, we used mice to test whether similar activities occur in vivo. We first examined whether the mouse galectin-4 (mGal-4) possesses similar ability to bind and kill BG B+ E. coli. mGal-4 recognized BG B+ E. coli and recognition was inhibited by lactose (Fig. 6a) and thiodigalactoside (TDG) (data not shown). Furthermore, mGal-4 recognition of BG B+ E. coli appeared to be specific to the BG B antigen as mGal-4 failed to recognize the WaaL− mutant (Fig. 6b), similar to human Gal-4 (data not shown). mGal-4 also displayed high binding toward blood group antigens (Fig. 6c). Importantly, mGal-4 recognition of BG B+ E. coli resulted in a substantial reduction in viability, which appeared to be specific to BG B antigen binding as mGal-4 failed to alter viability of the WaaL− mutant (Fig. 6d) and killing was inhibited by TDG (data not shown). However, mGal-4 killing was less potent when compared to human Gal-4, possibly due to the reduced affinity of mGal-4 for BG B when compared to BG A (Fig. 6c,d).
Figure 6. Gal-4 and Gal-8 specifically kill blood group B positive E. coli in vivo.
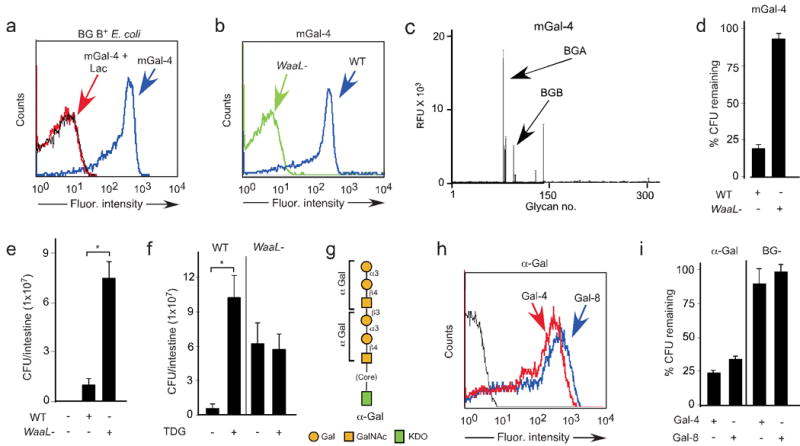
(a) Flow cytometric analysis following incubation of BG B+ E. coli with ∼0.1 μM mGal-4 with or without lactose. (b) Flow cytometric analysis following incubation of BG B+ E. coli and mutants WaaL− with ∼0.1 μM mGal-4. (c) mGal-4 binding to the CFG glycan microarray at 20 μg/ml (0.5 μM). BGB= blood group B glycans, BGA= blood group A glycans (See Supplementary Table 1). (d) Quantification of WT BG B+ and ΔwaaL mutant E. coli after incubation with ~5 μM mGal-4. Viable bacteria were quantified by dilution plating; n = 3 experiments; one representative experiment in duplicate over two dilutions is shown; error bars represent means ± s.d. (e) Live antibiotic-treated mice were fed PBS, Wild type (WT), or WaaL− mutant BG B+ E. coli. The number of viable bacteria in the intestine of mice sacrificed 24 h after feeding was quantified by dilution plating. * = p value 0.049. (f) Growth of WT and WaaL− mutant BG B+ E. coli in the presence and absence of TDG. * = p value 0.008. (g) Schematic of O antigen structures on α-Gal E. coli. (h) Flow cytometric analysis of α-Gal expressing bacteria after incubation of α-Gal–expressing bacteria with ~0.1 μM human Gal-4 or Gal-8. (i) Bar graph showing the percentage of α-Gal–expressing bacteria and BG B− bacteria remaining after incubation with 5 μM Gal-4 and Gal-8 as compared to PBS-treated control bacteria. Viable bacteria were quantified by dilution plating, n=3, representative experiment in duplicate over 2 dilutions shown, error bars=SD.
The selective killing of BG B+ E. coli by mGal-4 suggests that the WaaL− mutant should exhibit increased growth in vivo due to the inability of endogenous galectins to bind and kill these bacteria, whereas BG B+ E. coli should be limited in their growth due to killing by endogenous Gal-4 and Gal-8. It has been shown previously that Gal-4/Gal-8 are the only intestinal proteins that detectably bind β-galactosides22, but double knockout mice are not available for Gal-4/Gal-8 and such animals may not be viable. Thus, to specifically test the physiological functions of these intestinal galectins, we fed wild-type (WT) mice with BG B+ E. coli or WaaL− mutant. In this in vivo model, we first treated mice with streptomycin to deplete endogenous bacteria followed by feeding mice with the WT and WaaL− mutant strain bacteria. Importantly, the number of WT bacteria detected was significantly reduced in vivo compared to the WaaL− mutant (Fig. 6e) although both displayed equal growth kinetics in vitro (data not shown), which implicated a possible galectin-mediated process in vivo. The few bacteria isolated from mice inoculated with WT bacteria were positive for BG B antigen. Similarly, isolated bacteria were negative for the BG B antigen following introduction of the WaaL− mutant, indicating that the bacteria examined reflected those utilized during the inoculation (data not shown). To test the potential role of galectins in this process, we co-incubated BG B+ E. coli or the WaaL− with or without the inclusion of TDG, a non-metabolizable inhibitor of galectins, in vivo. Although TDG failed to alter BG B+ E. coli or the WaaL− in the absence of mGal-4 (data not shown), TDG significantly increased BG B+ E. coli viability in vivo while failing to alter the WaaL− viability (Fig. 6f). These results strongly suggest that endogenous galectins specifically alter BG B+ E. coli in vivo. While blood group antigens are expressed to some extent in glycosphingolipids and mucins of the gastrointestinal tract23, it has been found that they are susceptible to degradation by bacterial-derived glycosidases24,25 and in infants this bacterial-induced degradation of blood group antigens is observed soon after weaning26. Thus, it is not likely that host blood group antigens, which are expressed at low levels, can complex all of the galectins present, since Gal-4 and Gal-8 are highly expressed in the intestinal tract27,28.
Although Gal-4 and Gal-8 appear to specifically kill BG B+ E. coli, whether Gal-4 or Gal-8 possess the ability to recognize and kill bacteria expressing other types of blood group antigens remained unknown. To test this we examined whether Gal-4 and Gal-8 could recognize and kill bacteria expressing the α1-3Gal epitope (α–Gal E. coli), a common glycan moiety found in many mammalian species (Fig. 6g). Similar to BG B+ E. coli, α–Gal E. coli glycans were recognized by Gal-4 and Gal-8 (Fig. 6h) and recognition was inhibited by TDG (data not shown). Furthermore, Gal-4 and Gal-8 recognition of α–Gal E. coli resulted in a considerable reduction in viability (Fig. 6i), although killing of α–Gal E. coli by Gal-4 and Gal-8 was reduced when compared to Gal-4 and Gal-8-mediated killing of BG B+ E. coli, suggesting possible reduced binding affinity toward this glycan epitope. Consistent with this, Gal-4 and Gal-8 only recognized α–Gal epitopes on the glycan array when incubated at higher concentrations7 (data not shown). Taken together, these results demonstrate that Gal-4 and Gal-8 possess the ability to specifically kill bacteria expressing common blood group-associated mammalian antigens.
Many human pathogens decorate their surfaces with diverse carbohydrate structures and many of these structures have similarities to human antigens, a common mechanism of both commensal and pathogenic organisms, used to render themselves immunologically inert. However, mechanisms must also be in place to prevent the overgrowth of any potential pathogens that are shielded from normal adaptive immune responses. Thus, the ability of Gal-4 and Gal-8 to specifically kill BG B+ E. coli extends previous observations suggesting critical roles for galectins in innate immunity14 and may reflect a common but unrealized feature of other innate immune lectins that provide direct protection against pathogens displaying particular self-antigens, where adaptive immunity cannot.
Similar to many innate immune factors, the galectins represent an ancient family of proteins present in a wide variety of species20. As galectins evolved long before the selection of adaptive immunity, it is intriguing to speculate that the types of carbohydrate modifications on some self-antigens, such as blood group antigens, may reflect the binding specificity of pre-existing innate immune factors such as the galectins. The generation of ABO(H) antigen diversity in the human population has been proposed to facilitate pathogen evasion during human evolution1. For example, differential expression of blood group ABO antigens in host tissues can differentially affect pathogen adhesion and infection, as recently shown for Helicobacter pylori29. However, this diversity might have arisen with a significant fitness cost as development of these antigens precludes adaptive immune responses against blood group-bearing pathogens. The ability of galectins to recognize blood group baring pathogens may have facilitated the selection of ABO(H) expression on human erythrocytes rather than alternative antigens that did not have the same pre-existing innate immune protection. In contrast, the ability of Gal-4 and Gal-8 to also kill α-Gal expressing bacteria importantly demonstrates that galectin-mediated killing is not limited to human blood group expressing bacteria and suggests that galectins may significantly impact the composition of multiple populations of intestinal bacteria thereby significantly modulating the intestinal microbiome. Future studies will exposure these intriguing possibilities.
Methods
Preparation of recombinant human galectins
We prepared Gal-1, Gal-3, Gal-4, Gal-4 domains, Gal-8, Gal-8 domains, and Gal-8 mutants as outlined previously6,30. We generated Gal-8R69H and Gal-8R233H using appropriate primers as described previously. We purified galectins to apparent homogeneity by affinity chromatography on lactosyl-separose (Sigma) as observed by SDS-PAGE (Supplementary Figure 1). We derivatized all galectins by addition of EZ-link ™ Sulfo-NHS-LC-Biotin (Sulfosuccinimidyl-6-(biotinamido) hexanoate) (Pierce) as described previously for detection.
Glycan array preparation and analysis
We obtained from the Consortium for Functional Glycomics (http://www.functionalglycomics.org/) glycan microarrays prepared as described previously31. We determined galectin recognition of glycans on the printed glycan microarray at the indicated concentrations, 0.2 μM Gal-1, 0.2 μM Gal-3, 0.5 μM Gal-4, or 0.02 μM Gal-8, as outlined in Supplementary Methods.
Flow cytometric analysis
To examine potential binding by each galectin, we grew bacteria to mid-log phase in Luria Bertani (LB) media (Fisher), and resuspended 108 cells/ml in PBS pH 7.4 with biotinylated Gal-1, Gal-3, Gal-4, Gal-4 domains, Gal-8, Gal-8 domains or mutant Gal-4 at concentrations of ∼0.1 μM at 4 °C for 30 min. As a control we also incubated bacteria with 20 mM lactose along with galectins. Following incubation, we washed bacteria 3× and incubated with Alexa Fluor-488 streptavidin or Alexa Fluor-633 streptavidin (Molecular Probes) at 4 °C for 30 min. We washed bacteria 2×, and resuspended in 400 μL PBS for analysis by flow cytometry using a FACSCalibur flow cytometer (BD Biosciences). We analyzed results using CellQuest software (BD Biosciences).
Growth and treatment of bacteria
We received blood group B positive E. coli, E. coli O86 (BG B+ E. coli), as well as mutant strains and α-Gal E. coli, from Ohio State University. We obtained clinical reference strains, including the two blood group negative strains of E. coli (ATCC# 25922, ATCC# 35218), K. pneumoniae (ATCC#700603), P. aeruginosa (ATCC# 27853), and S. aureus (ATCC# 29213) from the Emory University Clinical Microbiology lab. When assaying potential anti-microbial effects of galectins, we grew all bacteria to mid-log phase in LB media (Fisher) and incubated 108 cells/ml with the indicated concentrations of each galectin for 2 h at 37 °C, unless otherwise indicated. Following incubation with each respective galectin, we determined the number of viable bacteria by dilution plating and CFU enumeration. We incubated 20 mM lactose or sucrose with the galectin as indicated for 10 min prior to incubation with bacteria.
Mixed population
To assess the specificity of Gal-8-mediated killing in a mixed population, we mixed BG B+ E. coli with GFP expressing P. aeruginosa (see Supplementary Methods) in a 4:1 ratio. We incubated this mixture in the presence or absence of Gal-8 for 2 h at 37 °C followed by dilution plating for CFU enumeration or flow cytometric examination of percent GFP positive cells.
Preparation for video analysis
We added to BG B+ E. coli, grown to log phase, either PBS control or 5 μM Gal-8 at 37 °C. We acquired videos at 100× magnification using an Olympus BX51 phase contrast microscope equipped with an Olympus DP71 camera (Olympus America Inc.). We captured movies using the Olympus movie sequence editor DP Controller 3.2.1.276 (Olympus America Inc.) and exported resulting images as a movie sequence.
Bacteria viability analysis
We incubated BG B+ E. coli for 30 min with PBS control or 5 μM Gal-8 at 37 °C. We added 20 mM lactose to halt treatment and reduce agglutination. We washed bacteria 1× with PBS and centrifuged at 15,000 rpm in a table-top centrifuge. We resuspended bacteria in PBS and added 1 μl of propidium iodide (PI) solution (1:20 PI from Live/Dead viability kit (Invitrogen) to PBS). We incubated bacteria in the dark at room temperature for 15 min and visualized by fluorescence microscopy at 100× magnification.
SEM and TEM imaging
We incubated BG B+ E. coli 30 min with PBS control or 5 μM Gal-8 at 37 °C then added 20 mM lactose to halt treatment and reduce agglutination. We washed bacteria 2× with PBS to remove debris. Following this we prepared the treated bacteria samples for analysis of morphological changes by either SEM or TEM as outlined in Supplementary Methods.
Animal Studies
We conducted all experiments in accordance with the guidelines of the Animal Care Committee from the University (process # 09.1.543.53.5) We obtained C57BL/6 Specific Pathogens Free (SPF) mice from the animal facilities of the Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Brazil. For in vivo experiments, we treated mice with streptomycin (5 g/L) for 48 h, as described in Supplementary Methods. Mice were then fed with either wild type or WaaL− BG B+ E. coli with or without addition of thiodigalactoside (TDG). After 24 h, mice were sacrificed and the number of viable bacteria in the intestine was determined by dilution plating and CFU enumeration.
Supplementary Material
Acknowledgments
We thank Sandy Cummings and Margaret Willard (Emory University) for technical assistance. We also thank Peng George Wang (Ohio State University) for providing the original E. coli O86:B7 strain, as well as mutant versions of this strain lacking expression of either the Wzy or WaaL genes. We obtained P. aeruginosa strain 8830 from Dr. Ananda Chakrabaty (University of Illinois College of Medicine) and plasmid pSMC21 encoding GFP from Dr. O'Toole (Dartmouth Medical School), We also thank the staff at the Robert P. Apkarian Integrated Electron Microscopy Core Facility (Emory University) for their help with electron microscopy. This work was supported by grants from the US National of Institutes of Health to R.D.C. (HL085607), D.F.S. (GM085448), and by resources from the Consortium for Functional Glycomics (Core D and Core H), funded by the US National Institute of General Medical Sciences/U.S. National of Institutes of Health (GM62116).
Footnotes
Author Contributions: S.R.S. and C.M.A. planned the project along with R.D.C. and carried out and analyzed the experiments, together with M.D.B, L.C.R., J.P.G., J.H.M., B.X., C.R., T.J., R.J.M., and D.F.S., who also helped to perform the experiments and provided critical support. S.R.S., C.M.A., J.H.M., and R.D.C. wrote the manuscript, which was additionally edited and commented on by the other authors.
Supplementary Information: Supplementary information accompanies this paper.
References
- 1.Rowe JA, et al. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proc Natl Acad Sci U S A. 2007;104:17471–6. doi: 10.1073/pnas.0705390104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamamoto F, Clausen H, White T, Marken J, Hakomori S. Molecular genetic basis of the histo-blood group ABO system. Nature. 1990;345:229–33. doi: 10.1038/345229a0. [DOI] [PubMed] [Google Scholar]
- 3.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–30. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol. 2002;2:77–84. doi: 10.1038/nri723. [DOI] [PubMed] [Google Scholar]
- 5.van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9:593–601. doi: 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- 6.Stowell SR, et al. Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J Biol Chem. 2008;283:10109–23. doi: 10.1074/jbc.M709545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stowell SR, et al. Dimeric Galectin-8 induces phosphatidylserine exposure in leukocytes through polylactosamine recognition by the C-terminal domain. J Biol Chem. 2008;283:20547–59. doi: 10.1074/jbc.M802495200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reeves P. Role of O-antigen variation in the immune response. Trends Microbiol. 1995;3:381–6. doi: 10.1016/s0966-842x(00)88983-0. [DOI] [PubMed] [Google Scholar]
- 9.Springer GF, Williamson P, Brandes WC. Blood Group Activity of Gram-Negative Bacteria. J Exp Med. 1961;113:1077–1093. doi: 10.1084/jem.113.6.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Springer GF, Horton RE. Blood group isoantibody stimulation in man by feeding blood group-active bacteria. J Clin Invest. 1969;48:1280–91. doi: 10.1172/JCI106094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garratty G. Blood groups and disease: a historical perspective. Transfus Med Rev. 2000;14:291–301. doi: 10.1053/tmrv.2000.16228. [DOI] [PubMed] [Google Scholar]
- 12.Yi W, et al. Escherichia coli O86 O-antigen biosynthetic gene cluster and stepwise enzymatic synthesis of human blood group B antigen tetrasaccharide. J Am Chem Soc. 2005;127:2040–1. doi: 10.1021/ja045021y. [DOI] [PubMed] [Google Scholar]
- 13.Wooters MA, Hildreth MB, Nelson EA, Erickson AK. Immunohistochemical characterization of the distribution of galectin-4 in porcine small intestine. J Histochem Cytochem. 2005;53:197–205. doi: 10.1369/jhc.4A6439.2005. [DOI] [PubMed] [Google Scholar]
- 14.Vasta GR. Roles of galectins in infection. Nat Rev Microbiol. 2009;7:424–38. doi: 10.1038/nrmicro2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohatsu L, Hsu DK, Jegalian AG, Liu FT, Baum LG. Galectin-3 induces death of Candida species expressing specific beta-1,2-linked mannans. J Immunol. 2006;177:4718–26. doi: 10.4049/jimmunol.177.7.4718. [DOI] [PubMed] [Google Scholar]
- 16.Thiel S, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997;386:506–10. doi: 10.1038/386506a0. [DOI] [PubMed] [Google Scholar]
- 17.Eshkar Sebban L, et al. The involvement of CD44 and its novel ligand galectin-8 in apoptotic regulation of autoimmune inflammation. J Immunol. 2007;179:1225–35. doi: 10.4049/jimmunol.179.2.1225. [DOI] [PubMed] [Google Scholar]
- 18.Hadari YR, et al. Galectin-8. A new rat lectin, related to galectin-4. J Biol Chem. 1995;270:3447–53. doi: 10.1074/jbc.270.7.3447. [DOI] [PubMed] [Google Scholar]
- 19.Ribi HO, Ludwig DS, Mercer KL, Schoolnik GK, Kornberg RD. Three-dimensional structure of cholera toxin penetrating a lipid membrane. Science. 1988;239:1272–6. doi: 10.1126/science.3344432. [DOI] [PubMed] [Google Scholar]
- 20.Houzelstein D, et al. Phylogenetic analysis of the vertebrate galectin family. Mol Biol Evol. 2004;21:1177–87. doi: 10.1093/molbev/msh082. [DOI] [PubMed] [Google Scholar]
- 21.Guo H, et al. Molecular analysis of the O-antigen gene cluster of Escherichia coli O86:B7 and characterization of the chain length determinant gene (wzz) Appl Environ Microbiol. 2005;71:7995–8001. doi: 10.1128/AEM.71.12.7995-8001.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gitt MA, et al. Galectin-4 and galectin-6 are two closely related lectins expressed in mouse gastrointestinal tract. J Biol Chem. 1998;273:2954–60. doi: 10.1074/jbc.273.5.2954. [DOI] [PubMed] [Google Scholar]
- 23.Hansson GC. Structural aspects of blood group glycosphingolipids in the gastrointestinal tract. Adv Exp Med Biol. 1988;228:465–94. doi: 10.1007/978-1-4613-1663-3_17. [DOI] [PubMed] [Google Scholar]
- 24.Hoskins LC, Boulding ET. Degradation of blood group antigens in human colon ecosystems. I. In vitro production of ABH blood group-degrading enzymes by enteric bacteria. J Clin Invest. 1976;57:63–73. doi: 10.1172/JCI108270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoskins LC, Boulding ET. Degradation of blood group antigens in human colon ecosystems. II. A gene interaction in man that affects the fecal population density of certain enteric bacteria. J Clin Invest. 1976;57:74–82. doi: 10.1172/JCI108271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larson G, Watsfeldt P, Falk P, Leffler H, Koprowski H. Fecal excretion of intestinal glycosphingolipids by newborns and young children. FEBS Lett. 1987;214:41–4. doi: 10.1016/0014-5793(87)80009-1. [DOI] [PubMed] [Google Scholar]
- 27.Huflejt ME, Leffler H. Galectin-4 in normal tissues and cancer. Glycoconj J. 2004;20:247–55. doi: 10.1023/B:GLYC.0000025819.54723.a0. [DOI] [PubMed] [Google Scholar]
- 28.Nagy N, et al. Galectin-8 expression decreases in cancer compared with normal and dysplastic human colon tissue and acts significantly on human colon cancer cell migration as a suppressor. Gut. 2002;50:392–401. doi: 10.1136/gut.50.3.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linden S, et al. Role of ABO secretor status in mucosal innate immunity and H. pylori infection. PLoS Pathog. 2008;4:e2. doi: 10.1371/journal.ppat.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stowell SR, et al. Galectin-1 induces reversible phosphatidylserine exposure at the plasma membrane. Mol Biol Cell. 2009;20:1408–18. doi: 10.1091/mbc.E08-07-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blixt O, et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc Natl Acad Sci U S A. 2004;101:17033–8. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.