

Summary
Matrix metalloproteinases (MMPs) are invariably upregulated in the stromal compartment of epithelial cancers and appear to promote invasion and metastasis. Here we report that phenotypically normal mammary epithelial cells with tetracycline-regulated expression of MMP3/stromelysin-1 (Str1) form epithelial glandular structures in vivo without Str1 but form invasive mesenchymal-like tumors with Str1. Once initiated, the tumors become independent of continued Str1 expression. Str1 also promotes spontaneous premalignant changes and malignant conversion in mammary glands of transgenic mice. These changes are blocked by coexpression of a TIMP1 transgene. The premalignant and malignant lesions have stereotyped genomic changes unlike those seen in other murine mammary cancer models. These data indicate that Str1 influences tumor initiation and alters neoplastic risk.
Introduction
Extracellular matrix (ECM)-degrading matrix metalloproteinases (MMPs) are universal features of carcinoma progression and are associated with tumor angiogenesis, invasion, and metastasis. MMPs not only foster invasion and spread by disrupting ECM barriers but also affect cellular signaling by several routes (Werb, 1997; Lukashev and Werb, 1998). Interestingly, most MMPs are synthesized not by the genetically altered cancer cells but by adjacent and intervening stromal cells (Coussens and Werb, 1996). There is also a growing awareness that stromal cells and the matrix microenvironment can influence initial tumor development (Jacoby et al., 1997; Howe et al., 1998; Hsieh et al., 1998; Jacobs et al., 1999). Thus, given their stromal origin, consistent upregulation, and signaling capacity, stromal MMPs may also contribute to the initial stages of cancer development.
MMP3/stromelysin-1 (Str1) is a candidate for a stromal MMP that can exert oncogenic effects. It can degrade numerous ECM substrates, including collagens III, IV, V, IX, X, and XI, laminins, elastin, entactin, fibronectin, fibrin, fibrillins, fibulin, link protein, osteonectin, tenascin, vitronectin, and ECM proteoglycans (reviewed in Sternlicht and Werb, 1999). Str1 can also release cell surface molecules, including E-cadherin, L-selectin, heparin-binding EGF-like growth factor, and TNF-α; it can activate other MMPs, including gelatinase B and the collagenases; and it can inactivate several serine proteinase inhibitors (reviewed in Sternlicht and Werb, 1999). Importantly, Str1 was originally cloned (Matrisian et al., 1985) and repeatedly recloned as a tumor-specific gene (Muller et al., 1988; Ostrowski et al., 1988).
Str1 is expressed in stromal cells throughout mammary development and is maximally expressed during involution when ECM remodeling and alveolar regression take place (Talhouk et al., 1992; Witty et al., 1995; Lund et al., 1996). Although stromal cells synthesize Str1 in vivo, the protein associates with the epithelium (Talhouk et al., 1992; Lund et al., 1996). There are two distinct responses to Str1 in mammary epithelium: proliferation and branching in ductal cells, and apoptosis in differentiated secretory alveolar cells (Sympson et al., 1994; Boudreau et al., 1995; Witty et al., 1995). There are also parallels between development and neoplasia. Expression of Str1 in the mammary epithelium of transgenic mice during development induces upregulation of endogenous Str1 and other MMPs in surrounding stromal fibroblasts and leads to fibrosis, neovascularization, and tenascin-C expression, all of which are hallmarks of the reactive stroma of involution (Alexander et al., 1996; Thomasset et al., 1998). Increased cell proliferation, an altered stroma, angiogenesis, and tenascin-C expression are also features of cancer progression (Borsi et al., 1992; Rønnov-Jessen et al., 1996).
Here, we have examined how Str1 affects tumor progression using two genetic approaches: phenotypically normal mammary epithelial cells that express Str1 in a tetracycline-regulated manner, and an Str1 transgene targeted to mouse mammary glands by the mouse whey acidic protein (WAP) gene promoter. Our results indicate that not only can Str1 induce an altered stromal environment, but as a product of such an environment, it can promote the phenotypic conversion and malignant transformation of mammary epithelial cells.
Results
Str1 Promotes Epithelial-to-Mesenchymal Transitions in Culture
Str1 has both ECM and cell surface targets (Sternlicht and Werb, 1999). Thus, its biologic effects could arise from its alteration of surrounding ECM or by its direct action on the epithelial cells themselves. Therefore, we treated an immortal but functionally normal mouse mammary epithelial cell line (Scp2), which responds to ECM signals but does not assemble its own ECM, with recombinant Str1 (rStr1). Scp2 cells grow as an epithelial sheet with E-cadherin-rich adherens junctions and are cytokeratin positive and vimentin negative. The rStr1 rapidly induced epithelial-to-mesenchymal transitions (EMT), characterized by loss of cell–cell interactions, acquisition of a scattered morphology, downregulation of epithelial cytokeratins, and upregulation of the mesenchymal marker vimentin (Figure 1). These changes did not occur when the synthetic MMP inhibitor GM6001 was also added (data not shown). This treatment of the cells in trans mimics the in vivo production of Str1 by fibroblasts acting on mammary epithelium.
Figure 1. Effect of Str1 on Morphology and Intermediate Filament Expression in Scp2 Cells.
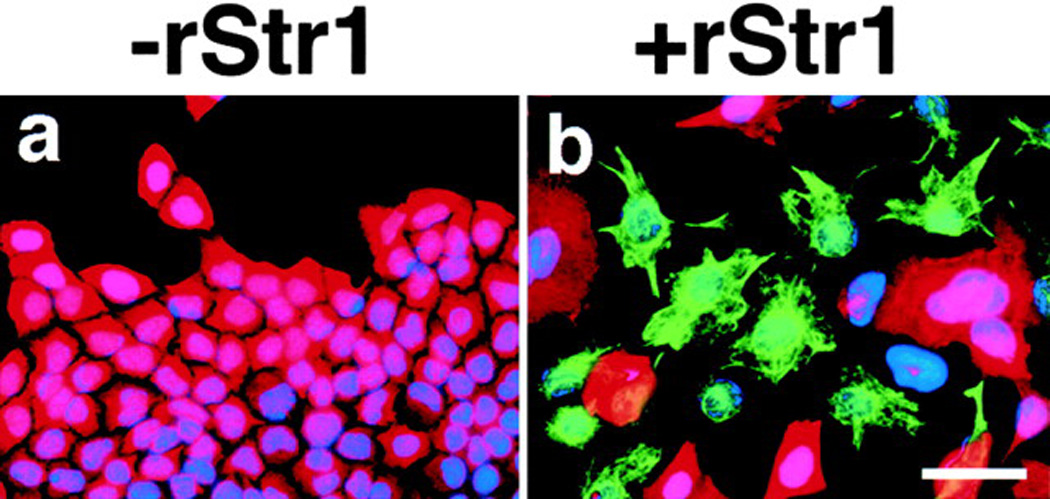
Cells were maintained for 6 days in the (a) absence or (b) presence of activated recombinant Str1 (rStr1) and stained by indirect immunofluorescence for cytokeratins (red) and vimentin (green). Nuclei were counterstained with DAPI (blue). Scale bar, 50 µm.
Str1 Promotes EMT and Tumorigenicity In Vivo
To examine the effect of Str1 on Scp2 cells in vivo, we used Scp2 cells stably transfected with an autoactivating rat Str1 cDNA under the control of a tetracycline (Tet)-repressible promoter (Lochter et al., 1997). These transfected cells grow as epithelial sheets when grown in medium containing Tet, whereas induction of Str1 expression by Tet withdrawal results in EMT, cleavage and loss of E-cadherin, and acquisition of the ability to form anchorage-independent colonies in agar and invade Matrigel (Lochter et al., 1997). For the present study, we used two clones with regulated Str1 expression (p2S7 and p2S10), a nonexpressing clone (p2S3), and parental Scp2 cells. When Scp2 cells and Str1-transfected cells grown in the presence of Tet were injected into surgically cleared (gland-free) mammary fat pads of scid/scid mice, they grew and formed relatively normal duct-like and pseudoglandular structures if the mice were given Tet in their drinking water (Figures 2Aa and 2Ab). These structures had cytokeratin-8-positive, vimentin-negative luminal cells but lacked smooth muscle actin-positive myoepithelial cells that normally surround luminal epithelium (Figures 2Ac and 2Ad). The ductal structures did not branch, which is consistent with the lack of myoepithelial cells that produce epimorphin, a morphogen required for branching (Hirai et al., 1998). This growth pattern persisted for at least 13 weeks. However, when Str1 expression was induced in vivo by withholding Tet from the drinking water, the p2S10 cells formed small tumors in 5 of 18 injected sites by 6 weeks (Figure 2B). The tumors were composed largely of vimentin-positive and cytokeratin-negative spindle-shaped (mesenchymal-like) cells and were invasive at their periphery (Figure 2A). Thus, induction of Str1 expression triggered EMT and rendered the cells tumorigenic and invasive in vivo.
Figure 2. Effect of Str1 on Mammary Epithelial Growth In Vivo.
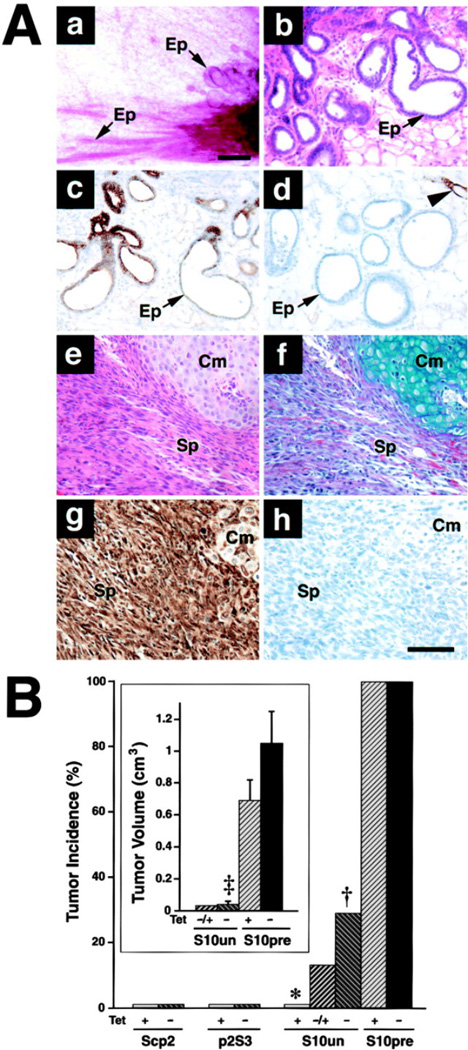
(A) Histologic appearance of p2S10 mammary epithelial cells grown in cleared mammary fat pads. (a–d) Appearance of epithelial ductal and gland-like structures (Ep) that form in the absence of Str1 expression as seen by (a) whole-mount, (b) H&E, (c) anti-cytokeratin-8, and (d) anti-smooth muscle actin staining. The arrowhead in (d) indicates vascular smooth muscle cells. (e–h) Appearance of spindle-cell tumors (Sp) that form when Str1 expression is induced in vivo or prior to injection as seen by (e) H&E, (f) Alcian blue, (g) anti-vimentin, and (h) anti-cytokeratin-8 staining. Areas of cartilage (chondroid metaplasia, Cm) are present in the upper right corner (e–h). Scale bars, (a) 200 µm and (b–h) 100 µm.
(B) Tumor incidence (percent of injected sites) and volume (cm3; mean ± SEM) following injection of parental (Scp2), nonexpressing (p2S3), uninduced (S10un), and preinduced (S10pre) cells into cleared mammary fat pads in mice maintained for 6 weeks with (+) or without (−) Tet in their drinking water or without Tet for the first 12 days only (−/+). *, p < 0.0001 versus S10pre cells in mice maintained with or without Tet (two-tailed Fisher’s exact test); †, p < 0.05 versus S10un cells in mice maintained with Tet and p < 0.001 versus S10pre cells in mice maintained with or without Tet (Fisher’s exact test); ‡, p < 0.005 and p < 0.001 versus S10pre cells in mice maintained with and without Tet, respectively (t test).
When Tet was withheld from the drinking water for 12 days after injecting uninduced p2S10 cells and then replaced for the remaining 4.3 weeks, tumor formation was still seen, albeit at a lower rate than when Tet was continuously absent (Figure 2B). This suggests that once tumorigenicity is achieved, it would not be blocked by repressing Str1 expression. To test this, we induced Str1 expression in culture for 2 months in p2S10 and p2S7 cells. When the preinduced cells were injected into cleared fat pads, large tumors grew at all injected sites regardless of the presence or absence of Tet in the drinking water (Figure 2B). The tumors were highly invasive and composed almost entirely of vimentin-positive spindle-shaped cells with fewer than 1% cytokeratin-positive cells (Figure 2Ae–h). Interestingly, several tumors had small areas of differentiation to a cartilage-like phenotype (Figure 2Ae–h). Such changes, called chondroid metaplasia, are also seen in human tumors with EMT and were first described three centuries ago (cited in Wargotz and Norris, 1989). Thus, once EMT is induced, converted cells no longer require Str1 transgene stimulation to elicit an altered program of gene expression and tumorigenesis.
The preinduced p2S10 cells also acquired the ability to form subcutaneous tumors with or without Tet in the drinking water (data not shown). These tumors were about one-tenth the size of the orthotopic tumors but had the same infiltrative spindle-cell morphology. No other cells grew subcutaneously. Taken together, these data indicate that Str1 can trigger tumor progression in an immortal but functional mammary epithelial cell line.
Str1 Promotes the Development of Premalignant and Malignant Mammary Lesions in Transgenic Mice
To examine the long-term effect of Str1 on normal mammary epithelial cells, we used WAP-Str1 transgenic mice. We had previously observed precocious lobuloalveolar development in 3- to 10-week-old WAP-Str1 transgenic mice (Sympson et al., 1994) and an altered reactive stroma in 2- to 4-month-old mice (Thomasset et al., 1998). Here we found that 6- to 24-month-old transgenic mice exhibited mammary abnormalities, pre-malignant lesions, and malignancies (Figure 3 and Figure 4). Of 163 mice from five independent transgenic lines (median age 18 months), 77% had moderate or severe fibrosis (collagen and fibroblast accumulation with loss), 64% had moderate or severe hyperplasia (epithelial cell accumulation), 53% had lymphocytic infiltrates, 20% had dysplasias (atypical proliferative lesions) or ductal carcinoma in situ, and 7.4% developed mammary carcinomas.
Figure 3. Incidence of Mammary Gland Pathologies in Str1 Transgenic Mice.
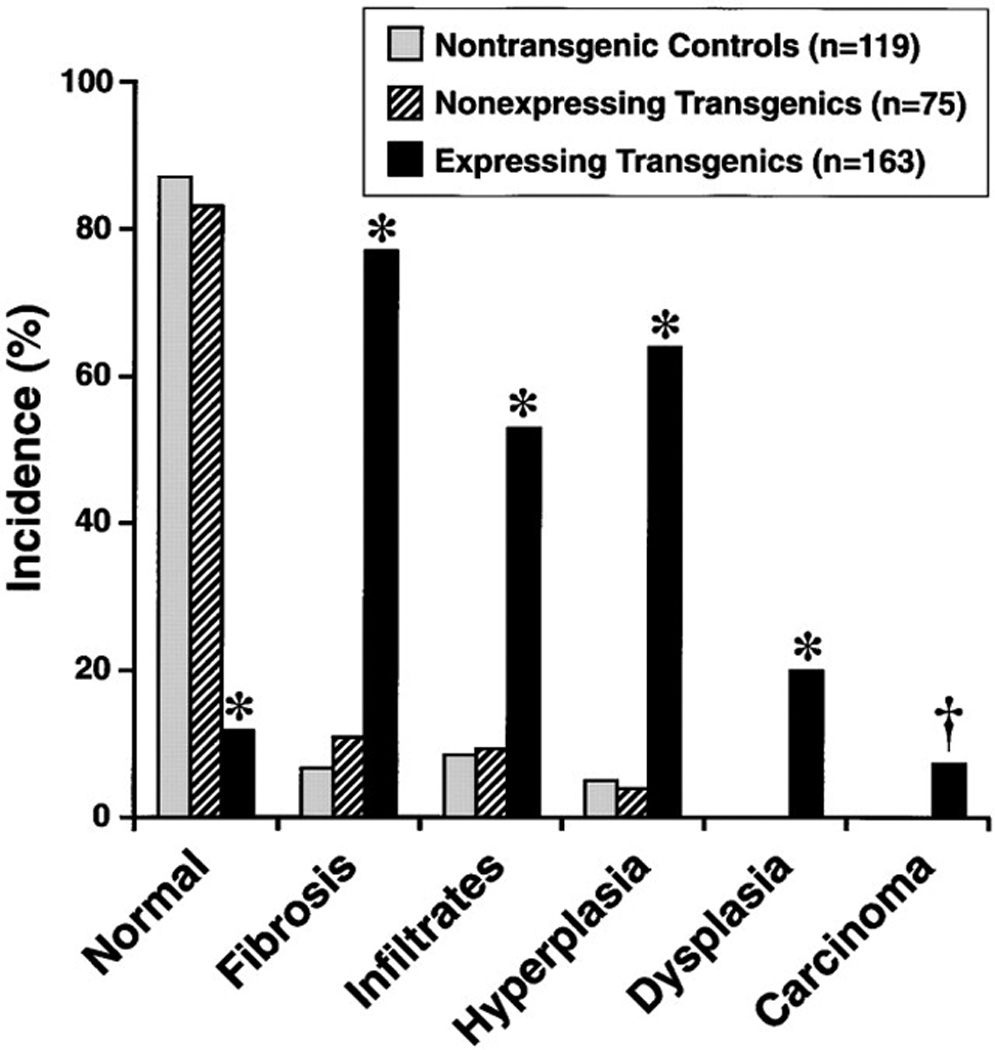
The Str1-expressing transgenics include 100, 31, 16, 9, and 7 mice from five independent transgenic lines, respectively. *, p < 0.00001 versus nontransgenic or nonexpressing transgenic controls (two-tailed Fisher’s exact test); †, p = 0.002 and p = 0.02 versus non-transgenic and nonexpressing transgenic controls, respectively (Fisher’s exact test).
Figure 4. Histologic Appearance of Normal and WAP-Str1 Mammary Glands.
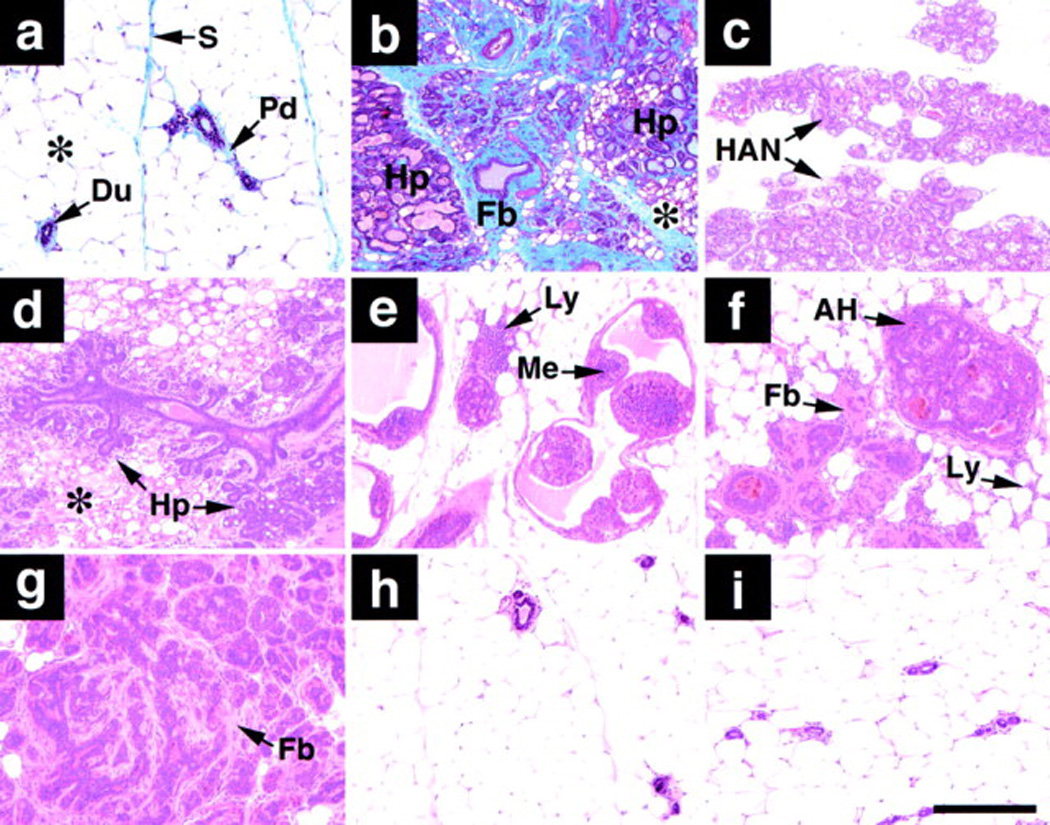
Histologic sections are from (a) nontransgenic, (b–g) WAP-Str1 transgene–expressing, (h) WAP-Str1 transgene–nonexpressing, and (i) Str1/TIMP1 double transgene–positive female mice.
(a) Normal mammary gland with resting ducts (Du), abundant adipose tissue (asterisk), and minimal periductal (Pd) and septal (S) collagen (stained blue).
(b) Severe hyperplasia (Hp) with considerable intervening fibrosis (Fb; stained blue) and multilocular adipocytes (asterisk).
(c) Hyperplastic alveolar nodule (HAN) with lipid droplets characteristic of secretory activity even though this gland comes from a nulliparous mouse.
(d) Multifocal alveolar hyperplasia (Hp) with eosinophilic (pink) fibrotic areas and multilocular adipocytes (asterisk).
(e) Intraductal papillary hyperplasia with lymphocytic infiltrates (Ly). The small hyperchromatic cells (Me) were cytokeratin-8 negative and smooth muscle actin positive, indicating the abnormal presence of myoepithelial cells within the severely distended ducts.
(f) Atypical hyperplasia (AH) with lymphocytic infiltrates (Ly) and mild fibrosis (Fb).
(g) Atypical hyperplasia with considerable fibrosis.
(h and i) Normal mammary histology seen with the loss of Str1 transgene expression or its inhibition by TIMP1, respectively.
(a and b) Masson’s trichrome. (c–i) Hematoxylin/eosin. Scale bar, 200 µm.
The incidence of the various lesions was 1.2- to 1.9-fold higher in parous mice than in virgin mice, and hyper-plastic and fibrotic changes were generally more severe in parous animals. Whereas increases in incidence and severity would be expected given the pregnancy-driven nature of the WAP promoter, their modesty may reflect the fact that the promoter is minimally active during each estrus cycle, so that parity only slightly increases lifetime Str1 exposure. Indeed, even low transgene expression during puberty causes increased ductal branching and precocious lobuloalveolar development (Sympson et al., 1994), thus increasing the number of potential target cells that are “at risk” over an extended period of time. Average tumor latency was 18.7 months, with the first tumor appearing in a 6-month-old parous mouse. The hyperplastic and fibrotic lesions tended to be more severe in older animals, and the incidence of hyperplasia increased from 46% at 6 months to 78% at 2 years of age. These data are consistent with a model of multistage neoplastic progression induced by Str1 expression.
Mammary carcinomas were seen in mice from three different founders (fewer than ten mice were studied in the other two lines). Nine of the tumors were moderately well-differentiated adenocarcinomas (Figure 5a–c), and three were undifferentiated tumors with evidence of EMT. Two of these were carcinosarcomas with cytokeratin-positive and vimentin-negative epithelial-like populations and distinct vimentin-positive and cytokeratin-negative fibroblast-like populations (Figure 5g–i). The third undifferentiated tumor gave rise to lung and kidney metastases and expressed both cytokeratins and vimentin (Figure 5d–f). A cell line derived from the primary mammary tumor also coexpressed cytokeratin and vimentin and formed invasive tumors that were cytokeratin and vimentin positive in vivo (data not shown).
Figure 5. Histologic Appearance of Malignant Mammary Tumors from Str1 Transgenic Mice.

(a–c) Moderately differentiated adenocarcinoma (Ad) with adjacent and intervening vimentin-positive stromal cells (St).
(d–f) Renal metastasis (Met) from an undifferentiated mammary carcinoma. Normal kidney (Kid) is present on the right.
(g–i) Carcinosarcoma with distinct carcinomatous (Ca) and sarcomatous (Sa) areas exhibiting epithelial and mesenchymal features, respectively.
H&E, hematoxylin/eosin stains. Scale bar, 200 µm.
In contrast to the Str1-expressing transgenic mice, nontransgenic littermate controls (median age 18 months) exhibited a low incidence of mild hyperplasia, fibrosis, and lymphocytic infiltration (Figure 3). These low incidence rates were similar for virgin and parous controls. We also isolated two sublines of Str1 transgenic mice in which expression of the transgene was silenced and could not be detected by RT-PCR, presumably due to transgene methylation as determined by altered sensitivity to restriction endonucleases. The nonexpressing transgenic mice (median age 13 months) also showed a low incidence of mild lesions (Figure 3). These data indicate that expression of the Str1 transgene is required to promote neoplastic progression.
TIMP1 Inhibits the Development of Mammary Hyperplasias in Str1 Transgenic Mice
If the induction of neoplasia by Str1 is due to its proteolytic activity, then overexpression of its endogenous inhibitor, tissue inhibitor of metalloproteinases-1 (TIMP1), should quench this phenotype. Thus, we mated WAP-Str1 mice with mice carrying a human TIMP1 transgene under the control of the same WAP promoter to examine the effect of TIMP1 on the development of mammary lesions in WAP-Str1 mice. Mammary hyperplasias were examined as a surrogate endpoint in 10- to 16-month-old female offspring (Figure 6). Only 3 of 16 mice (19%) carrying both Str1 and TIMP1 transgenes had mild mammary hyperplasias, whereas 8 of 11 mice (73%) carrying the Str1 transgene alone had moderate to severe hyperplasias. A similar frequency (66%) was seen in the cohort of 100 WAP-Str1 females from the same transgenic founder (M2-5). Mice carrying only the TIMP1 transgene had a slightly altered involution phenotype but were otherwise normal. Nontransgenic littermates had entirely normal mammary glands. These data indicate that active Str1 is required for mammary lesions to develop.
Figure 6. Effect of a TIMP1 Transgene on Str1 Transgene–Induced Mammary Hyperplasias as Seen by Whole-Mount Staining.
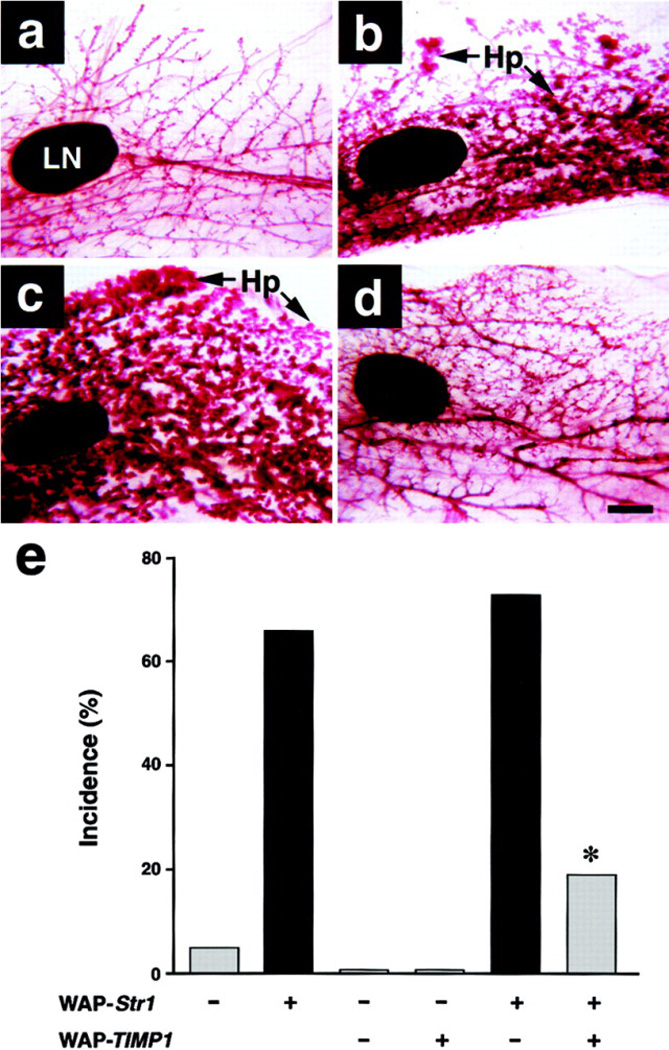
(a) 16-month-old nontransgenic control.
(b) 16-month-old WAP-Str1 transgenic mouse with multifocal alveolar hyperplasia (Hp).
(c) 12-month-old WAP-Str1-positive/WAP-TIMP1-negative mouse with diffuse alveolar hyperplasia.
(d) 12-month-old Str1/TIMP1 double transgenic mouse from the same litter as the mouse in (c). The mammary gland in (d) was judged to be within normal limits by whole-mount and hematoxylin/eosin staining. The glands shown are from mice that had undergone a single pregnancy and lactation at least 5 months prior to sacrifice. LN, lymph node. Scale bar, 500 µm.
(e) Incidence of mammary hyperplasias in double and single transgenic mice. The single transgenic mice are from the related M2-5 line. Gray and black bars indicate mild and moderate to severe hyperplasias, respectively. *, p < 0.02 versus littermates carrying the Str1 transgene alone and p < 0.0006 versus the large cohort of M2-5 Str1 transgenics, respectively (two-tailed Fisher’s exact test).
Str1 Expression Promotes Stereotyped Genomic Changes
Because Str1 acts extracellularly, we wanted to determine if tumorigenicity was accompanied by genomic reorganization. Analysis of ten mammary lesions from three Str1 transgenic lines by comparative genomic hybridization (CGH) revealed DNA losses in specific regions of mouse chromosomes 4 and 7 in both premalignant and malignant lesions (Figure 7A). In addition, the three tumors with EMT and a severe hyperplasia had DNA copy number gains on chromosomes 6 and 15. Because these gains were associated with EMT, we separately analyzed the epithelial- and mesenchymal-like populations of one carcinosarcoma after laser microdissection. The chromosome 15 amplification was only seen in microdissected fibroblast-like areas that had undergone EMT, whereas other CGH changes were seen throughout the tumor. Because the DNA from the severe hyperplasia with gains on chromosomes 6 and 15 came from an area of tissue that was not expressly visualized and microdissected, its genomic changes may reflect those of an occult tumor with EMT. CGH profiles for nonneoplastic tissues from the same ten mice (data not shown) and from two histologically normal mammary glands from separate transgenic lines were invariably normal. Thus, even with this sample size and despite using a statistic (Fisher’s exact test) that ignores consistency in direction and subchromosomal localization, the genomic changes on chromosomes 4, 7, 6, and 15 were remarkably nonrandom (p < 0.001, 0.01, 0.05, and 0.05, respectively).
Figure 7. CGH Profiles.
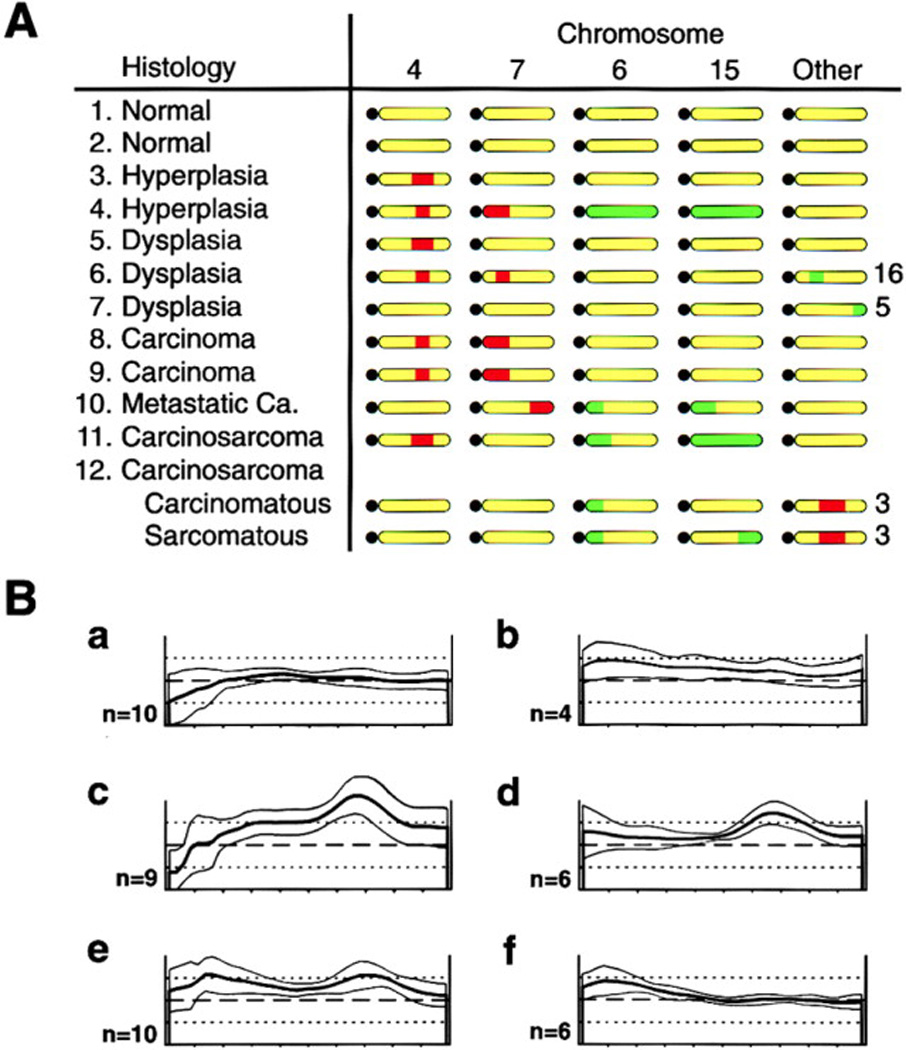
(A) Genomic changes seen in the mammary glands of 12 individual Str1 transgenic mice. Samples 1, 3, 10, and 11 were from one transgenic founder line; samples 4 and 8 were from another line; and the remaining samples were from a third independent line. Approximate locations of macroscopic DNA gains (green) and losses (red) are indicated along otherwise unaltered (yellow) chromosomes, with black circles representing acrocentric centromeres. Sample 12, a carcinosarcoma, was microdissected and its carcinomatous and sarcomatous regions analyzed separately. All adjacent stromal and nonmammary control tissues had normal CGH profiles.
(B) Normalized fluorescence intensity profiles for chromosome 15 obtained with DNA isolated from (a) parental Scp2 cells, (b) a p2S7 cell–derived tumor, (c) preinduced p2S10 cells, (d) microdissected spindle-cell areas from a tumor derived from the same preinduced cells as in (c), (e) chondroid areas from the same tumor as in (d), and (f) normal stroma adjacent to the tumor in (d) and (e). Average green:red fluorescence ratios (heavy lines) ± 1 standard deviation (thin lines) are shown for the number of metaphase chromosomes examined (n). Dashed horizontal lines and upper and lower dotted lines indicate fluorescence ratios of 1, 1.5, and 0.5, respectively.
Independent evidence for an association of mouse chromosome 15 with EMT was obtained from the p2S cells and their tumors. DNA gains in chromosome 15 were seen in the preinduced p2S10 and p2S7 cells and their tumors but not in the parental cells (Figure 7B). Identical gains in the middistal portion of chromosome 15 were seen in both microdissected spindle-cell and cartilage-like areas of p2S10 tumors but not in adjacent normal stroma, indicating that both areas arose from injected rather than host cells and that the cartilage-like areas represent a further manifestation of EMT. The preinduced cells and their tumors also showed amplifications on chromosomes 3, 5, and 11. Unlike the WAP-Str1 mammary tumors, no changes were seen on chromosomes 4, 7, or 6. Thus, although we studied relatively few samples, our data further implicate mouse chromo some 15 as significant for Str1-induced tumors that have undergone EMT.
Discussion
Stromal MMPs Alter Neoplastic Risk
An altered stromal environment appears to presage cancer development. In the case of WAP-Str1 transgenic mice, stromal defects appear (Thomasset et al., 1998) well before neoplastic changes are observed. In humans, cancer susceptibility is increased in certain fibrotic and chronic inflammatory conditions (Hsieh et al., 1998; Jacobs et al., 1999). Moreover, some inherited cancer syndromes result from gene defects that occur in stromal cells and induce stromal changes before epithelial abnormalities ever appear (Jacoby et al., 1997; Howe et al., 1998). These states of stromal remodeling and inflammation are precisely the conditions in which MMPs become upregulated (Mehindate et al., 1996). Thus, injury and inflammation may contribute to tumor development through MMPs that then promote the effects of carcinogens and preexisting gene defects. Likewise, the tumor promoter activity of phorbol esters may, in part, stem from their ability to upregulate stromal MMP expression (Gack et al., 1994).
Here we have shown that Str1 induces tumors in animals with and without an intact immune system. Using mammary epithelial cells that express Str1 in a Tet-regulated manner, we found that Str1 converts functionally normal mammary epithelial cells into highly infiltrative mesenchymal-like tumors in vivo. In an independent set of experiments, we demonstrated that Str1 induces spontaneous neoplastic progression in the mammary glands of WAP-Str1 transgenic mice. Although WAP-Str1 and MMTV-Str1 transgenic mice show similar proliferative changes in puberty (Sympson et al., 1994; Witty et al., 1995), neoplasms have not been described in MMTV-Str1 mice. This may reflect the reported quenching of MMTV-driven transgene expression during pregnancy, strain differences, or the long latency and low incidence of tumor formation that one would expect to see based on our own results. In WAP-Str1 mice, however, neoplastic changes arose without carcinogens or preexisting mutations, were mild or absent in the absence of Str1 expression, and were quenched by TIMP1 expression. Thus, Str1 can promote mammary carcinogenesis by virtue of its proteolytic activity.
Several other observations support the participation of stromal MMPs early in cancer development. Wild-type fibroblasts foster the growth of human breast cancer cells in nude mice, yet fibroblasts lacking MMP11/stromelysin-3 do not (Masson et al., 1998). MMP1/collagenase-1 transgenic mice show increased sensitivity to chemical carcinogens (D’Armiento et al., 1995), while MMP11 null mice have a reduced sensitivity to carcinogens (Masson et al., 1998). In addition, lack of either MMP9/gelatinase B or Str1 slows the development of squamous carcinomas in human papilloma virus-16 transgenic mice (L. M. Coussens, D. Hanahan, and Z. W., unpublished observations). Epithelial MMPs may also contribute to tumorigenesis. Indeed, the lack of MMP7/matrilysin slows intestinal adenoma formation in mice carrying the Apcmin mutation (Wilson et al., 1997), and its overexpression within mammary glands accelerates mammary tumorigenesis in mice carrying an MMTV-neu transgene (Rudolph-Owen et al., 1998). Thus, several MMPs may contribute to early neoplastic progression.
EMT Is Related to Invasive and Migratory Behavior and MMP Expression
Our results also indicate that Str1 can trigger EMT in culture and in vivo. EMT occurs during normal embryonic development (Hay, 1995) and wound repair (SundarRaj et al., 1992) when adherent epithelia become migratory and invasive. Under such conditions, MMPs are highly expressed by adjacent mesenchymal cells (Chin and Werb, 1997). EMT also occurs in high-grade cancers (Birchmeier et al., 1996; Gilles and Thompson, 1996). The most aggressive human breast cancers undergo EMT, so they lack E-cadherin (Sommers et al., 1994), coexpress cytokeratins and vimentin (Domagala et al., 1990; Sommers et al., 1994), and express MMPs, such as Str1, that are otherwise confined to stromal cells (Ahmad et al., 1998; Martorana et al., 1998). Carcinosarcomas, which are among the most aggressive cancers, represent a compelling example of EMT (Gilles and Thompson, 1996). They are extremely uncommon in mice (Squartini and Pingitore, 1994) and account for only about 0.1% of all human breast cancers (Fisher et al., 1975), yet their rate of occurrence in Str1 transgenic mice appears unusually high. As their name implies, carcinosarcomas are composed of distinct malignant cell populations that exhibit epithelial and mesenchymal features, respectively. Thus, it is not surprising that the mesenchymal-like cells of carcinosarcomas express MMP11 (Ahmad et al., 1998), which is normally confined to adjacent stromal cells. The acquisition of mesenchymal features is also consistent with the induced expression of MMP genes, including endogenous Str1, in Str1-transfected cells that have undergone EMT (Lochter et al., 1997). Altered E-cadherin and β-catenin expression are also hallmarks of EMT (Kim et al., 1998; Sun et al., 1998). Thus, the E-cadherin cleavage and β-catenin re-distribution seen following Str1 induction (Lochter et al., 1997) may have signaling implications for both EMT and tumorigenicity (Christofori and Semb, 1999).
How Do MMPs Foster Tumorigenicity?
MMPs are not mutagens. Therefore, they must promote tumor development by virtue of their ability to affect cellular signaling (Werb, 1997). MMPs can alter cell–cell and cell–ECM interactions and release bioactive fragments (Lukashev and Werb, 1998; Noe et al., 1999). For example, Str1 cleaves cell surface proteins, including E-cadherin (Lochter et al., 1997), a tumor suppressor (Christofori and Semb, 1999). MMPs also release growth factors, angiogenic factors, and their inhibitors from the ECM and cell surface (Patterson and Sang, 1997; Suzuki et al., 1997) and cleave growth factor binding proteins (Fowlkes et al., 1994) and receptors (Levi et al., 1996). They can induce a reactive stroma and cause recruitment of other host cells (Thomasset et al., 1998), and they generate cleavage products that may compromise cellular cytotoxicity (Kataoka et al., 1999). Thus, there are several ways in which MMPs can influence all stages of cancer progression, including initiation.
We favor the hypothesis that MMPs act to trigger the E-cadherin/β-catenin pathway, which can be linked to several aspects of cancer, including EMT, invasion, and genomic instability (Tlsty, 1998; Noe et al., 1999). In support of our hypothesis, the E-cadherin cleavage and β-catenin redistribution seen with Str1-induced EMT are accompanied by upregulation of cyclin D1 (M. E. Lukashev and Z. W., unpublished results), a β-catenin-regulated oncogene (Tetsu and McCormick, 1999). Thus, Str1-induced E-cadherin cleavage may trigger both normal developmental and abnormal neoplastic changes.
MMPs also exert comparable effects in development and cancer. It is noteworthy that Str1 stimulates ductal proliferation and branching during puberty (Sympson et al., 1994; Witty et al., 1995) but induces apoptosis in anchorage-dependent secretory epithelium during pregnancy (Alexander et al., 1996; Thomasset et al., 1998). These seemingly contradictory actions can be reconciled by noting that the normal developmental fates of the target cells differ. Mammary ducts contain stem cells that are triggered to divide during branching morphogenesis. They also persist throughout involution, whereas alveolar cells do not. These effects are also consistent with the process of neoplastic transformation. The normal function of Str1 in inducing ductal proliferation and invasion during puberty is precisely what Str1 does to transformed mammary cells. Furthermore, although the induction of apoptosis in alveolar cells may defy tumorigenesis, it could also provide pressure for the selection of anchorage-independent, apoptosis-resistant clones and thereby foster tumorigenicity. Taken together with the propensity of Str1 to foster an altered stromal microenvironment (Thomasset et al., 1998), Str1 has the hallmarks of a multifactorial tumor promoter.
Do MMPs Stimulate Genomic Instability?
The presence of spontaneous tumors in Str1 transgenic mice indicates that Str1 promotes either the accumulation of mutations or the survival of mutant cells. By altering cellular adhesion, MMPs could conceivably alter cell cycle checkpoint controls and promote genomic instability (Tlsty, 1998). The presence of recurrent DNA losses in both premalignant and malignant mammary lesions in Str1 transgenic mice and of consistent DNA copy number gains in undifferentiated tumors suggests that these loci contain recessive- and dominant-acting genes that contribute to early and late cancer progression, respectively. They also support the hypothesis that MMPs can produce an abnormal stromal environment within which clones of epithelial cells containing selected mutations may accumulate. It is intriguing that the WAP-Str1 tumors are histologically diverse and arise late, suggesting a stochastic evolution, yet they exhibit stereotyped genomic changes, suggesting a common tumorigenic pathway. Several tumors did, however, exhibit the unifying feature of EMT together with consistent DNA gains on chromosomes 6 and 15. Although c-myc is located on mouse chromosome 15 (Adolph et al., 1987), its expression failed to correlate with DNA changes (M. D. S. et al., unpublished results). Thus, other relevant genes may reside at these loci.
Most of the genomic changes in the WAP-Str1 transgenic mice were distinct from those in other transgenic mammary tumor models. For example, p53-deficient Wnt-1 transgenicmice exhibit recurrent genomic changes on several chromosomes, including 4, but not 6, 7, or 15 (Donehower et al., 1995). Likewise, 82% of mammary tumors in MMTV-neu transgenic mice exhibit loss of heterozygosity on chromosome 4 (Ritland et al., 1997), and chromosome 4 deletions were also particularly prevalent in our own study. Thus, our results further implicate the middistal region of chromosome 4 as a potential tumor suppressor locus. Mammary tumors in SV40 T-antigen transgenic mice show consistent DNA gains in the telomeric region of chomosome 6 (Liu et al., 1998), rather than at its centromeric end as we observed in high-grade tumors of Str1 transgenic mice. Thus, our data also indicate a number of novel loci of potential importance.
Implications for Human Cancer
If MMPs function in human cancer as they do in mice, then MMP mutations, amplifications, or polymorphisms may be associated with tumor development. Few studies have addressed this possibility despite the frequent cloning of MMPs as tumor-specific genes. Interestingly, a polymorphism in the human MMP1 gene promoter that creates a transcription-enhancing Ets site occurs more often in tumor cell lines than in the general population (Rutter et al., 1998). This suggests that enhanced MMP1 transcription may contribute to cancer susceptibility and supports the enhanced skin carcinogenesis seen in MMP1 transgenic mice (D’Armiento et al., 1995). Epigenetic inactivation of the TIMP3 promoter, seen often in human cancers (Bachman et al., 1999), could have similar implications. A functional polymorphism has also been found in the Str1 promoter (Ye et al., 1996); however, its role in cancer remains unexplored.
Our results may also have implications concerning the therapeutic use of MMP inhibitors. Inhibition of Str1 by overexpression of TIMP1 quenched its ability to promote neoplasia in transgenic mice, indicating that active Str1 is required and that neoplasia can be suppressed if its activity is inhibited early on. Thus, a compelling argument could be made for inhibiting Str1 during any stage of tumor progression. However, once the neoplastic process was triggered in Str1-transfected cells, tumors could still form without continued Str1 expression. Thus, our results also indicate that once Str1 effects alter the phenotype and genotype of mammary cells, its activity is no longer required for tumorigenicity. Likewise, cells induced to express Str1 for 6 days in culture continue to undergo progressive EMT despite addition of Tet and an MMP inhibitor (Lochter et al., 1997). Thus, the converted cells may perpetuate further EMT by an MMP-independent feedback mechanism. This “hit-and-run” action of Str1 is also in keeping with the capacity of MMPs to affect signaling. Thus, although MMPs are expressed throughout tumor progression, and although MMP inhibitors may defy invasion, other stages of progression may become resistant to anti-proteinase therapy targeting Str1. Whether this is true for other enzymes remains to be determined.
Our findings thus indicate that Str1 can promote early neoplastic changes, stereotyped genomic changes, and late phenotypic conversions associated with aggressive tumor behavior. They also support the hypothesis that an altered stromal environment can promote neoplastic transformation. Elucidation of the pathways downstream from Str1 will be critical for defining new molecular targets.
Experimental Procedures
Cell Culture
Recombinant human Str1 (0.8 mg/ml; a gift from Dr. M. Navre, Affymax Research Institute) was prepared as described previously (Lochter et al., 1999) and activated by treatment with trypsin for 30 min at 37°C, followed by addition of soybean trypsin inhibitor. Scp2 cells were treated every other day for 6 days with 1 µg/ml activated Str1 in serum-free DMEM/F12 medium containing 5 µg/ml insulin, 5 µg/ml transferrin, 5 ng/ml selenium, and 50 µg/ml gentamicin with or without the hydroxamic acid metalloproteinase inhibitor GM6001 (10 µM; a gift from Dr. R. Galardy, Glycomed Corp.). All other culture and immunocytochemistry methods were performed as previously described (Lochter et al., 1997).
Tumorigenicity Assay
The developing epithelial parenchyma of abdominal (#4) mammary glands was removed from weanling scid/scid mice (DeOme et al., 1959), and 1 × 106 Scp2 or p2S cells in serum-free medium were injected into residual gland-free mammary fat pads or subcutaneously at the nape of the neck. Mice were maintained for 6 or more weeks with or without 10 µg/ml Tet in their drinking water. Inhibition of enzymatic activity by intraperitoneal injection of GM6001 (100 mg/kg/day) was not pursued, due to inhibited wound repair and postsurgical morbidity that was not seen for the carrier (4% carboxy-methylcellulose in PBS). Tumor volumes were calculated as length × width2/2.
Transgenic Mice
CD1 mice with an autoactivating rat Str1 transgene targeted to mammary epithelium by the murine WAP gene promoter were generated as described (Sympson et al., 1994). Five independent transgenic founder lines (M2-5, M2-20, M2-21, M2-25, and M1-9), their nontransgenic littermates, and two transgenic sublines that had lost expression of the transgene (M2-5N and M2-21N) were analyzed. All mice were housed under similar conditions, and a similar fraction from each group (approximately one-third) was carried through pregnancy and lactation. The CA10 WAP gene promoter (Sympson et al., 1994) was also used to generate transgenic mice overexpressing a human TIMP1 transgene (Alexander et al., 1996). These mice expressed human TIMP1 protein primarily during pregnancy and lactation (data not shown). WAP-TIMP1 mice were crossed with one line (M2-5) of WAP-Str1 mice to generate double transgenics. Half of these were carried through at least one pregnancy and lactation.
Histopathology
Mammary whole mounts (Sympson et al., 1994) were photodocumented and reprocessed for paraffin embedment. Hematoxylin/eosin, Masson’s trichrome, and Alcian blue staining were by standard methods. Antigen retrieval was by brief 0.4 µg/ml proteinase K digestion for vimentin or by microwave heating in citrate buffer. Before adding peroxidase (HRP)-conjugated reagents, endogenous peroxidase activity was blocked with a methanol/H2O2 solution. Immunolocalization was by rat anti-mouse cytokeratin-8 (a gift from Dr. R. Kemler; 1:50) and biotinylated rabbit anti-rat IgG (Vector Laboratories; 1:200), HRP-conjugated mouse anti-bovine vimentin (DAKO; prediluted), or biotinylated rat anti-mouse smooth muscle actin (a gift from Dr. L. R. Lund; 1:50). Biotinylated antibodies were detected with avidin-biotin-HRP complexes. HRP activity was visualized with diaminobenzidine, and nuclei were counterstained with Meyer’s hematoxylin.
Comparative Genomic Hybridization
DNAs were extracted from cultured cells, frozen tissues, or paraffin blocks by standard methods or from lightly stained paraffin sections after laser microdissection (Emmert-Buck et al., 1996). Reference and test DNAs labeled with Texas red-5-dCTP and fluorescein-12-dCTP, respectively, were hybridized to normal metaphase chromosome spreads; chromosomes were identified by 4,6-diamino-2-phenylindole (DAPI) counterstaining; and green:red fluorescence intensity profiles were obtained as previously described (Bain et al., 1997).
Acknowledgments
We thank R. Boudreau, J. Xie, D. R. Williams, Y.-P. Shi, and Drs. R. D. Cardiff, M. E. Lukashev, S. Galosy, and H. Sanchez for their assistance. This work was supported by grants from the National Cancer Institute (CA57621, CA72006, and CA64786), the UCSF Breast Cancer SPORE (Bishop Fund), the United States Army Medical Research and Materiel Command (DAMD17-97-1-7246), the California Breast Cancer Research Program (to A. L.), the Association pour la Recherche Contre le Cancer (to J.-P. R.), and the United States Department of Energy (DE-AC03-76-SF00098).
References
- Adolph S, Bartram CR, Hameister H. Mapping of the oncogenes Myc, Sis, and int-1 to the distal part of mouse chromosome 15. Cytogenet. Cell Genet. 1987;44:65–68. doi: 10.1159/000132345. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Hanby A, Dublin E, Poulsom R, Smith P, Barnes D, Rubens R, Anglard P, Hart I. Stromelysin 3: an independent prognostic factor for relapse-free survival in node-positive breast cancer and demonstration of novel breast carcinoma cell expression. Am. J. Pathol. 1998;152:721–728. [PMC free article] [PubMed] [Google Scholar]
- Alexander CM, Howard EW, Bissell MJ, Werb Z. Rescue of mammary epithelial cell apoptosis and entactin degradation by a tissue inhibitor of metalloproteinases-1 transgene. J. Cell Biol. 1996;135:1669–1677. doi: 10.1083/jcb.135.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman KE, Herman JG, Corn PG, Merlo A, Costello JF, Cavenee WK, Baylin SB, Graff JR. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggests a suppressor role in kidney, brain, and other human cancers. Cancer Res. 1999;59:798–802. [PubMed] [Google Scholar]
- Bain G, Engel I, Robanus Maandag EC, te Riele HP, Voland JR, Sharp LL, Chun J, Huey B, Pinkel D, Murre C. E2A deficiency leads to abnormalities in alphabeta T-cell development and to rapid development of T-cell lymphomas. Mol. Cell. Biol. 1997;17:4782–4791. doi: 10.1128/mcb.17.8.4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Brand-Saberi B. Epithelial-mesenchymal transitions in cancer progression. Acta Anat. 1996;156:217–226. doi: 10.1159/000147848. [DOI] [PubMed] [Google Scholar]
- Borsi L, Carnemolla B, Nicol G, Spina B, Tanara G, Zardi L. Expression of different tenascin isoforms in normal, hyper-plastic and neoplastic human breast tissues. Int. J. Cancer. 1992;52:688–692. doi: 10.1002/ijc.2910520504. [DOI] [PubMed] [Google Scholar]
- Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JR, Werb Z. Matrix metalloproteinases regulate morphogenesis, migration and remodeling of epithelium, tongue skeletal muscle and cartilage in the mandibular arch. Development. 1997;124:1519–1530. doi: 10.1242/dev.124.8.1519. [DOI] [PubMed] [Google Scholar]
- Christofori G, Semb H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Bio-chem. Sci. 1999;24:73–76. doi: 10.1016/s0968-0004(98)01343-7. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Matrix metalloproteinases and the development of cancer. Chem. Biol. 1996;3:895–904. doi: 10.1016/s1074-5521(96)90178-7. [DOI] [PubMed] [Google Scholar]
- D’Armiento J, DiColandrea T, Dalal SS, Okada Y, Huang MT, Conney AH, Chada K. Collagenase expression in transgenic mouse skin causes hyperkeratosis and acanthosis and increases susceptibility to tumorigenesis. Mol. Cell. Biol. 1995;15:5732–5739. doi: 10.1128/mcb.15.10.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeOme KB, Faulkin LJJ, Bern HA, Blair PE. Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland-free mammary fat pads of female C3H mice. Cancer Res. 1959;19:515–520. [PubMed] [Google Scholar]
- Domagala W, Lasota J, Bartkowiak J, Weber K, Osborn M. Vimentin is preferentially expressed in human breast carcinomas with low estrogen receptor and high Ki-67 growth fraction. Am. J. Pathol. 1990;136:219–227. [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Godley LA, Aldaz CM, Pyle R, Shi YP, Pinkel D, Gray J, Bradley A, Medina D, Varmus HE. Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes Dev. 1995;9:882–895. doi: 10.1101/gad.9.7.882. [DOI] [PubMed] [Google Scholar]
- Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA, Liotta LA. Laser capture microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- Fisher ER, Gregorio RM, Fisher B, Redmond C, Vellios F, Sommers SC. The pathology of invasive breast cancer. A syllabus derived from findings of the National Surgical Adjuvant Breast Project (protocol no. 4) Cancer. 1975;36:1–85. doi: 10.1002/1097-0142(197507)36:1<1::aid-cncr2820360102>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Fowlkes JL, Enghild JJ, Suzuki K, Nagase H. Matrix metalloproteinases degrade insulin-like growth factor-binding protein-3 in dermal fibroblast cultures. J. Biol. Chem. 1994;269:25742–25746. [PubMed] [Google Scholar]
- Gack S, Vallon R, Schaper J, Ruther U, Angel P. Phenotypic alterations in fos-transgenic mice correlate with changes in Fos/Jun-dependent collagenase type I expression. Regulation of mouse metalloproteinases by carcinogens, tumor promoters, cAMP, and Fos oncoprotein. J. Biol. Chem. 1994;269:10363–10369. [PubMed] [Google Scholar]
- Gilles C, Thompson EW. The epithelial to mesenchymal transition and metastatic progression in carcinoma. Breast J. 1996;2:83–96. [Google Scholar]
- Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- Hirai Y, Lochter A, Galosy S, Koshida S, Niwa S, Bissell MJ. Epimorphin functions as a key morphoregulator for mammary epithelial cells. J. Cell Biol. 1998;140:159–169. doi: 10.1083/jcb.140.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe JR, Roth S, Ringold JC, Summers RW, Järvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res. 1998;58:3942, 3945. [PubMed] [Google Scholar]
- Jacobs TW, Byrne C, Colditz G, Connolly JL, Schnitt SJ. Radial scars in benign breast-biopsy specimens and the risk of breast cancer. N. Engl. J. Med. 1999;340:430–436. doi: 10.1056/NEJM199902113400604. [DOI] [PubMed] [Google Scholar]
- Jacoby RF, Schlack S, Cole CE, Skarbek M, Harris C, Meisner LF. A juvenile polyposis tumor suppressor locus at 10q22 is deleted from nonepithelial cells in the lamina propria. Gastroenterology. 1997;112:1398–1403. doi: 10.1016/s0016-5085(97)70156-2. [DOI] [PubMed] [Google Scholar]
- Kataoka H, Uchino H, Iwamura T, Seiki M, Nabeshima K, Koono M. Enhanced tumor growth and invasiveness in vivo by a carboxyl-terminal fragment of α-proteinase inhibitor generated by matrix metalloproteinases: a possible modulatory role in natural killer cytotoxicity. Am. J. Pathol. 1999;154:457–468. doi: 10.1016/s0002-9440(10)65292-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Daniels KJ, Hay ED. Tissue-specific expression of β-catenin in normal mesenchyme and uveal melanomas and its effect on invasiveness. Exp. Cell Res. 1998;245:79–90. doi: 10.1006/excr.1998.4238. [DOI] [PubMed] [Google Scholar]
- Levi E, Fridman R, Miao HQ, Ma YS, Yayon A, Vlodavsky I. Matrix metalloproteinase 2 releases active soluble ectodomain of fibroblast growth factor receptor 1. Proc. Natl. Acad. Sci. USA. 1996;93:7069–7074. doi: 10.1073/pnas.93.14.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ML, Von Lintig FC, Liyanage M, Shibata MA, Jorcyk CL, Ried T, Boss GR, Green JE. Amplification of Kiras and elevation of MAP kinase activity during mammary tumor progression in C3(1)/SV40 Tag transgenic mice. Oncogene. 1998;17:2403–2411. doi: 10.1038/sj.onc.1202456. [DOI] [PubMed] [Google Scholar]
- Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 1997;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter A, Muschler J, Navre M, Werb Z, Bissell MJ. α1 and α2 integrins mediate invasive activity of mouse mammary carcinoma cells through regulation of stromelysin-1 expression. Mol. Biol. Cell. 1999;10:271–282. doi: 10.1091/mbc.10.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashev ME, Werb Z. ECM signaling: orchestrating cell behaviour and misbehaviour. Trends Cell Biol. 1998;8:437–441. doi: 10.1016/s0962-8924(98)01362-2. [DOI] [PubMed] [Google Scholar]
- Lund LR, Rømer J, Thomasset N, Solberg H, Pyke C, Bissell MJ, Danø K, Werb Z. Two distinct phases of apoptosis in mammary gland involution: proteinase-independent and-dependent pathways. Development. 1996;122:181–193. doi: 10.1242/dev.122.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martorana AM, Zheng G, Crowe TC, O’Grady RL, Lyons JG. Epithelial cells up-regulate matrix metalloproteinases in cells within the same mammary carcinoma that have undergone an epithelial-mesenchymal transition. Cancer Res. 1998;58:4970–4979. [PubMed] [Google Scholar]
- Masson R, Lefebvre O, Noël A, Fahime ME, Chenard MP, Wendling C, Kebers F, LeMeur M, Dierich A, Foidart JM, et al. In vivo evidence that the stromelysin-3 metalloproteinase incontributes in a paracrine manner to epithelial cell malignancy. J. Cell Biol. 1998;140:1535–1541. doi: 10.1083/jcb.140.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrisian LM, Glaichenhaus N, Gesnel MC, Breathnach R. Epidermal growth factor and oncogenes induce transcription of the same cellular mRNA in rat fibroblasts. EMBO J. 1985;4:1435–1440. doi: 10.1002/j.1460-2075.1985.tb03799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehindate K, al-Daccak R, Aoudjit F, Damdoumi F, Fortier M, Borgeat P, Mourad W. Interleukin-4, transforming growth factor beta 1, and dexamethasone inhibit superantigen-induced prostaglandin E2-dependent collagenase gene expression through their action on cyclooxygenase-2 and cytosolic phospholipase A2. Lab. Invest. 1996;75:529–538. [PubMed] [Google Scholar]
- Muller D, Quantin B, Gesnel MC, Millon-Collard R, Abecassis J, Breathnach R. The collagenase gene family in humans consists of at least four members. Biochem. J. 1988;253:187–192. doi: 10.1042/bj2530187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noe V, Willems J, Vandekerckhove J, Van Roy F, Bruyneel E, Mareel M. Inhibition of adhesion and induction of epithelial invasion by HAV-containing E-cadherin-specific peptides. J. Cell Sci. 1999;112:127–135. doi: 10.1242/jcs.112.1.127. [DOI] [PubMed] [Google Scholar]
- Ostrowski LE, Finch J, Krieg P, Matrisian L, Patskan G, O’Connell JF, Phillips J, Slaga TJ, Breathnach R, Bowden GT. Expression pattern of a gene for a secreted metalloproteinase during late stages of tumor progression. Mol. Carcinog. 1988;1:13–19. doi: 10.1002/mc.2940010106. [DOI] [PubMed] [Google Scholar]
- Patterson BC, Sang QXA. Angiostatin-converting enzyme activities of human matrilysin (MMP-7) and gelatinase B/type IV collagenase (MMP-9) J. Biol. Chem. 1997;272:28823–28825. doi: 10.1074/jbc.272.46.28823. [DOI] [PubMed] [Google Scholar]
- Ritland SR, Rowse GJ, Chang Y, Gendler SJ. Loss stromely of heterozygosity analysis in primary mammary tumors and lung metastases of MMTV-MTAg and MMTV-neu transgenic mice. Cancer Res. 1997;57:3520–3525. [PubMed] [Google Scholar]
- Rønnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol. Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- Rudolph-Owen LA, Chan R, Muller WJ, Matrisian LM. The matrix metalloproteinase matrilysin influences early-stage mammary tumorigenesis. Cancer Res. 1998;58:5500–5506. [PubMed] [Google Scholar]
- Rutter JL, Mitchell TI, Butticé G, Meyers J, Gusella JF, Ozelius LJ, Brinckerhoff CE. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter creates an Ets binding site and augments transcription. Cancer Res. 1998;58:5321–5325. [PubMed] [Google Scholar]
- Sommers CL, Byers SW, Thompson EW, Torri JA, Gelmann EP. Differentiation state and invasiveness of human breast cancer cell lines. Breast Cancer Res. Treat. 1994;31:325–335. doi: 10.1007/BF00666165. [DOI] [PubMed] [Google Scholar]
- Squartini F, Pingitore R. Tumours of the mammary gland. In: Turusov VS, Mohr U, editors. Pathology of Tumours in Laboratory Animals. Lyon, France: IARC Scientific Publications No. 111; 1994. pp. 47–100. [PubMed] [Google Scholar]
- Sternlicht MD, Werb Z. ECM proteinases. In: Kreis T, Vale R, editors. Guide-book to the Extracellular Matrix and Adhesion Proteins. New York: Oxford University Press; 1999. pp. 503–562. [Google Scholar]
- Sun D, McAlmon KR, Davies JA, Bernfield M, Hay ED. Simultaneous loss of expression of syndecan-1 and E-cadherin in the embryonic palate during epithelial-mesenchymal transformation. Int. J. Dev. Biol. 1998;42:733–736. [PubMed] [Google Scholar]
- SundarRaj N, Rizzo JD, Anderson SC, Gesiotto JP. Expression of vimentin by rabbit corneal epithelial cells during wound repair. Cell Tissue Res. 1992;267:347–356. doi: 10.1007/BF00302973. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Raab G, Moses MA, Fernandez CA, Klagsbrun M. Matrix metalloproteinase-3 releases active heparin-binding EGF-like growth factor by cleavage at a specific juxtamembrane site. J. Biol. Chem. 1997;272:31730–31737. doi: 10.1074/jbc.272.50.31730. [DOI] [PubMed] [Google Scholar]
- Sympson CJ, Talhouk RS, Alexander CM, Chin JR, Clift SM, Bissell MJ, Werb Z. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J. Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. Erratum: J. Cell Biol. 132(4), 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhouk RS, Bissell MJ, Werb Z. Coordinated expression of extracellular matrix-degrading proteinases and their inhibitors regulates mammary epithelial function during involution. J. Cell Biol. 1992;118:1271–1282. doi: 10.1083/jcb.118.5.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Thomasset N, Lochter A, Sympson CJ, Lund LR, Williams DR, Behrendtsen O, Werb Z, Bissell MJ. Expression of autoactivated stromelysin-1 in mammary glands of transgenic mice leads to a reactive stroma during early development. Am. J. Pathol. 1998;153:457–467. doi: 10.1016/S0002-9440(10)65589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty TD. Cell-adhesion-dependent influences on genomic instability and carcinogenesis. Curr. Opin. Cell Biol. 1998;10:647–653. doi: 10.1016/s0955-0674(98)80041-0. [DOI] [PubMed] [Google Scholar]
- Wargotz ES, Norris HJ. Metaplastic carcinomas of the breast. I. Matrix-producing carcinoma. Hum. Pathol. 1989;20:628–635. doi: 10.1016/0046-8177(89)90149-4. [DOI] [PubMed] [Google Scholar]
- Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- Wilson CL, Heppner KJ, Labosky PA, Hogan BL, Matrisian LM. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc. Natl. Acad. Sci. USA. 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty JP, Wright JH, Matrisian LM. Matrix metalloproteinases are expressed during ductal and alveolar mammary morphogenesis, and misregulation of stromelysin-1 in transgenic mice induces unscheduled alveolar development. Mol. Biol. Cell. 1995;6:1287–1303. doi: 10.1091/mbc.6.10.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S, Eriksson P, Hamsten A, Kurkinen M, Humphries SE, Henney AM. Progression of coronary atherosclerosis is associated with a common genetic variant of the human stromelysin-1 promoter which results in reduced gene expression. J. Biol. Chem. 1996;271:13055–13060. doi: 10.1074/jbc.271.22.13055. [DOI] [PubMed] [Google Scholar]