

Abstract
The transcriptional corepressor SnoN is a critical regulator of axonal morphogenesis, but how SnoN drives axonal growth is unknown. Here, we report that gene-profiling analyses in cerebellar granule neurons reveal that the large majority of genes altered upon SnoN knockdown are surprisingly downregulated, suggesting that SnoN may activate transcription in neurons. Accordingly, we find that the transcriptional coactivator p300 interacts with SnoN, and p300 plays a critical role in SnoN-induced axon growth. We also identify the gene encoding the signaling scaffold protein Ccd1 as a critical target of SnoN in neurons. Ccd1 localizes to the actin cytoskeleton, is enriched at axon terminals in neurons, and activates the axon growth-promoting kinase JNK (c-Jun N-terminal protein kinase). Knockdown of Ccd1 in neurons reduces axonal length and suppresses the ability of SnoN to promote axonal growth. Importantly, Ccd1 knockdown in rat pups profoundly impairs the formation of granule neuron parallel fiber axons in the rat cerebellar cortex in vivo. These findings define a novel SnoN–Ccd1 link that promotes axonal growth in the mammalian brain, with important implications for axonal development and regeneration.
Introduction
The growth of axons is a fundamental biological process that is a prerequisite for the establishment of neuronal connectivity in the developing nervous system. Neurons lose the capacity to grow axons in the mature mammalian CNS, a property that contributes significantly to the inability of the CNS to recover after injury and disease. Therefore, elucidation of the mechanisms that control the growth of axons is critical for our understanding of brain development and disease.
Although the extrinsic factors that control axon guidance and growth have been the subject of intense scrutiny, growing evidence suggests that cell-intrinsic mechanisms also play a central role in the regulation of axonal growth (Butler and Tear, 2007; Polleux et al., 2007). Among cell-intrinsic mechanisms, the transcriptional regulator SnoN has emerged as a critical regulator of axonal morphogenesis in mammalian neurons (Stegmüller et al., 2006, 2008; Kim and Bonni, 2007). SnoN drives the growth of axons in postmitotic rat cerebellar granule neurons and hippocampal neurons. Importantly, SnoN promotes the development of axons in the mammalian brain as demonstrated by the dramatic impairment of granule neuron parallel fiber axons in the cerebellar cortex in rats subjected to SnoN knockdown in vivo (Stegmüller et al., 2006).
The mechanisms that regulate SnoN function in postmitotic neurons are beginning to be characterized. The E3 ubiquitin ligase Cdh1–APC stimulates the ubiquitination and consequent degradation of SnoN in neurons and thereby inhibits the growth of axons (Stegmüller et al., 2006). In addition, TGFβ–Smad2 signaling operates in a shared pathway with Cdh1–APC to promote SnoN degradation and thereby limits axonal growth (Stegmüller et al., 2008). Collectively, these studies suggest that SnoN plays a critical role in orchestrating neuronal responses to signals that influence axonal growth.
A major question that has remained to be addressed is how SnoN drives axonal growth and development. As a transcriptional regulator, SnoN is anticipated to control a program of genes whose products directly control the morphology of axons. Investigations of SnoN in cells outside the nervous system have highlighted SnoN′s transcriptional corepressor activity in promoting cell proliferation and oncogenic transformation (Luo, 2004; Pot and Bonni, 2008). However, growing evidence suggests that SnoN may also operate as a transcriptional coactivator to mediate TGFβ-induced transcription and cell cycle arrest in proliferating cells (Sarker et al., 2005, 2008). These observations raise the important question of whether SnoN represses or activates a program of genes in neurons to drive axonal growth. Importantly, although SnoN has been implicated in the control of diverse biological responses including the promotion of axonal growth, a function for a SnoN target gene in the biological effects of SnoN has remained to be identified.
In this study, we identify a mechanism by which SnoN promotes axonal growth in the mammalian brain. In microarray analyses of granule neurons in which SnoN knockdown is triggered, we find that the expression of a large number of genes is downregulated, suggesting that SnoN may activate transcription in neurons. Consistent with the conclusion that SnoN can activate transcription, we find that SnoN forms a complex with the transcriptional coactivator p300. Importantly, we find a requirement for p300 in promoting axon growth downstream of SnoN in granule neurons. Among the SnoN-dependent genes subjected to further analysis, we identify the gene encoding the actin-binding protein Ccd1 as a critical SnoN target gene in neurons. Knockdown of SnoN or p300 significantly reduces Ccd1 promoter-mediated transcription in granule neurons, suggesting that SnoN and p300 are required for transcription of the Ccd1 gene in neurons. We also find that Ccd1 is enriched at the axon terminal in neurons and activates the protein kinase JNK (c-Jun N-terminal protein kinase), a signaling molecule that promotes axon growth (Oliva et al., 2006). Knockdown of Ccd1 significantly reduces axon length in primary granule neurons and dramatically impairs the formation of granule neuron parallel fibers in the rat cerebellar cortex, suggesting that Ccd1 phenocopies the effect of SnoN knockdown on axonal development. Ccd1 knockdown also suppresses SnoN-induced axon growth. Together, these findings define the signaling scaffold protein Ccd1 as a key target of SnoN that may mediate the ability of SnoN to drive axonal morphogenesis in the brain.
Materials and Methods
Plasmids and antibody.
Ccd1 was cloned from rat brain cDNA into pCMV5 with Flag tag to construct Flag–Ccd1 overexpression plasmid. Ccd1 was also cloned into pEGFP–C1 for GFP–Ccd1 expression plasmid. The U6/ccd1i RNAi plasmid was generated with two primers (5′-AGACAGAGGAATGCCAGTGTttcaagcttACACTGGCATTCCTCTGTCTcttttg-3′ and 5′-aattcaaaagAGACAGAGGAATGCCAGTGTaagcttgaaACACTGGCATTCCTCTGTCT-3′), here capitalized letters indicate Ccd1-targeting sequence. The U6/p300i1 RNAi plasmid was generated using two primers: (5′-GACTACCCTATCAAGTAAAcaagttaacTTTACTTGATAGGGTAGTCctttttg-3′ and 5′-aattcaaaaagGACTACCCTATCAAGTAAAgttaacttgTTTACTTGATAGGGTAGTC-3′); capitalized letters indicate the p300-targeting sequence. The U6/p300i2 RNAi plasmid was generated using two primers: (5′-CACGAACTAGGAAAGAAAcaagttaacTTTCTTTCCTAGTTCGTGctttttg-3′ and 5′-aattcaaaaagCACGAACTAGGAAAGAAAgttaacttgTTTCTTTCCTAGTTCGTG-3′); capitalized letters indicate the p300-targeting sequence. The upstream 1kb region of the rat Ccd1 gene was cloned into pGL3basic (Promega) to generate the Ccd1–luciferase reporter gene using PCR amplification with two primers (5′-GTCATGCTCGAGTGTATCCAAGCAGAACAATCCTTTA-3′ and 5′-TGCGATAAGCTTTTCAGGAAGCCTGCTTCCCTG-3′). The Ccd1 antibody has been described previously (Shiomi et al., 2005). The Flag M2 (Sigma), hemagglutinin (HA, Covance), GFP (Invitrogen), ERK1/2 (Cell Signaling), dsRed (Clontech), SnoN (Santa Cruz), and 14-3-3β (Santa Cruz) antibody were purchased.
Microarray analysis.
To generate lentiviruses, 293T cells were transfected with the control pll3.7-cmvGFP vector or the pll3.7/snoni-cmvGFP plasmid together with plasmids encoding RRE, REV/RSV, and VSVG. Supernatant was harvested and viruses were concentrated by ultracentrifugation. Primary neurons were transduced at 0 d in vitro (DIV0) and exposed to virus for 5 d. RNA was isolated using Trizol (Invitrogen), subjected to additional purification using RNeasy (Qiagen), and subjected to microarray analyses using rat expression array 230 2.0 (Affymetrix) chips.
RT-PCR.
Superscript III First-Strand Synthesis system (Invitrogen) was used for cDNA synthesis. Lightcycler 480 and SYBR green master reagent (Roche) were used for quantitative PCR. The following primers were used for the RT-PCR analysis: RTccd1-Fwd (5′-catgaccctcagcatcacac-3′), RTccd1-Rev (5′-agctctgggagtgaggcaac-3′), RTtropomyosin3-Fwd (5′-ctgaggtggcctccttgaac-3′), RTtropomyosin3-Rev (5′-tgcctcttctgcaatgtgct-3′), RTcks2-Fwd (5′-accggcatgtcatgttaccc-3′), RTcks2-Rev (5′-ttttggaagaggccgtctaaag-3′), RThnRNPA1-Fwd (5′-agccagagaggtcgaagtgg-3′), RThnRNPA1-Rev (5′-agcttccaccacctccaaaa-3′), RTCXCL12-Fwd (5′-gtgacggtaagccagtcagc-3′), RTCXCL12-Rev (5′-tttcgggtcaatgcacactt-3′).
Immunoprecipitation.
293T cell line was maintained in DMEM (Invitrogen) with high glucose and l-glutamine, supplemented with 10% FBS. 293T cells were transiently transfected using the calcium phosphate method. Two days later, cells were lysed in 50 mm Tris/HCl, 150 mm NaCl, 1 mm EDTA (TNE) buffer containing 0.5% Triton X-100 with protease and phosphatase inhibitors. Total lysates were cleared by centrifugation at 14,000 × g for 10 min at 4°C, and protein content was determined by the Bradford assay. Ninety percent of the supernatant was subjected to immunoprecipitation using mouse anti-Flag or anti-GFP antibody. The protein compositions of total cell lysate and immunoprecipitation samples were resolved by SDS-PAGE followed by immunoblotting.
Primary neuron cultures and transfections.
Granule neurons were prepared from isolated cerebella of P6 Long–Evans rat pups as described previously (Shalizi et al., 2003). For morphological assays, granule neurons were transfected either 8 h after plating or at DIV2 with a modified calcium phosphate method as described previously (Gaudillière et al., 2004; Konishi et al., 2004).
Luciferase assays.
Luciferase assays were performed as described previously (Shalizi et al., 2006) with minor modifications. Granule neurons were transfected with Ccd1–luciferase reporter plasmid or the control vector pGL3basic and renilla luciferase-elongation factor (EF) promoter and the indicated RNAi plasmids, and an expression construct for Bcl-XL at 2 d in vitro. Cells were lysed 72 h after transfection. Firefly- and renilla-luciferase activities were determined using a dual-luciferase assay kit (Promega) according to the instructions of the manufacturer.
Morphometry.
The morphology of axons of cerebellar granule neurons was performed by capturing images of the neurons in a blinded manner using a Nikon eclipse TE2000 epifluorescence microscope. Measurements of axon length were performed using SPOT imaging software as described previously (Gaudillière et al., 2004; Konishi et al., 2004).
In vivo electroporation.
In vivo electroporation of P3 Sprague Dawley rat pups was performed as described previously (Konishi et al., 2004).
Results
Identification of SnoN-regulated genes in neurons
We investigated the mechanism by which SnoN promotes the growth of axons in granule neurons of the rat cerebellar cortex. The development of granule neurons in the cerebellar cortex follows a stereotypical pattern (Hatten and Heintz, 1995; Altman and Bayer, 1997). Granule neurons are generated in the external granule layer (EGL) of the developing cerebellar cortex, where they begin to extend axons along the coronal plane. As granule neurons migrate inward toward the internal granule layer (IGL), their axons form “T”-shaped parallel fibers in the molecular layer. The anatomical layout of granule neuron axons provides a straightforward system for the study of axonal morphogenesis in the brain. In primary cultures of granule neurons, axons are easily identified based on morphology and markers (Gaudillière et al., 2004; Konishi et al., 2004). Because granule neurons are found in large numbers, primary cultures also facilitate the analysis of mechanisms of neuronal development in a relatively homogeneous population of neurons.
To characterize the mechanism by which SnoN promotes the growth of axons, we performed gene-profiling analyses of primary rat cerebellar granule neurons in which SnoN knockdown was triggered. We generated a lentiviral construct that encodes short hairpin RNAs (shRNAs) that induce the knockdown of rat SnoN. We confirmed that lentiviral infection of primary rat cerebellar granule neurons with the SnoN RNAi construct dramatically reduced the expression of endogenous SnoN as determined by immunoblotting (Fig. 1A). We next subjected total RNA isolated from control lentiviral-infected and SnoN shRNA-expressing granule neurons to Affymetrix rat 230 2.0 arrays. SnoN knockdown was confirmed in the microarray analyses (Fig. 1C). Surprisingly, we found that the large majority of altered genes were downregulated upon SnoN knockdown (Fig. 1B). An annotated list of some of these genes is shown in Figure 1C. A subset of these genes was examined and validated to undergo downregulation upon SnoN knockdown in primary granule neurons (Fig. 1D). These results suggest that SnoN, commonly known for its transcriptional repressive activity, may activate transcription of genes in neurons.
Figure 1.
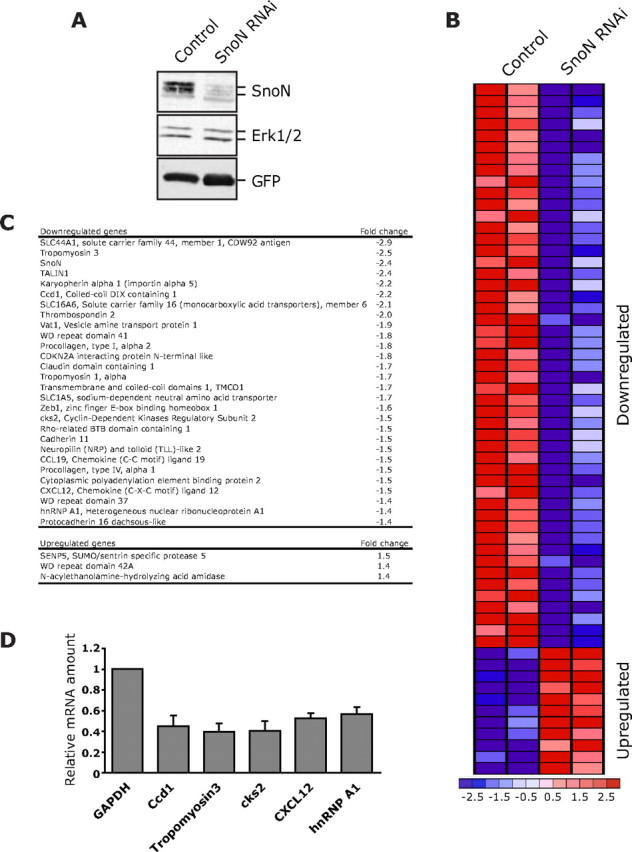
Identification of SnoN-regulated genes in neurons. A, Lentiviral-mediated SnoN RNAi induced knockdown of endogenous SnoN in neurons. Lysates of rat cerebellar granule neurons transduced with a SnoN RNAi lentivirus that also encodes GFP or the control GFP virus were subjected to immunoblotting using the SnoN, ERK1/2, or GFP antibody. ERK1/2 served as a loading control. B, A heat map of genes altered by >1.2-fold upon SnoN knockdown compared with control virus in granule neurons. The majority (80%) of genes altered upon SnoN knockdown were downregulated. C, Selected downregulated genes (top) and upregulated genes (bottom) upon SnoN knockdown in granule neurons are listed with fold change in their expression level. D, Lysates of granule neurons transfected by a nucleofection method with the SnoN RNAi plasmid (U6/snoni) or control U6 plasmid were subjected to real-time RT-PCR to measure the mRNA levels of the indicated subset of genes. The mRNA level for each gene was normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and shown as the relative ratio of normalized mRNA levels in SnoN knockdown neurons compared with control-transfected neurons. The value from three independent experiments is shown as mean + SEM. SnoN knockdown triggered the downregulation of the indicated genes.
Notably, a number of genes that were downregulated in SnoN knockdown neurons, including thrombospondin 2, procollagen type I, α 2, and Zeb1 are transcriptionally responsive to TGFβ signaling (Chang and Goldberg, 1995; Negoescu et al., 1995; Shirakihara et al., 2007). Consistent with this observation, we found that SnoN stimulates the expression of a TGFβ-responsive reporter gene and conversely SnoN knockdown reduces the expression of the reporter gene in granule neurons (Sarker et al., 2008). Together, our results suggest that SnoN may activate the expression of TGFβ-responsive genes in neurons.
Since SnoN′s transcriptional repressive activity is reportedly mediated via its association with other transcriptional corepressors (Luo, 2004; Pot and Bonni, 2008), we asked whether SnoN has the capacity to associate with transcriptional coactivators. In coimmunoprecipitation analyses, we found that SnoN robustly forms a physical complex with the transcriptional coactivator p300 (Fig. 2A), suggesting that SnoN can activate transcription. To determine if association with p300 is relevant to SnoN function in the promotion of axon growth, we assessed the effect of p300 knockdown in granule neurons. The p300 gene is expressed in neurons including granule neurons of the cerebellar cortex as determined by RT-PCR and in situ analyses (data not shown; Allen Brain Atlas). We used a plasmid-based method of RNAi to induce the acute knockdown of p300 (Gaudilliere et al., 2002). Expression of shRNAs targeting distinct regions of p300 induced the knockdown of p300 (Fig. 2B). We next monitored the effect of p300 knockdown on the development of axons and dendrites in granule neurons. As established in our previous studies, axons and dendrites are easily distinguishable based on their morphological appearance and process-specific markers (Gaudillière et al., 2004; Konishi et al., 2004). We found that p300 knockdown led to a reduction in the length of axons (Fig. 2C). Morphometric analyses confirmed that total axon length in p300 knockdown neurons was significantly lower than that in control transfected neurons (Fig. 2D). Thus, p300 knockdown phenocopies the effect of SnoN knockdown on axon growth in neurons (Stegmüller et al., 2006). In contrast to the inhibitory effect of p300 knockdown on axonal length, p300 knockdown had little effect on total dendrite length or neuronal survival (Fig. 2E) (data not shown). In other experiments, we found that overexpression of p300 had little effect on axon length, suggesting that p300 overexpression is not sufficient to induce axon growth (data not shown). Together, these results show that p300 at its endogenous levels plays a critical role in axon growth in neurons.
Figure 2.
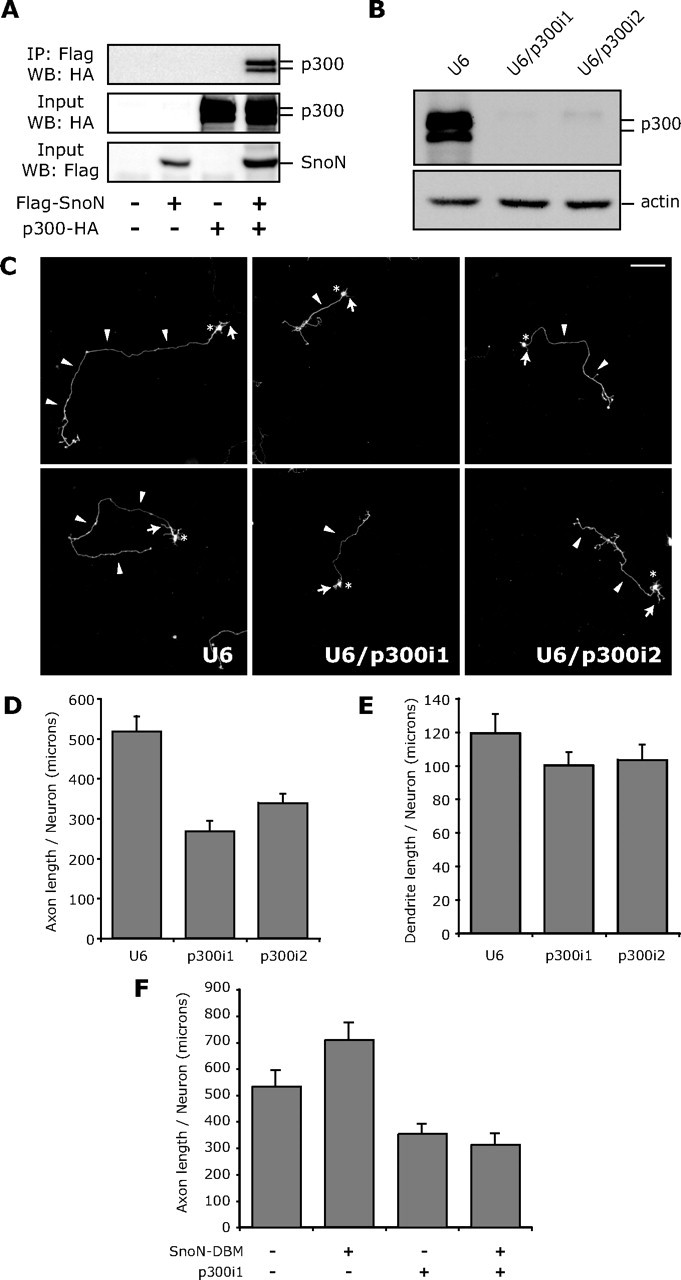
The transcriptional coactivator p300 associates with SnoN and promotes axon growth. A, Lysates of 293T cells transfected with an expression plasmid encoding Flag-tagged SnoN or the control vector alone or together with an expression plasmid encoding HA-tagged p300 were immunoprecipitated (IP) using the Flag antibody followed by immunoblotting (IB) with the HA antibody. Input lysates were also immunoblotted with the Flag or HA antibody. SnoN robustly interacted with p300. B, Lysates of 293T cells transfected with the U6, U6/p300i1, or U6/p300i2 RNAi plasmid together with a plasmid expressing p300–HA were immunoblotted with the HA or actin antibody. C, Granule neurons were transfected with the control U6 (left), U6/p300i1 (middle), or U6/p300i2 RNAi plasmid (right) together with a GFP expression plasmid. Four days after transfection, granule neurons were subjected to immunocytochemistry using the GFP antibody. Neurons in which p300 knockdown was induced had shorter axons than control-transfected neurons. Arrowheads, arrows, and asterisks indicate axons, dendrites, and cell bodies, respectively. Scale bar, 100 μm. D, Morphometric analyses of granule neurons transfected and analyzed as in C. Quantification of axon length is shown as mean + SEM. Axon length was significantly reduced in p300 knockdown neurons compared with control-transfected neurons (ANOVA, p < 0.05). Total number of neurons measured = 267. E, Granule neurons transfected with the U6, U6/p300i1, or U6/p300i2 RNAi plasmid together with the GFP expression plasmid were subjected to immunocytochemistry using the GFP antibody followed by morphometric analysis of total dendrite length. No significant difference in total dendrite length between p300 knockdown and control-transfected neurons was found. Total number of neurons measured = 149. F, Granule neurons transfected with the control U6 or U6/p300i1 RNAi plasmid together with the SnoN–DBM expression plasmid or control vector were analyzed as in D. SnoN–DBM expression increased axon length compared with control-transfected neurons (ANOVA; p < 0.05). Knockdown of p300 significantly reduced axon length in the background of SnoN–DBM expression (ANOVA; p < 0.05). Total number of neurons measured = 218 neurons. These results show that p300 knockdown overrides the ability of SnoN–DBM to stimulate axon growth.
To determine the relationship of SnoN with p300 in the control of axonal growth, we tested the effect of p300 knockdown on the ability of a constitutively active form of SnoN, in which the Cdh1-recognition motif is mutated [SnoN–D-box mutant (DBM)] (Stegmüller et al., 2006), to promote axonal growth. As expected, expression of SnoN–DBM in granule neurons induced the growth of axons (Fig. 2F) (Stegmüller et al., 2006). However, knockdown of p300 significantly reduced axon length in neurons in which SnoN–DBM was expressed to levels that were similar to those in p300 knockdown neurons (Fig. 2F). Thus, p300 knockdown overrides the ability of SnoN–DBM to stimulate axonal growth. These results suggest that the transcriptional coactivator p300 is required for SnoN-induced axon growth. Collectively, our findings support the model that SnoN may activate transcription because of its association with p300 and thereby promotes a program of gene expression dedicated to the growth of axons.
Identification of Ccd1 as a downstream target gene of SnoN in neurons
Among the SnoN-downregulated genes, we focused on the gene encoding the actin-binding protein Ccd1 (Wang et al., 2006). Ccd1 contains a coiled-coil domain and a DIX domain that is also found in Dishevelled and Axin (Shiomi et al., 2003). Ccd1 has been suggested to act as a scaffold-signaling protein that cooperates with Dishevelled to promote early embryonic zebrafish neural patterning (Shiomi et al., 2003). We asked whether Ccd1 might mediate the ability of SnoN to promote axonal morphogenesis in the mammalian brain. We first characterized the expression of Ccd1 in neurons.
Ccd1 mRNA was found in primary granule neurons as determined by RT-PCR and in situ analyses (data not shown; Allen Brain Atlas). In addition, immunoblotting of primary granule neurons revealed a Ccd1-immunoreactive band that was deemed to be specific based on knockdown by RNAi (Fig. 3A,B). Corroborating these results, Ccd1 mRNA has been reported to be expressed widely in neurons during development and adulthood by in situ analyses (Soma et al., 2006). Collectively, these observations show that Ccd1 is expressed in postmitotic neurons in the mammalian brain. In addition to demonstrating that Ccd1 mRNA expression is dependent on SnoN expression (Fig. 1D), we also confirmed that SnoN knockdown led to the reduction of Ccd1 protein in granule neurons (Fig. 3C).
Figure 3.

Identification of Ccd1 as a SnoN-regulated downstream target gene. A, Lysates of granule neurons cultured for the indicated DIV were immunoblotted with the Ccd1 or 14-3-3β antibody. Cultured days in vitro are indicated. B, Lysates of granule neurons transfected by nucleofection method with the Ccd1 RNAi plasmid (pSuper/ccd1i) or control pSuper plasmid were immunoblotted with the Ccd1 or ERK1/2 antibody. Ccd1 RNAi reduced the Ccd1-immunoreactive band, specifically. C, Lysates of granule neurons transfected by nucleofection with the SnoN RNAi (U6/snoni) or control U6 plasmid were immunoblotted with the SnoN, Ccd1, or 14-3-3β antibody. SnoN knockdown triggered downregulation of Ccd1 protein in neurons. D, Granule neurons were transfected with the SnoN RNAi (U6/snoni) or control U6 plasmid together with the Ccd1–luciferase reporter or the control vector pGL3basic and the EF renilla reporter, the latter serving as an internal control for transfection efficiency. The level of Ccd1–luciferase reporter gene expression was significantly reduced in SnoN knockdown neurons compared with control-transfected neurons (ANOVA; p < 0.01; n = 3). E, Granule neurons were transfected with the p300 RNAi (U6/p300i1) or control U6 plasmid together with the Ccd1–luciferase reporter or the control vector pGL3basic and the EF renilla reporter, the latter serving as an internal control for transfection efficiency. The level of Ccd1–luciferase reporter gene expression was significantly reduced in p300 knockdown neurons compared with control-transfected neurons (ANOVA; p < 0.05; n = 3).
We next determined if SnoN regulates the expression of Ccd1 at the level of transcription. We generated a luciferase reporter gene that is controlled by the Ccd1 promoter (Ccd1–luciferase) and measured the effect of knockdown of SnoN or knockdown of the associated transcriptional coactivator p300 on Ccd1 promoter-mediated transcription. SnoN knockdown significantly reduced the expression of the Ccd1–luciferase reporter gene in granule neurons (Fig. 3D). Likewise, knockdown of p300 significantly reduced the expression of the Ccd1–luciferase reporter gene in neurons (Fig. 3E). Collectively, these results suggest that SnoN induces transcription of the Ccd1 gene in neurons.
Ccd1 is localized in axon terminal and activates the axon growth promoting kinase JNK
To understand how Ccd1 functions in neurons, we first characterized the subcellular localization of Ccd1. Immunoblotting of fractionated granule neuron lysates revealed that Ccd1 is predominantly in the cytoplasmic fraction (Fig. 4A). To corroborate the subcellular fractionation experiments, we characterized the subcellular localization of Ccd1 by immunocytochemistry. Although the Ccd1 antibody recognized specific Ccd1 immunoreactivity by immunoblotting (Fig. 3B), the antibody did not perform effectively in immunocytochemistry experiments (data not shown). Therefore, we generated a plasmid encoding a GFP–Ccd1 fusion protein to aid in the visualization of Ccd1's subcellular localization. We expressed GFP–Ccd1 in COS cells and evaluated the subcellular localization of Ccd1 by GFP immunostaining. Consistent with immunoblotting results of fractionated neurons, GFP–Ccd1 was enriched in the cytoplasm (Fig. 4B). GFP–Ccd1 immunoreactivity colocalized with phalloidin staining, suggesting that GFP–Ccd1 colocalizes with F-actin in the cytoplasm (Fig. 4B).
Figure 4.

Ccd1 is localized in the cytoplasm and enriched at axon terminals in neurons. A, Lysates of granule neurons were fractionated into cytoplasmic (Cyto) and nuclear (Nuc) fractions and then immunoblotted with the Ccd1, SnoN, or α-tubulin antibody. SnoN and α-tubulin served as markers of the nuclear and the cytoplasmic fractions, respectively. B, COS cells were transfected with the GFP–Ccd1 expression plasmid and subjected to immunocytochemistry using the GFP antibody (left), rhodamine–phalloidin staining (middle), or the DNA dye bisbenzimide (Hoechst, merged with GFP and phalloidin staining in right panels). Boxed regions in the top left panel are enlarged in second and third row. GFP–Ccd1 colocalizes with phalloidin-positive fibers, suggesting that Ccd1 is localized at the actin cytoskeleton as indicated with arrowheads. Scale bar, 10 μm. C, Hippocampal neurons transfected with the GFP–Ccd1 and mCherry expression plasmids were subjected to immunocytochemistry using the GFP antibody (left), dsRed antibody (middle), or the DNA dye bisbenzimide (Hoechst, merged with GFP and dsRed staining in right panel). Quantification of GFP–Ccd1 staining intensity is shown in the graph. The mCherry immunofluorescence signal was used to label the entire neuron. GFP–Ccd1 was present in the soma and enriched in the axon terminals. Scale bar, 100 μm.
We next examined the localization of GFP–Ccd1 in neurons. We expressed GFP–Ccd1 in primary hippocampal neurons, which are large in size and thus facilitated the visualization of the subcellular localization of GFP–Ccd1 immunocytochemically. We coexpressed dsRed with GFP–Ccd1 to label transfected neurons. We found that GFP–Ccd1 immunoreactivity was cytoplasmic. In addition to a high-level of staining in the soma, GFP–Ccd1 immunoreactivity was enriched in the axon, in particular at the axon terminal (Fig. 4C). Collectively, these results suggest that the subcellular localization of Ccd1 in axons, and in particular in the axon terminal, is consistent with a role for Ccd1 in regulating axon growth.
Having identified a subcellular localization of Ccd1 that is consistent with a function in axon growth, we next asked whether Ccd1 regulates a signal known to influence axon growth. Because of its DIX domain and association with Dishevelled, Ccd1 reportedly positively regulates the Wnt-signaling pathway (Shiomi et al., 2003). However, in our experiments, we were unable to show that Ccd1 alters the expression of a β-catenin-responsive reporter gene that reflects the activity of canonical Wnt signaling (data not shown). Thus, we examined whether Ccd1 might influence the activity of the JNK-signaling pathway, representing a noncanonical Wnt pathway. First, we confirmed that Ccd1 forms a physical complex with Dishevelled and Axin. In coimmunoprecipitation analyses, Ccd1 robustly interacted with Dishevelled and Axin (Fig. 5A). We next asked if Ccd1 might also bind to proteins that act upstream of the JNK-signaling pathway. We found that Ccd1 associates with the protein kinase mixed lineage kinase 3 (MLK3), suggesting that Ccd1 might influence the activity of the JNK-signaling pathway (Fig. 5B). To assess the role of Ccd1 in JNK signaling, we first expressed both Ccd1 and JNK in 293T cells and measured the phosphorylation of JNK at sites that reflect its activation. Expression of Ccd1 stimulated the phosphorylation of JNK (Fig. 5C). We also assessed the role of endogenous Ccd1 in JNK phosphorylation in neuronal cells. Knockdown of endogenous Ccd1 in Neuro2A neuronal cells reduced the phosphorylation of endogenous JNK (Fig. 5D). These data suggest that Ccd1 promotes the activation of JNK. Since activated JNK is reported to stimulate axon growth (Oliva et al., 2006), these data supported the possibility that Ccd1 might promote the growth of axons.
Figure 5.

Ccd1 activates JNK signaling. A, Lysates of 293T cells transfected with an expression plasmid encoding GFP–Ccd1 or its control vector together with an expression plasmid encoding myc–Dvl or myc–Axin were immunoprecipitated (IP) using the GFP antibody followed by immunoblotting (IB) with the myc or GFP antibody. Input lysates were also immunoblotted with the myc or GFP antibody. Ccd1 strongly interacted with Dvl and Axin. B, Lysates of 293T cells transfected with the GFP–Ccd1 expression plasmid or its vector together with an expression plasmid encoding Flag–MLK3 were IP using the Flag antibody followed by IB with the Flag or GFP antibody. Input lysates were immunoblotted with the Flag or GFP antibody. Ccd1 robustly interacted with MLK3. C, Lysates of 293T cells transfected with an expression vector encoding Flag–JNK together with the Flag–Ccd1 expression plasmid or its control vector were immunoblotted with phospho-JNK (pJNK) or Flag antibody. Ccd1 induced the phosphorylation of JNK. D, Lysates of Neuro2A cells transfected with the Ccd1 RNAi plasmid (pSuper/ccd1i) or control pSuper plasmid were immunoblotted with the phospho-JNK, JNK, or Ccd1 antibody. Ccd1 knockdown reduced the phosphorylation of JNK.
Ccd1 plays a critical role in axon growth
We next directly determined the function of the SnoN downstream target gene Ccd1 in the development of axons. To ascertain the function of Ccd1 in axonal morphogenesis, we induced the acute knockdown of Ccd1. Expression of shRNAs targeting distinct regions of Ccd1 induced the knockdown of Ccd1 (Fig. 6A). We next monitored the effect of Ccd1 knockdown in granule neurons on the development of axons and dendrites in cohorts of granule neurons at 3 and 5 d after transfection. We found that Ccd1 knockdown led to a reduction in the length of axons (Fig. 6B,D). Morphometric analyses confirmed that total axon length in Ccd1 knockdown neurons was significantly lower than that in control-transfected neurons (Fig. 6C,E). In addition, whereas axonal length increased in control-transfected neurons over time, axonal length failed to increase in Ccd1 knockdown neurons (Fig. 6C,E). Ccd1 knockdown had little effect on total dendrite length (Fig. 6F). In addition, Ccd1 knockdown had little effect on the survival of neurons (data not shown). Collectively, these results suggest that Ccd1 plays an important and specific role in the growth of axons.
Figure 6.
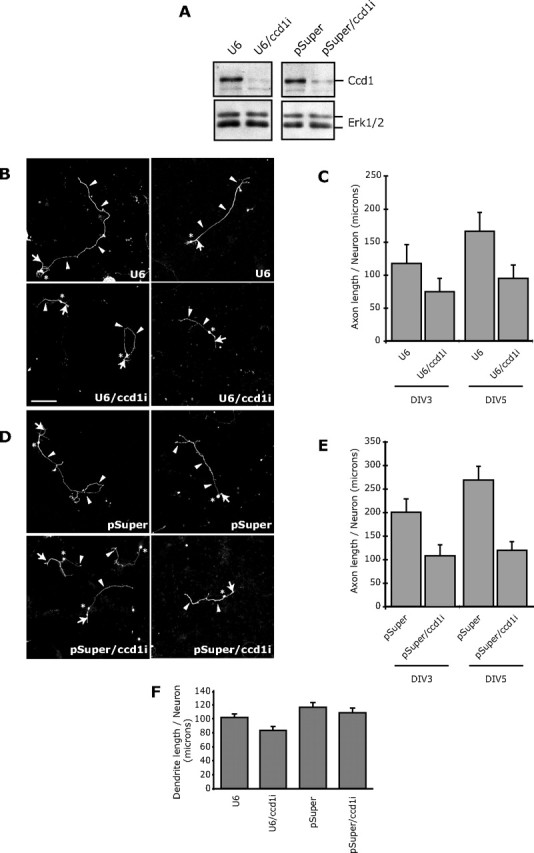
Ccd1 promotes axon growth. A, Lysates of 293T cells transfected with the U6, U6/ccd1i, pSuper, or pSuper/ccd1i RNAi plasmid together with a plasmid expressing Flag–Ccd1 were immunoblotted with the Flag or ERK1/2 antibody. Expression of shRNAs targeting distinct Ccd1 sequences (U6/ccd1i and pSuper/ccd1i) induced knockdown of Ccd1. B, Granule neurons were transfected with the control U6 (top) or U6/ccd1i RNAi plasmid (bottom) together with a GFP expression plasmid. Five days after transfection, granule neurons were subjected to immunocytochemistry using the GFP antibody. Ccd1 knockdown neurons had shorter axons than control-transfected neurons. Arrowheads, arrows, and asterisks indicate axons, dendrites, and cell bodies, respectively. Scale bar, 100 μm. C, Morphometric analyses of granule neurons transfected and analyzed as in B 3 and 5 d after transfection. Quantification of axon length is shown as mean + SEM. Axon length was significantly reduced in Ccd1 knockdown neurons compared with control-transfected neurons at 5 d in vitro (ANOVA; p < 0.05). Total number of neurons measured = 501. D, Granule neurons were transfected with the control pSuper plasmid (top) or the pSuper/ccd1i RNAi plasmid (bottom) together with the GFP expression plasmid and analyzed as in B. Ccd1 knockdown neurons had shorter axons than control-transfected neurons. E, Morphometric analyses of neurons transfected and analyzed as in D 3 and 5 d after transfection. Axon length was significantly reduced in Ccd1 knockdown neurons compared with control-transfected neurons at 3 and 5 d in vitro (ANOVA; p < 0.05). Total number of neurons measured = 345. F, Granule neurons transfected with the U6, U6/ccd1i, pSuper, or pSuper/ccd1i RNAi plasmid together with the GFP expression plasmid were subjected to immunocytochemistry using the GFP antibody followed by morphometric analysis of total dendrite length. No significant difference in total dendrite length between Ccd1 knockdown or control-transfected neurons was found. Total number of neurons measured = 360.
To assess the relationship of Ccd1 function with SnoN in the control of axonal growth, we tested the effect of combining SnoN RNAi together with Ccd1 RNAi on axonal length in neurons. We found that both SnoN and Ccd1 RNAi significantly reduced axon length to a similar extent (Fig. 7A,B). Importantly, the combination of SnoN and Ccd1 RNAi did not further reduce axon length beyond SnoN or Ccd1 RNAi alone (Fig. 7A,B), suggesting that SnoN and Ccd1 operate in a shared pathway to promote axonal growth.
Figure 7.

SnoN promotes axon growth in a Ccd1-dependent manner. A, B, Granule neurons transfected with the Ccd1 RNAi or control pSuper plasmid together with the SnoN RNAi plasmid or control U6 plasmid were analyzed as in Figure 6B–E. Ccd1 and SnoN knockdown each led to significant reduction in axon length compared with control-transfected neurons (ANOVA; p < 0.001). Ccd1 knockdown in the background of SnoN RNAi did not lead to additional reduction of axon length. Total number of neurons measured = 285. C, D, Granule neurons transfected with the Ccd1 RNAi or control pSuper plasmid together with the SnoN–DBM expression plasmid or control vector were analyzed as in Figure 6B–E. SnoN–DBM expression increased axon length compared with control-transfected neurons (ANOVA; p < 0.001). Ccd1 knockdown significantly reduced axon length in the background of SnoN–DBM expression (ANOVA; p < 0.001). Total number of neurons measured = 413 neurons. These results show that Ccd1 knockdown suppresses the ability of SnoN–DBM to stimulate axon growth.
We next tested the effect of Ccd1 knockdown on the ability of the constitutively active form of SnoN (SnoN–DBM) (Stegmüller et al., 2006) to promote axonal growth in granule neurons. Expression of SnoN–DBM induced the growth of axons (Fig. 7C,D) (Stegmüller et al., 2006). However, Ccd1 knockdown significantly reduced the length of axons in neurons in which SnoN–DBM was expressed to levels that were similar to those in Ccd1 knockdown (Fig. 7C,D), suggesting that Ccd1 knockdown suppresses the ability of SnoN–DBM to stimulate axonal growth. Collectively, our results suggest that SnoN promotes axonal growth in a Ccd1-dependent manner.
Ccd1 plays an essential role in parallel fiber development in vivo
The identification of Ccd1 as a critical SnoN-regulated gene in neurons that links SnoN function in the nucleus to the local control of axon growth led us next to determine the function of Ccd1 in the context of the intact developing brain. We assessed the effect of Ccd1 knockdown on the development of axons in postnatal rat pups. Using an in vivo electroporation procedure (Konishi et al., 2004; Shalizi et al., 2006; Stegmüller et al., 2006), we transfected postnatal day 3 (P3) rat pups with a Ccd1 RNAi plasmid that also encoded GFP (pSuper/ccd1i–cmvGFP) or the control plasmid pSuper–cmvGFP (Fig. 8A). Transfected rat pups were returned to moms and were examined 5 d later at P8. Cerebella were isolated from transfected pups and subjected to immunohistochemistry using a GFP antibody.
Figure 8.
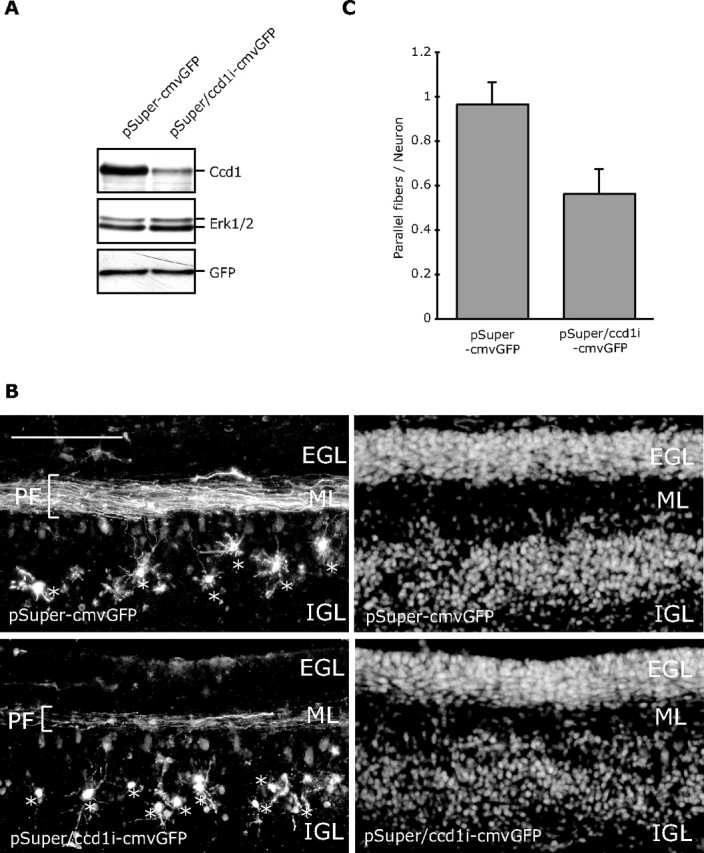
Ccd1 is required for parallel fiber morphogenesis in postnatal rat pups in vivo. A, Lysates of 293T cells transfected with the Ccd1 RNAi plasmid that also encodes GFP (pSuper/ccd1i–cmvGFP) or the control pSuper–cmvGFP plasmid together with a plasmid encoding Flag–Ccd1 were immunoblotted with the Flag, ERK1/2, or GFP antibody. B, P3 rat pups were injected and electroporated with the pSuper–cmvGFP or pSuper/ccd1i–cmvGFP plasmid into the cerebellum. Electroporated rat pups were returned to moms and were killed 5 d later at P8. Coronal sections (40 μm) of cerebellum were prepared and subjected to immunohistochemistry with the GFP antibody. Representative images of cerebellar sections from pups electroporated with the control pSuper–cmvGFP (top) or pSuper/ccd1i–cmvGFP RNAi plasmid (bottom) are shown. Granule neurons were visualized with GFP immunostaining (left) and Hoechst staining (right). The parallel fibers (PF), EGL, molecular layer (ML), and IGL are indicated. Asterisks indicate GFP-expressing granule neurons in transfected animals. Scale bar, 100 μm. C, Quantification of parallel fibers in cerebellar sections imaged in B. Granule neurons in the IGL and parallel fibers in the molecular layer in cerebellar sections from Ccd1 knockdown or control-transfected rat pups were measured. Graph indicates the percentage of granule neurons that were associated with parallel fibers. Parallel fiber number in granule neurons was significantly reduced in Ccd1 knockdown animals compared with control-transfected animals (t test; p < 0.05). Total number of neurons measured = 498.
In control-transfected animals, granule neurons appeared normal in the IGL with neatly stacked parallel fiber axons in the molecular layer (Fig. 8B). In Ccd1 knockdown animals, granule neurons were present in the IGL with normal appearing dendrites (Fig. 8B). However, there was a striking parallel fiber phenotype in the Ccd1 knockdown animals (Fig. 8B). In particular, there were fewer parallel fiber axons in Ccd1 knockdown animals compared with control-transfected animals. Quantitation of the number of parallel fibers in the molecular layer compared with the number of neurons in the IGL revealed a nearly one-to-one ratio of the number of parallel fibers to granule neurons in control-transfected animals (Fig. 8C). In contrast, the ratio of parallel fibers associated with granule neurons was reduced by nearly 45% in Ccd1 knockdown animals (Fig. 8C). These results suggest that Ccd1 plays a critical role in the development of parallel fiber axons in the cerebellar cortex. The effect of Ccd1 knockdown on parallel fiber development observed here closely phenocopies the effect of SnoN knockdown in the cerebellar cortex (Stegmüller et al., 2006). Collectively, our results suggest that Ccd1 operates downstream of SnoN to promote the morphogenesis of axons in the mammalian brain.
Discussion
In this study, we have elucidated a mechanism by which the transcriptional modulator SnoN promotes axonal growth. We found that upon SnoN knockdown in primary neurons, the large majority of altered genes are downregulated, suggesting that SnoN may activate transcription in neurons. In agreement with this conclusion, SnoN associates with the transcriptional coactivator p300 and stimulates axon growth in a p300-dependent manner. We also identified a novel function for the SnoN target gene encoding the signaling scaffold protein Ccd1 in the growth of axons. Ccd1 is enriched at axon terminals and activates the axon growth-promoting protein kinase JNK. Knockdown of Ccd1 impairs the growth of axons in primary granule neurons and importantly impairs the formation of granule neuron parallel fibers in the rat cerebellar cortex in vivo. Epistatic analyses suggest that Ccd1 operates downstream of SnoN to promote axon growth. Collectively, these findings define SnoN and Ccd1 as components of a pathway that drives axonal morphogenesis in the mammalian brain.
SnoN is best known for its function as a transcriptional corepressor (Luo, 2004). Our findings suggest that SnoN may predominantly activate transcription in neurons. Although the possibility remains that all of the downregulated genes in SnoN knockdown neurons may have resulted from upregulation of a transcriptional repressor protein, this seems unlikely since the large majority of altered genes were downregulated, and we did not observe the induction of a transcriptional repressor in SnoN knockdown neurons. Recent studies suggest that SnoN does not always act as a transcriptional corepressor and may also operate as a transcriptional coactivator in specific cell types including neurons (Sarker et al., 2005, 2008; Pot and Bonni, 2008). Interestingly, a number of genes that were found downregulated in SnoN knockdown neurons are transcriptionally responsive to TGFβ signaling (Chang and Goldberg, 1995; Negoescu et al., 1995; Shirakihara et al., 2007), suggesting that SnoN may activate transcription of TGFβ-responsive genes in neurons. Consistent with this interpretation, we found that SnoN activates the expression of a TGFβ-responsive reporter gene in granule neurons (Sarker et al., 2008). In addition, we have shown that SnoN associates with the transcriptional coactivator p300, and p300 plays a critical role in SnoN-dependent axon growth. Importantly, we also found that SnoN and p300 both are required for Ccd1 promoter-mediated transcription in neurons. Collectively, these observations suggest that SnoN can activate transcription in neurons. Thus, SnoN is a versatile transcriptional regulator that may activate or repress transcription in a cell- and gene-specific manner (Sarker et al., 2005, 2008; Pot and Bonni, 2008). The upregulation of some genes in SnoN knockdown granule neurons suggests that transcriptional repression might also be relevant to SnoN functions in neurons. Beyond Ccd1, it will be important in future studies to determine the function of other SnoN-regulated genes in the control of axonal development.
The identification of Ccd1 as a critical target gene of SnoN in neurons provides mechanistic insight into how SnoN influences axonal development. We found that Ccd1 is localized to the actin cytoskeleton and enriched at axon terminals in neurons. Thus, Ccd1 may couple SnoN function in the nucleus to the regulation of the actin cytoskeleton at the axon terminal. As a DIX domain protein, Ccd1 has been suggested to influence Wnt signaling. We were unable to show that Ccd1 alters the activity of canonical Wnt signaling (data not shown). In contrast, we provide evidence that Ccd1 associates with components of the JNK-signaling pathway and activates JNK in neuronal cells. Since JNK plays a critical role in axon growth (Oliva et al., 2006), Ccd1 activation of JNK might represent one route by which Ccd1 couples the axon growth-promoting signal from SnoN in the nucleus to the local regulation of axon growth in neuronal processes.
The elucidation of a novel SnoN–Ccd1 pathway in neurons has important implications for both SnoN and Ccd1 function in diverse biological settings including brain development and pathology. In addition to driving axonal morphogenesis, SnoN has been implicated in the control of TGFβ signaling and oncogenesis (Luo, 2004; Pot and Bonni, 2008). Therefore, our data raise the interesting question of whether beyond axon development, Ccd1 might also contribute to the ability of SnoN in regulating TGFβ signaling and tumorigenesis.
SnoN function in neurons is regulated by the ubiquitin–proteasome system. The ubiquitin ligase Cdh1–APC promotes the ubiquitination and consequent degradation of SnoN and thereby inhibits axonal growth (Konishi et al., 2004; Stegmüller and Bonni, 2005; Stegmüller et al., 2006; Kim and Bonni, 2007). Consequently, inhibition of Cdh1–APC stimulates axonal growth and even overrides the potent axon-inhibitory cue myelin that is associated with injury in the nervous system (Konishi et al., 2004; Stegmüller et al., 2008). Since SnoN is a critical substrate of neuronal Cdh1–APC (Stegmüller et al., 2006), our study suggests that levels of Ccd1 expression might be a critical readout of the nuclear Cdh1–APC/SnoN ubiquitination pathway in neurons and might thus reflect the capacity of axons to grow. It will be interesting to consider the possibility that augmenting Ccd1 expression in neurons might stimulate the ability of axons to grow and regenerate after injury in the nervous system.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS051255 (A.B.), the Harvard Medical School–Merck-Sponsored Research Program (A.B.), the Alberta Cancer Research Institute (ACRI; S.B.), the Uehara Memorial Foundation (Y.I.), the Naito Foundation (Y.I.), the Human Frontier Science Program (Y.I.), a Charles A. King Trust (Bank of America, Cotrustee) Postdoctoral Fellowship (J.S.), and NIH Training Grant GM077226 (M.A.H.). S.B. is an Alberta Heritage Foundation for Medical Research (AHFMR) Scholar. S.N. is an AHFMR and ACRI Postdoctoral Fellow. We thank Samantha Keough for technical assistance, Genevieve Konopka for helpful discussions, Sheng-Cai Lin for providing the pSuper/ccd1i plasmid, and Zhen-Ge Luo for providing the Dvl–myc expression plasmid.
References
- Altman J, Bayer S. Development of the cerebellar system: in relation to its evolution, structure, and functions. New York: CRC; 1997. [Google Scholar]
- Butler SJ, Tear G. Getting axons onto the right path: the role of transcription factors in axon guidance. Development. 2007;134:439–448. doi: 10.1242/dev.02762. [DOI] [PubMed] [Google Scholar]
- Chang E, Goldberg H. Requirements for transforming growth factor-beta regulation of the pro-alpha 2(I) collagen and plasminogen activator inhibitor-1 promoters. J Biol Chem. 1995;270:4473–4477. doi: 10.1074/jbc.270.9.4473. [DOI] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A. RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem. 2002;277:46442–46446. doi: 10.1074/jbc.M206653200. [DOI] [PubMed] [Google Scholar]
- Gaudillière B, Konishi Y, de la Iglesia N, Yao G, Bonni A. A CaMKII-NeuroD signaling pathway specifies dendritic morphogenesis. Neuron. 2004;41:229–241. doi: 10.1016/s0896-6273(03)00841-9. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- Kim AH, Bonni A. Thinking within the D box: initial identification of Cdh1-APC substrates in the nervous system. Mol Cell Neurosci. 2007;34:281–287. doi: 10.1016/j.mcn.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Stegmüller J, Matsuda T, Bonni S, Bonni A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- Luo K. Ski and SnoN: negative regulators of TGF-beta signaling. Curr Opin Genet Dev. 2004;14:65–70. doi: 10.1016/j.gde.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Negoescu A, Lafeuillade B, Pellerin S, Chambaz EM, Feige JJ. Transforming growth factors beta stimulate both thrombospondin-1 and CISP/thrombospondin-2 synthesis by bovine adrenocortical cells. Exp Cell Res. 1995;217:404–409. doi: 10.1006/excr.1995.1103. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Atkins CM, Copenagle L, Banker GA. Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci. 2006;26:9462–9470. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Ince-Dunn G, Ghosh A. Transcriptional regulation of vertebrate axon guidance and synapse formation. Nat Rev Neurosci. 2007;8:331–340. doi: 10.1038/nrn2118. [DOI] [PubMed] [Google Scholar]
- Pot I, Bonni S. SnoN in TGF-beta signaling and cancer biology. Curr Mol Med. 2008;8:319–328. doi: 10.2174/156652408784533797. [DOI] [PubMed] [Google Scholar]
- Sarker KP, Wilson SM, Bonni S. SnoN is a cell type-specific mediator of transforming growth factor-beta responses. J Biol Chem. 2005;280:13037–13046. doi: 10.1074/jbc.M409367200. [DOI] [PubMed] [Google Scholar]
- Sarker KP, Kataoka H, Chan A, Netherton SJ, Pot I, Huynh MA, Feng X, Bonni A, Riabowol K, Bonni S. ING2 as a novel mediator of transforming growth factor-beta-dependent responses in epithelial cells. J Biol Chem. 2008;283:13269–13279. doi: 10.1074/jbc.M708834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi A, Lehtinen M, Gaudilliere B, Donovan N, Han J, Konishi Y, Bonni A. Characterization of a neurotrophin signaling mechanism that mediates neuron survival in a temporally specific pattern. J Neurosci. 2003;23:7326–7336. doi: 10.1523/JNEUROSCI.23-19-07326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi A, Gaudillière B, Yuan Z, Stegmüller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- Shiomi K, Uchida H, Keino-Masu K, Masu M. Ccd1, a novel protein with a DIX domain, is a positive regulator in the Wnt signaling during zebrafish neural patterning. Curr Biol. 2003;13:73–77. doi: 10.1016/s0960-9822(02)01398-2. [DOI] [PubMed] [Google Scholar]
- Shiomi K, Kanemoto M, Keino-Masu K, Yoshida S, Soma K, Masu M. Identification and differential expression of multiple isoforms of mouse Coiled-coil-DIX1 (Ccd1), a positive regulator of Wnt signaling. Brain Res. 2005;135:169–180. doi: 10.1016/j.molbrainres.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol Biol Cell. 2007;18:3533–3544. doi: 10.1091/mbc.E07-03-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma K, Shiomi K, Keino-Masu K, Masu M. Expression of mouse Coiled-coil-DIX1 (Ccd1), a positive regulator of Wnt signaling, during embryonic development. Gene Expr Patterns. 2006;6:325–330. doi: 10.1016/j.modgep.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Stegmüller J, Bonni A. Moving past proliferation: new roles for Cdh1-APC in postmitotic neurons. Trends Neurosci. 2005;28:596–601. doi: 10.1016/j.tins.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Stegmüller J, Konishi Y, Huynh MA, Yuan Z, Dibacco S, Bonni A. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron. 2006;50:389–400. doi: 10.1016/j.neuron.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Stegmüller J, Huynh MA, Yuan Z, Konishi Y, Bonni A. TGFbeta-Smad2 signaling regulates the Cdh1-APC/SnoN pathway of axonal morphogenesis. J Neurosci. 2008;28:1961–1969. doi: 10.1523/JNEUROSCI.3061-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zheng L, Zeng Z, Zhou G, Chien J, Qian C, Vasmatzis G, Shridhar V, Chen L, Liu W. DIXDC1 isoform, l-DIXDC1, is a novel filamentous actin-binding protein. Biochem Biophys Res Commun. 2006;347:22–30. doi: 10.1016/j.bbrc.2006.06.050. [DOI] [PubMed] [Google Scholar]