

Abstract
Niemann-Pick Type C (NPC) disease is a fatal, neurodegenerative disorder, caused in most cases by mutations in the late endosomal protein NPC1. A hallmark of NPC disease is endosomal cholesterol accumulation and an impaired cholesterol homeostatic response, which might affect cholesterol transport to mitochondria and, thus, mitochondrial and cellular function. This study aimed to characterize mitochondrial cholesterol homeostasis in NPC disease. Using wild-type and NPC1-deficient Chinese hamster ovary cells, stably transfected with a CYP11A1 complex to assess mitochondrial cholesterol import by pregnenolone production, we show that cholesterol transport to the mitochondrial inner membrane is not affected by loss of NPC1. However, mitochondrial cholesterol content was higher in NPC1-deficient than in wild-type cells. Cholesterol transport to the mitochondrial inner membrane increased markedly upon exposure of cholesterol-deprived cells to lipoproteins, indicating transport of endosomal cholesterol to mitochondria. Reduction of endosomal metastatic lymph node protein 64 (MLN64) by RNA interference decreased cholesterol transport to the mitochondrial inner membrane and reduced mitochondrial cholesterol levels in NPC1-deficient cells, suggesting that MLN64 transported cholesterol to mitochondria even in the absence of NPC1. In summary, this study describes a transport pathway for endosomal cholesterol to mitochondria that requires MLN64, but not NPC1, and that may be responsible for increased mitochondrial cholesterol in NPC disease.
Keywords: mitochondrial cholesterol, STAR-related lipid transfer domain, steroidogenic acute regulatory protein, pregnenolone, steroids, oxysterols, metastatic lymph node protein 64
Niemann-Pick Type C (NPC) disease is a fatal, neurodegenerative disorder caused by mutations in NPC1 or NPC2 (1, 2). NPC1 is a late endosomal transmembrane protein with a sterol-sensing domain homologous to the cholesterol sensor sterol regulatory element binding protein cleavage activating protein (3, 4); NPC2 is a small lumenal protein in late endosomes/lysosomes (5, 6). Both NPC1 and NPC2 can bind cholesterol (5–8); NPC1 also has affinity for oxysterols (9). All NPC1-deficient cells accumulate cholesterol in late endosomal multivesicular bodies and have an impaired homeostatic response of the sterol regulatory element binding protein pathway to exogenous cholesterol (10). Based on these observations, it was proposed that NPC1 acts as a cholesterol sensor or transporter and is required for cholesterol egress from endosomes to plasma membrane and to the regulatory pool in the endoplasmic reticulum (7, 10). NPC1-deficient cells also sequester a variety of other lipids in their endosomes (11, 12), and the possibility remains that cholesterol accumulation is not the primary defect. The exact function of NPC1 and how its loss leads to the observed neuropathology remain unknown.
Regardless of the primary storage material in NPC1-deficient endosomes, the sequestration of cholesterol has a widespread impact on cellular cholesterol distribution. Recently, it has been proposed that changes in mitochondrial cholesterol contribute to NPC pathology (13). Cholesterol is required as a precursor for steroid and oxysterol synthesis at the mitochondrial inner membrane and as a component of mitochondrial membranes. Increased mitochondrial cholesterol can lead to mitochondrial dysfunction, including reduced fluidity of mitochondrial membranes (14, 15), reduced ATP generation (16), and decreased mitochondrial glutathione import (15).
In line with the theory that cholesterol entrapment in NPC1-deficient endosomes limits its availability to the rest of the cell, it was found that steroid and oxysterol levels were decreased in NPC1-deficient murine brain and fibroblasts (17–22). In seeming contradiction, several groups have reported increased cholesterol in mitochondria isolated from brain or liver of NPC1-deficient mice (15, 16, 23).
The mechanisms by which cholesterol is transported to the mitochondrial outer and inner membranes under basal conditions are not well defined. In steroidogenic cells, cholesterol transport to the mitochondrial inner membrane, which is rate limiting for steroid synthesis, is mediated by the steroidogenic acute regulatory (StAR) protein in conjunction with the translocator protein (formerly known as peripheral benzodiazepine receptor) (24, 25). However, StAR-mediated transport is low under basal, nonstimulated conditions and in nonsteroidogenic cells, which do not express significant amounts of StAR. Since even under these conditions, cholesterol is needed for the upkeep of mitochondrial membranes and in some cases for oxysterol synthesis, other mechanisms of mitochondrial cholesterol import must exist. Recently, it was proposed that plasma membrane cholesterol transported via cytosolic transport proteins served as a source for mitochondrial oxysterol production (26), but the mechanism was not defined in detail. Other potential mediators of mitochondrial cholesterol import include proteins that contain a lipid-binding domain homologous to the C terminus of StAR [START proteins (27)]. Among these, endosomal metastatic lymph node protein 64 (MLN64) is of particular interest, since its START domain binds cholesterol and it has been shown to transport cholesterol to mitochondria when expressed as a soluble protein lacking the transmembrane N terminus of MLN64 (28–30).
In view of the strong link between mitochondrial cholesterol and mitochondrial function and the central role of mitochondria in neurodegenerative disease (31), this study was designed to elucidate mechanisms of basal mitochondrial cholesterol import in wild-type and NPC1-deficient cells. We show that mitochondrial cholesterol import does not require functional NPC1 and that MLN64 can mediate cholesterol transport from endosomes to mitochondria under basal conditions, its contribution likely depending on the relative availability of cholesterol from different pools. Furthermore, our data indicate that in NPC1-deficient cells, transfer of cholesterol from outer to inner mitochondrial membrane may become rate limiting, leading to cholesterol buildup in mitochondrial outer membranes.
MATERIALS AND METHODS
Materials
Cell culture media, FBS, and supplements were obtained from Invitrogen. Geneticin was from Wisent Inc. Trilostane and 22R-hydroxycholesterol (22-OH-Chol) were purchased from Steraloids. The rabbit anti-pregnenolone antibody was purchased from MP Biologicals. Charcoal-coated dextran, filipin complex III, and mouse antitubulin antibodies were purchased from Sigma. Rabbit anti-human NPC1 antibodies that fully cross-react with NPC1 from mouse and hamster were obtained from Novus Biologicals. MLN64 antibodies were from Affinity Bioreagents or Abcam, goat anti-actin and rabbit anti-mouse Tom20 antibodies were from Santa Cruz Biotechnologies, and mouse anti-protein disulphide isomerase was from Assay Designs. Rabbit antibodies directed against the voltage-dependent anion channel (VDAC) or against lysosome-associated membrane protein (LAMP1), which cross-react with hamster VDAC or LAMP1, and rabbit anti-ferredoxin reductase antibodies were obtained from Abcam. Mitotracker Red CMXRos and Dynabeads M500 Subcellular were purchased from Invitrogen. Short interfering RNA (siRNA) sequences and transfection agent (dharmaFECT 4) were obtained from Dharmacon. [7-3H(N)]pregnenolone (1 mCi/ml and 11.5 Ci/mmol) was obtained from Perkin-Elmer Life Sciences. All other chemicals were from Sigma or Fisher Scientific. Human LDL was isolated from EDTA-plasma of apparently healthy, normolipidemic volunteers by KBr density gradient ultracentrifugation (32). Informed consent was obtained from all human volunteers, and the protocol was approved by the Human Research Ethics Board, Dalhousie University. Lipoprotein-deficient serum (LPDS) was prepared by density ultracentrifugation from FBS (32). All protein assays were performed with the BCA kit (Pierce).
Cell lines and expression vector
Wild-type Chinese hamster ovary (CHO) and NPC1-deficient 4-4-19 cell lines were a generous gift from L. Liscum (Tufts University, Boston, MA) and have been described and characterized previously (33, 34). The 4-4-19 cell line expresses a nonfunctional NPC1 protein with a point mutation (Gly660Arg) in the sterol-sensing domain (L. Liscum, personal communication). To simplify the description of these cell lines together with the murine model carrying a null mutation in NPC1, we use the term “NPC1-deficiency” to include NPC1 dysfunction and loss of NPC1 protein. The expression vector F2-pcDNA3 encodes a fusion protein (F2) of human CYP11A1 (P450 side chain cleavage complex), ferredoxin reductase, and ferredoxin 1 (35, 36) and was a kind gift from W. L. Miller (University of California, San Francisco, CA). The open reading frame was excised from the pcDNA3 backbone using the restriction enzymes EcoRI and KpnI and inserted into the pcDNA3.1(+) backbone (Invitrogen). To create cell lines stably expressing F2-pcDNA3.1, CHO and 4-4-19 cells were transfected with the F2-pcDNA3.1 vector using electroporation (Microporator; Montreal Biotech). Monoclonal colonies were selected for their survival in 500 µg/ml geneticin following serial dilution. Cells were cultured in Ham's F12 medium containing 5% FBS, antibiotics, and 300 µg/ml geneticin to keep the cells under selection pressure. Hereafter, cells stably expressing the F2-fusion protein will be designated with the prefix “F2.” Expression of F2-fusion complex was measured by RT-PCR. Total RNA was extracted from F2 cells using Trizol and reverse transcribed using Superscript II (Invitrogen), and cDNA was amplified using primers directed against cyclophilin and F2 [cyclophilin forward, 5′-TCTTCTTGCTGGTCTTGCCATTCC-3′; reverse, 5′-TCCAAAGACAGCAGAAAACTTTCG-3′; F2 forward (P450scc), 5′-AGTGGCCATCTATGCTCTGG-3′; reverse (ferredoxin reductase), 5′-ATGTCCGTTCTCTCCAGGTG-3′]. PCR products and DNA size standards (Fermentas) were separated on a 1.5% agarose gel and visualized with GelRed (Biotium).
In vitro CYP11A1 activity assay in lysed cells and subfractions
Cells were harvested into PBS and homogenized in 1 ml ice-cold mitochondria isolation (MI) buffer (5 mM HEPES, 250 mM mannitol, 1 mM EGTA, 70 mM sucrose, pH 7.4, and protease inhibitors) with 30 strokes in a Dounce homogenizer. Cell homogenates were centrifuged at 800 g, 4°C for 5 min to remove nuclei and unbroken cells, followed by centrifugation of the supernatant at 12,000 g, 4°C for 15 min to yield crude mitochondria. Samples were used immediately or frozen at −20°C for no longer than 1 week. CYP11A1 activity was determined essentially as described (37). Cell homogenates, crude mitochondria, or the 12,000 g supernatant were incubated for 1 h at 37°C in reaction buffer (250 mM sucrose, 20 mM KCl, 15 mM Tris-EDTA-HCl, 10 mM KH2PO4, and 5 mM MgCl2, pH 7.2) with 2 mM NADPH, 2 mM malate/pyruvate, 5 μM 22-OH-Chol, 0.3% Tween, and an NADPH regenerating system (BD Biosciences). Trilostane (10 μM) was added during all incubations for pregnenolone measurement to inhibit conversion of pregnenolone to downstream steroids (38). Reaction mixtures were assayed for pregnenolone by radioimmunoassay (RIA) according to the protocol provided by MP Biologicals.
Measurement of cholesterol transport to the mitochondrial inner membrane
Cells were plated at a density of 200 cells/mm2 and grown for 48 h in growth medium. Cells were then washed twice in phenol red-free, serum-free Ham's F12/DMEM (1:1, v/v) (import medium) and incubated for 6 or 24 h in import medium containing 10 µM trilostane. In experiments to determine the maximum rate of pregnenolone formation, 5 µM 22-OH-Chol was added to the import medium. Import medium was collected for measurement of pregnenolone by RIA. Each assay included a standard curve of known amounts of pregnenolone and a positive control of import medium with trilostane spiked with 300 pg pregnenolone to test recovery. A negative control of import medium (containing trilostane) that was not incubated with cells was routinely included but did not give readings above background. Where indicated, cells were incubated for 48 h in Ham's F12 medium containing 5% LPDS with or without 50 µg/ml LDL and then washed and incubated for 24 h in import medium with 10 µM trilostane with or without 50 µg/ml LDL. Import medium and cells were collected and analyzed as above.
Immunolocalization of the F2 fusion complex
CHO cells grown on coverslips were transiently transfected with F2-pcDNA3.1 vector by electroporation. Two days after transfection, cells were incubated with 50 nM Mitotracker Red CMXRos (Invitrogen) according to the manufacturer's protocol and then washed and fixed with 4% paraformaldehyde. Cells were permeabilized with 0.1% Triton X-100 for 5 min and blocked with 1% BSA in PBS followed by sequential incubation with antiferredoxin reductase antibody (1:100) and Cy2-conjugated affinity-purified donkey anti-rabbit IgG (Jackson Immunoresearch). Images were acquired on a Nikon TE2000 epifluorescence microscope equipped with a CCD camera (Orca-AG; Hamamatsu) at filter settings of 474/23 nm and 585/29 nm (excitation), dual-band dichroic, and 572/42 nm and 645/49 nm (emission), using a 60× oil immersion objective. Using these conditions, Mitotracker staining alone did not yield measurable fluorescence in the green channel.
Filipin staining
Cells grown on glass coverslips were fixed and stained with 50 µg/ml filipin as described (39). Images were acquired on a Nikon TE2000 epifluorescence microscope with CCD camera at filter settings of 387/11 nm (excitation) and 447/60 nm (emission) using a 20× objective.
Measurement of cholesterol biosynthesis
Cells were incubated for 48 h in Ham's F12 medium containing 5% LPDS, with or without 50 µg/ml LDL, washed twice in import medium, and incubated for 24 h in import medium containing 1 µCi/ml [14C]acetate, with or without 50 µg/ml LDL. Cellular lipids were extracted and separated by TLC with the solvent phase cyclohexane/ethyl acetate (3:2, v/v) (39). Radioactivity was determined in the band corresponding to unesterified cholesterol by liquid scintillation counting.
RNA interference
Transfection with siRNA was performed according to the manufacturer's protocol using DharmaFECT 4 (Dharmacon). siRNA (Dharmacon) was added to the cells at a final concentration of 50 nM for siNT or siMLN64, or 25 nM each for transfection with a mixture of three siRNAs against NPC1, using DharmaFECT 4 at a final concentration of 1:250 (v/v). The cells were grown with the siRNA for 24 h, and then medium was replaced with growth medium for 24 h, before import medium with trilostane was added for 24 h. Pregnenolone formation was measured by RIA. siRNA sequences were as follows: hamster NPC1: NPC1-1, sense 5′-GGAAAGAGUUCAUGAAAUUUU-3′ and antisense 5′-AAUUUCAUGAACUCUUUCCUU-3′; NPC1-2, sense 5′-GGGAAAGAGUUCAUGAAAUUU-3′ and antisense 5′-AUUUCAUGAACUCUUUCCCUU-3′; NPC1-3, sense 5′-CCGAGUAAGCCGAGCAGAAUU-3′ and antisense 5′-UUCUGCUCGGCUUACUCGGUU-3′; MLN64, siGENOME duplex (1), D-048833-01; siGENOME duplex (3), D-048833-03; nontargeting siRNA: siCONTROL nontargeting siRNA #1, D-001210-01-05. The MLN64 siGENOME sequences were predesigned against mouse MLN64, and sequences 1 and 3 were effective against hamster MLN64. The degree of protein depletion was tested by immunoblotting.
Isolation of mitochondria from cells and measurement of cholesterol content
Cells were harvested into PBS, collected by centrifugation, resuspended in 5 ml ice-cold MI buffer, and ruptured by nitrogen cavitation (40) (1,500 psi, 11 min on ice, cell disruption bomb 4639; Parr Instrument Co.). Unbroken cells were removed by centrifugation at 800 g, 4°C, 10 min. Centrifugation of the supernatant for 15 min, 4°C at 12,000 g yielded a crude mitochondrial fraction. Where indicated, crude mitochondria were treated with 0.1% trypsin (T9935; Sigma) for 10 min at room temperature, then soybean trypsin inhibitor (Sigma) was added at 0.5 mg/ml. Crude mitochondria were resuspended in 250 µl MI buffer and overlaid on a step gradient of 50, 22.5, and 10% Percoll in MI buffer (40). The gradients were centrifuged for 20 min, 4°C at 35,000 g in an SW61 rotor (Beckman). The mitochondria band was collected from the 50/22.5% Percoll interface, diluted 20-fold with MI buffer, and centrifuged at 12,000 g, 4°C for 15 min to yield purified mitochondria (40). A band visible at lower densities of the Percoll gradient was collected as an endosome/lysosome-enriched fraction. Cholesterol was measured with the Amplex Red assay (Invitrogen) without the addition of cholesterol esterase to detect only unesterified cholesterol. In the following, all data reported for cholesterol represent unesterified cholesterol. The purity of the fractions was tested by immunoblotting using antibodies against endosomal and mitochondrial markers, as indicated.
Immunoisolation of mitochondria from murine brain
Wild-type (Npc1+/+) and NPC1-deficient (Npc1−/−) mice were taken from an in-house breeding colony of BALB/cNctr-Npcm1N/J mice (Jackson Lab, stock number 003092). Animals were genotyped by PCR as described (41). All procedures were approved by the Dalhousie University Committee on Laboratory Animals (protocol 08-006). For isolation of mitochondria, cerebellum, cortex, and hippocampus dissected from 8-week-old Npc1+/+ and Npc1−/− mice were homogenized in MI buffer and centrifuged at 800 g to remove cell debris. The supernatants were centrifuged for 15 min, 4°C at 14,000 g. The pellet fraction was overlaid on a Percoll gradient constructed of 8.5 ml 30% Percoll in gradient buffer (25 mM HEPES, 225 mM mannitol, 1 mM EGTA, and protease inhibitors, pH 7.4) and centrifuged for 30 min at 95,000 g, 4°C in a swing out rotor (16). Mitochondria (100 µg) were incubated with 4 µg rabbit anti-Tom20 antibodies or unspecific control antibodies (rabbit anti-flag; Sigma F7425) in 800 µl MI buffer at 4°C overnight. Samples were centrifuged at 14,000 g for 20 min at 4°C, and the resulting pellets of mitochondria decorated with antibodies were washed, resuspended in 300 µl MI buffer, and incubated at 4°C overnight in a total volume of 1 ml with 107 Dynabeads M-500 Subcellular (coated with secondary anti-rabbit antibodies according to the manufacturer's protocol). Beads were washed in MI buffer and transferred to a fresh tube prior to elution of bound material (pure mitochondria) into PBS with 0.1% SDS. Cholesterol and protein were measured as above.
RESULTS
Measurement of cholesterol transported to the mitochondrial inner membrane by its conversion to pregnenolone
To measure cholesterol transport to the mitochondrial inner membrane, we used the conversion of cholesterol to pregnenolone by the P450 side chain cleavage enzyme (CYP11A1). Traditional radiotracer or fluorescent imaging techniques are unsuitable to monitor cholesterol trafficking into mitochondria due to the low levels of mitochondrial cholesterol. Since pregnenolone is only formed by CYP11A1, at a rate determined by availability of cholesterol substrate at the mitochondrial inner membrane, pregnenolone production reflects cholesterol transport to the mitochondrial inner membrane (42, 43). This approach has previously been used to investigate the role of StAR in steroid synthesis (35, 36). We introduced CYP11A1 activity into CHO cells using an expression vector encoding a fusion protein (F2-protein) of CYP11A1, ferredoxin reductase, and ferredoxin-1 with the CYP11A1 mitochondrial targeting sequence (35, 36). Immunostaining of CHO cells transiently transfected with the F2-fusion protein using antibodies against ferredoxin reductase yielded a distinctly mitochondrial fluorescence pattern (Fig. 1A), which colocalized with mitotracker (Fig. 1B), whereas untransfected cells showed little fluorescence from antiferredoxin reductase immunostaining. Thus, the F2-fusion protein is correctly targeted to mitochondria even when highly overexpressed.
Fig. 1.

Expression of F2-fusion protein of CYP11A1 complex confers the ability to convert cholesterol to pregnenolone in mitochondria. A, B: Localization of the F2 fusion protein to mitochondria. CHO cells transiently transfected with F2 were fixed and immunostained with antiferredoxin reductase antibodies (A) following incubation with Mitotracker Red (B). Images are representative of three independent experiments. Bars = 20 µm. C: CYP11A1 enzymatic activity was determined in crude mitochondria (mitochondria) and in the remaining cell fraction (supernatant) from CHO and F2-CHO cells (clone F2-CHO #6) as pregnenolone production from 22-OH-Chol in the presence of detergent and an NADPH regenerating system. Data represent means ± SEM of quadruplicate measurements.
We verified that enzymatic activity of CYP11A1 was restricted to mitochondria by subcellular fractionation and an in vitro enzyme activity assay (37) using monoclonal CHO cell lines stably expressing the F2-fusion protein (F2-CHO). Mitochondria isolated from F2-CHO cells, but not mitochondria from parental CHO cells, produced pregnenolone from the membrane-permeable precursor 22-OH-Chol (Fig. 1C). No significant pregnenolone formation was observed in the cell fraction remaining after removal of mitochondria (Fig. 1C), showing that pregnenolone in F2-cells is made in mitochondria. Due to the high sensitivity of the pregnenolone radioimmunoassay and the elimination of the need for subcellular fractionation, even the low levels of basal cholesterol transport to the mitochondrial inner membrane could be measured.
Cholesterol is transported to the mitochondrial inner membrane in the absence of functional NPC1
To investigate the role of NPC1 in mitochondrial cholesterol import, we generated monoclonal wild-type (F2-CHO) or NPC1-deficient (F2-4-4-19) cell lines stably transfected with the F2-protein. We selected two clones each of wild-type F2-CHO and NPC1-deficient F2-4-4-19 cells with comparable, moderate F2-complex mRNA expression and CYP11A1 enzyme activity in lysed cells (Fig. 2A, B). To prevent general effects on cholesterol homeostasis, we avoided clones with very high F2-complex expression. Filipin staining of endosomal cholesterol remained unaltered in F2-clones (Fig. 2C).
Fig. 2.

F2-CHO and F2-4-4-19 cells grown in the presence of serum transport the same amount of cholesterol to the mitochondrial inner membrane. A: F2-complex and cyclophilin mRNA in four monoclonal CHO cell lines stably expressing F2 and in parental CHO cells measured by RT-PCR, with each cell line in triplicate (clone #8 in duplicate). Lane 1, 1 kB ladder; lane 16, 100 bp ladder. B: CYP11A1 enzymatic activity in cell lysates of the four F2 clones shown as pregnenolone production from 22-OH-Chol in the presence of detergent and an NADPH regenerating system. Data are means ± SEM of four independent experiments in duplicate. C: Representative CHO, F2-CHO, 4-4-19, and F2-4-4-19 cells were fixed and stained with filipin to visualize unesterified cholesterol. Bars = 40 µm. D: Pregnenolone formation from endogenous cholesterol sources during 6 or 24 h in intact, adherent F2-CHO and F2-4-4-19 cells. E: Maximum rate of pregnenolone formation from excess membrane-permeable precursor 22-OH-Chol during 24 h in intact, adherent F2-CHO and F2-4-4-19 cells. In D and E, data are means ± SEM, three independent experiments in triplicate.
Pregnenolone formation was similar in F2-CHO and F2-4-4-19 clones during 6 or 24 h, indicating that under these conditions the amount of cholesterol reaching the mitochondrial inner membrane was unchanged by NPC1 deficiency (Fig. 2D). Addition of 22-OH-Chol increased pregnenolone production in all cell lines to a similar extent and at least 10-fold (Fig. 2E). As a membrane-permeable precursor for CYP11A1-catalyzed pregnenolone formation, 22-OH-Chol does not rely on active transport to reach the mitochondrial inner membrane (44); therefore, the amount of pregnenolone found in the medium of cells treated with an excess of 22-OH-Chol reflects maximum CYP11A1 activity. These results confirmed that pregnenolone formation is not limited by the rate of cholesterol oxidation but determined by cholesterol at the mitochondrial inner membrane.
A caveat of using two separate cell lines (CHO and 4-4-19) to investigate the role of NPC1 is that slight differences in expression levels of the F2-fusion protein, a divergence of CHO and 4-4-19 cell lines, or residual function of the mutated NPC1 protein in the 4-4-19 background might have enabled mitochondrial cholesterol import in the absence of functional NPC1. We therefore used RNA interference to suppress NPC1 expression in F2-CHO. The siRNA-mediated reduction of NPC1 protein levels by nearly 95% (Fig. 3A) was sufficient to induce the characteristic intracellular cholesterol accumulation within 3 days (Fig. 3B, C). In spite of the endosomal sequestration of cholesterol, depletion of NPC1 in F2-CHO cells had no effect on pregnenolone formation (Fig. 3D), indicating that normal levels of cholesterol reached the mitochondrial inner membrane in the absence of NPC1.
Fig. 3.

Levels of cholesterol transported to the inner mitochondrial membrane are unchanged by siRNA-mediated depletion of NPC1. A: NPC1 protein levels in F2-CHO cells transfected with nontargeting siRNA (siNT) or siRNA directed against NPC1 (siNPC1), analyzed by immunoblotting. Tubulin was used as a loading control. Shown is one representative blot of five independent experiments. B, C: F2-CHO cells transfected with nontargeting siRNA (siNT; B) or siRNA directed against NPC1 (siNPC1; C) were stained with filipin to visualize cholesterol accumulation. Bars = 40 µm. D: Pregnenolone formed during 24 h from endogenous cholesterol sources in intact, adherent F2-CHO cells grown in the presence of serum and transfected with nontargeting siRNA (siNT) or siRNA directed against NPC1 (siNPC1) was measured as ng pregnenolone per mg cell protein and is depicted as percentage of the average of siNT. Mean ± SEM, five independent experiments [total n = 13 (siNT) or 16 (siNPC1)].
Endosomal and endogenously synthesized cholesterol are transported to the mitochondrial inner membrane
It is commonly assumed that in NPC1 deficiency, cholesterol egress from the endosome is severely impaired, which might suggest that cholesterol transported to the mitochondrial inner membrane did not come from endosomes. However, it has also been postulated that excessive transport of accumulated endosomal cholesterol leads to increased mitochondrial cholesterol in NPC1-deficient cells (16, 23). In the absence of suitable radiotracer techniques to monitor the low levels of mitochondrial cholesterol import, it is difficult to directly follow the trafficking of separate cholesterol pools. Thus, to investigate potential cholesterol transport pathways from endosomes to mitochondria, we induced changes in the relative abundance of endogenously synthesized and endosomal cholesterol by incubating F2-CHO and F2-4-4-19 cells in the presence or absence of LDLs. Pregnenolone formation and cholesterol biosynthesis were then measured during an ensuing period of acute cholesterol deprivation (serum-free medium) or refeeding (serum-free medium with LDL).
In F2-CHO cells, pregnenolone formation was the same during a 24 h incubation in serum-free medium without exogenous cholesterol as in medium with LDL following growth in the presence of LDL (Fig. 4A). Cholesterol biosynthesis was upregulated 5.5-fold by cholesterol deprivation under the same conditions (Fig. 4B), indicating that depending on availability, either endocytosed or endogenously synthesized cholesterol were transported to the mitochondrial inner membrane. When cells were first deprived of cholesterol for 48 h in LPDS medium, and then incubated in the presence of LDL for 24 h (LDL refeeding), cholesterol biosynthesis was downregulated nearly 7-fold in F2-CHO cells. During the same time period of refeeding, we observed a 30% increase of pregnenolone formation, suggesting that LDL-derived endosomal cholesterol moved to the mitochondrial inner membrane.
Fig. 4.

Endosomal and endogenously synthesized cholesterol is transported into mitochondria. A, C: Pregnenolone formation in intact, adherent F2-CHO (A) or F2-4-4-19 (C) cells grown in medium containing 5% LPDS and 50 µg/ml LDL for 48 h, then incubated in the presence (LDL→LDL) or absence (LDL→SF) of LDL in serum-free (SF) import medium for 24 h, or grown in medium containing 5% LPDS without LDL for 48 h, then incubated in the presence (LPDS→LDL) or absence (LPDS→SF) of LDL in serum-free import medium for 24 h. Pregnenolone was determined in import medium as ng pregnenolone per mg cell protein and is depicted as percentage of the average of the LDL→LDL incubation. B, D: Cholesterol biosynthesis in F2-CHO (B) or F2-4-4-19 (D) cells grown under the same conditions as in A and C with the addition of 1 µCi/ml [14C]acetate to the import medium. Cellular lipid extracts were separated by thin-layer chromatography, and radioactivity was counted in the band corresponding to unesterified cholesterol. Data are expressed as dpm per mg cell protein. All data are means ± SEM of three independent experiment in triplicate with two clones each F2-CHO and F2-4-4-19 cells (n = 18). * P < 0.05.
In NPC1 deficiency, most egress of cholesterol from endosomes is impaired. To test whether this is also the case for the movement of endosomal cholesterol to mitochondria, we performed the same cholesterol deprivation and refeeding experiments in F2-4-4-19 cells (Fig. 4C, D). Cholesterol biosynthesis in F2-4-4-19 cells incubated in the presence of LDL was lower than in F2-CHO cells under the same conditions and was upregulated to a much lesser extent during a 24 h cholesterol deprivation period than in F2-CHO cells (Fig. 4D). Still, F2-4-4-19 cells produced the same amount of pregnenolone in the absence or presence of LDL (Fig. 4C) and also produced as much pregnenolone as F2-CHO cells (Fig. 2), in spite of their low rates of cholesterol biosynthesis, suggesting that endosomal cholesterol partially met the requirement for cholesterol in mitochondria. [14C]acetate incorporation into cholesteryl esters was very low in F2-4-4-19 cells (data not shown), ruling out this source of cholesterol in F2-4-4-19 cells. Moreover, following a 48 h period of cholesterol deprivation, F2-4-4-19 cells responded to LDL-refeeding with a 30% increase in pregnenolone formation compared with cells in the continued absence of LDL (Fig. 4C). This response of cholesterol-depleted F2-4-4-19 cells to addition of LDL was comparable to the response of F2-CHO cells. Since, as demonstrated in Fig. 2B, the maximal catalytic activity of the F2-fusion protein by far exceeded the rates of pregnenolone formation under basal conditions, we assume that neither in F2-CHO nor in F2-4-4-19 cells the increase in cholesterol transport to the mitochondrial inner membrane was greater than detected in our assay. Arrival at the mitochondrial outer membrane alone would, however, not be detected by the F2 complex. These observations strongly suggest that endosomal cholesterol was transported to the mitochondrial inner membrane in the absence of functional NPC1. However, cholesterol depletion for 48 h had the unexpected effect that pregnenolone formation in F2-CHO, but not F2-4-4-19 cells, was increased compared with cells preincubated in the presence of LDL prior to the measurement period.
MLN64 mediates transport of cholesterol to mitochondria
A prime candidate for mediating cholesterol trafficking from endosomes to mitochondria is MLN64, which resides in the same endosomal population as NPC1 and has a cholesterol-binding START domain on the cytosolic side (28). To test whether MLN64 contributes to mitochondrial cholesterol import, we transfected F2-CHO cells with siRNA directed against MLN64 either alone or together with siRNA against NPC1. MLN64 and NPC1 protein levels were reduced by 80–90% as shown by immunoblotting (Fig. 5A). MLN64 depletion by two different siRNA sequences caused a significant decrease in pregnenolone formation in cholesterol-fed cells either alone or in combination with siRNA-mediated depletion of NPC1 (Fig. 5B). To test whether MLN64 mediates transport of LDL-derived cholesterol, we measured pregnenolone formation during refeeding of LDL to cholesterol-deprived cells. RNA interference against MLN64 or against both MLN64 and NPC1 caused a 30% reduction of pregnenolone formation under these conditions (Fig. 5C), which was similar to the increase in pregnenolone formation that we observed in cells given LDL following cholesterol deprivation compared with cells in the continued absence of LDL (Fig. 4). Pregnenolone formation was never completely inhibited by MLN64 depletion, indicating that, as expected, MLN64 is not the only pathway for mitochondrial cholesterol import.
Fig. 5.
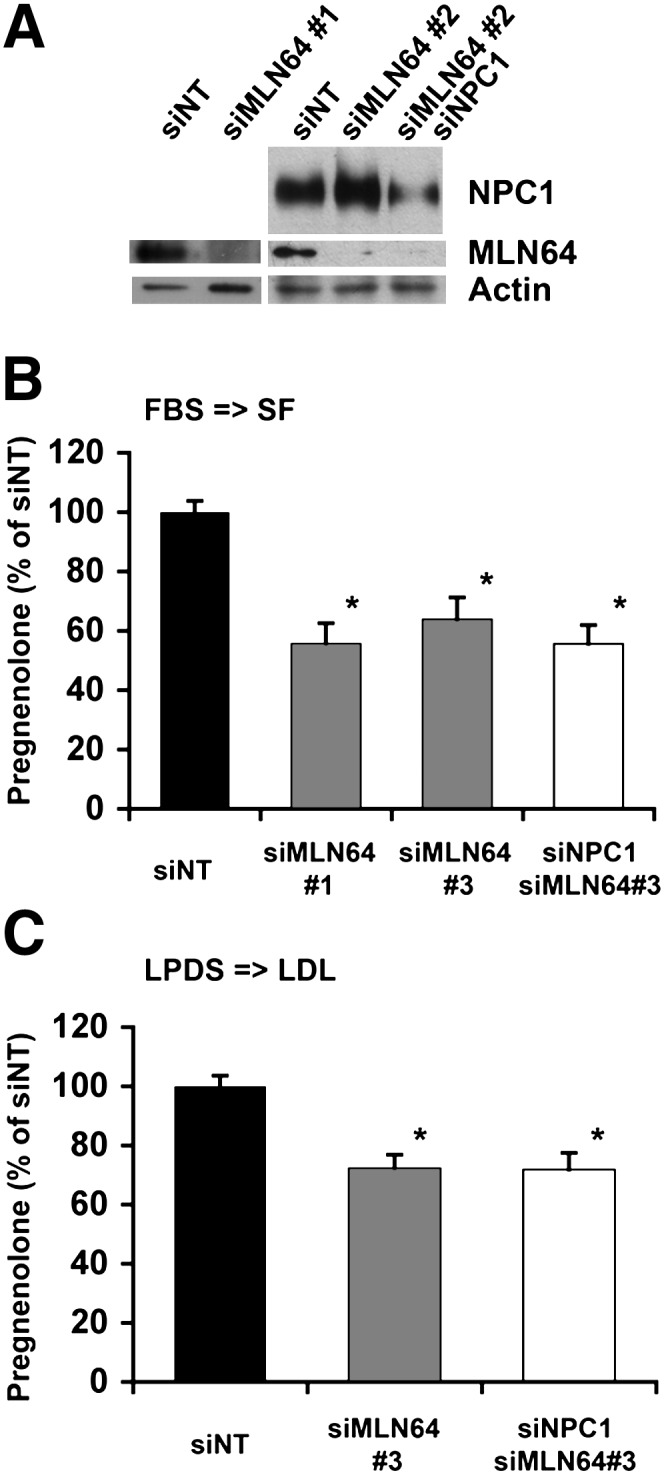
siRNA-mediated downregulation of MLN64 expression decreases cholesterol transport to the mitochondrial inner membrane. F2-CHO cells were transfected with nontargeting (siNT) siRNA, siRNA directed against MLN64 (siMLN64 #1 or siMLN64#3), or siMLN64#3 combined with siRNA against NPC1 (siMLN64/siNPC1) as indicated. A: Cell lysates analyzed by immunoblotting with antibodies directed against MLN64 and NPC1. Actin was used as loading control. B: Cells were grown in serum-containing medium; pregnenolone formation was measured in intact, adherent cells during 24 h in serum-free import medium. C: Cells were cholesterol deprived in medium with LPDS for 48 h; pregnenolone formation was measured in intact, adherent cells during 24 h in import medium containing LDL. Data are expressed as percentage of the average of siNT (measured as ng pregnenolone per mg cell protein), means ± SEM, four independent experiment with duplicate or triplicate measurements. * P < 0.05 compared with siNT.
Mitochondrial cholesterol content is increased in NPC1-deficient cells
Several studies have reported increased mitochondrial cholesterol content in NPC1-deficient cells or tissue. We found a similar increase in cholesterol in the mitochondrial fraction isolated by density gradient ultracentrifugation of lysed CHO and 4-4-19 cells and in mitochondria further purified by immunoisolation with antibody-coated magnetic beads (Fig. 6A). Due to species reactivity of the antibody and the large amount of material required, these mitochondria were isolated from wild-type and NPC1-deficient murine brain. The purity of fractions was verified by immunoblotting against endosomal and mitochondrial markers (Fig. 6B). However, our observation of MLN64-mediated transport of cholesterol from endosomes to mitochondria suggests the existence of sites of close contact between endosomes and mitochondria, since the N-terminal domain of MLN64 is anchored in the endosomal perimeter membrane, and the C-terminal START domain likely needs to come in contact with the mitochondrial outer membrane if direct cholesterol transfer takes place (27). If parts of endosomes were attached to mitochondria, even a thorough isolation of mitochondria by density fractionation or immunoisolation would copurify some endosomal membranes. To test whether this is the explanation for the reported increase in mitochondrial cholesterol, we disrupted potential protein-mediated contact sites by mild trypsin treatment of the crude mitochondria fraction containing mitochondria and endosomes/lysosomes and separated the trypsinized fraction by density ultracentrifugation. Trypsin treatment did not markedly affect mitochondrial ultrastructure (Fig. 6C) nor did it change sedimentation in the Percoll gradient (data not shown). Endosomal LAMP1 and the mitochondrial outer membrane protein VDAC were partially degraded by trypsin treatment, as shown by the presence of lower molecular weight bands in the immunoblot, whereas cytochrome c oxidase in the mitochondrial inner membrane remained intact (Fig. 6D). For unknown reasons, trypsin degradation of VDAC seemed stronger in the 4-4-19-derived fractions. Immunoblotting with an antibody against the cytosolic domain of MLN64 did not detect any bands in trypsinized endosomes or mitochondria (Fig. 6D). We therefore conclude that protein contact sites were disrupted. Indeed, Percoll density ultracentrifugation following mild trypsinization yielded a mitochondrial fraction free of endosomal proteins in the immunoblot (Fig. 6D). Some VDAC was detected in the endosome-enriched upper band of the density gradient, indicating that the mitochondria isolation was not quantitative (Fig. 6D). Importantly, cholesterol levels were increased in mitochondrial fractions from NPC1-deficient 4-4-19 compared with wild-type CHO cells even after artificial disruption of potential contact sites (Fig. 6E), strongly indicating that cholesterol builds up in the mitochondrial membranes and is not merely a contamination with endosomes. Further support for this conclusion comes from the observation that imipramine treatment, which is known to cause endosomal cholesterol accumulation, did not lead to increased cholesterol in mitochondrial fractions (Fig. 6E).
Fig. 6.

Increased mitochondrial cholesterol content in 4-4-19 cells. A, B: Mitochondria were isolated from CHO and 4-4-19 cells grown in serum-containing medium by Percoll density ultracentrifugation or from wild-type and Npc1−/− murine brain by immunoisolation as described in Materials and Methods. A: Unesterified cholesterol in mitochondria determined with the Amplex Red assay. B: Whole-cell homogenate (cells), crude mitochondria (crude mito), and pure mitochondria (pure mito) analyzed by immunoblotting with antibodies against VDAC (mitochondria), protein disulphide isomerase (endoplasmic reticulum), and NPC1 (late endosomes/lysosomes). C–F: Mitochondria were isolated from CHO and 4-4-19 cells grown in serum-containing medium by Percoll density ultracentrifugation after mild trypsinization. Where indicated, CHO cells were treated with 40 µM imipramine for 48 h prior to analysis. C: Electron micrograph of nontrypsinized and trypsinized mitochondria, 40,000× magnification. Bars = 500 nm. D: Immunoblot of crude mitochondria, and the mitochondrial and endosome-enriched fractions isolated by Percoll density ultracentrifugation following brief incubation with or without trypsin as described in Materials and Methods (pure mitochondria and endosomes). Fractions were prepared from CHO and 4-4-19 cells. Antibodies used as indicated. E: Unesterified cholesterol in pure mitochondria isolated from CHO, 4-4-19, or CHO cells treated with imipramine (imi), determined with the Amplex Red assay. Cholesterol data are standardized to mitochondrial protein and represent means ± SEM of three independent experiments (* P < 0.05). Immunoblots are representative of three independent experiments.
Since the arrival of cholesterol at the mitochondrial inner membrane was not increased in NPC1-deficient cells (Figs. 2 and 3), these data suggest that cholesterol transport from the outer to the inner membrane becomes rate-limiting under certain conditions. Interestingly, imipramine significantly increased cholesterol transport to the mitochondrial inner membrane by 44 ± 2.8% (data not shown) compared with untreated CHO cells, which might suggest that in this case, cholesterol buildup was prevented by increased movement from mitochondrial outer to inner membrane.
Depletion of MLN64 reduces mitochondrial cholesterol buildup in NPC1-deficient cells
If cholesterol building up in mitochondria of NPC1-deficient cells was transported by MLN64 from the endosomes, suppression of MLN64 expression should decrease mitochondrial cholesterol. As expected, reduction of MLN64 protein levels in CHO and 4-4-19 cells by RNA interference (Fig. 7A) significantly decreased mitochondrial cholesterol levels in 4-4-19 cells compared with 4-4-19 cells transfected with nontargeting siRNA (Fig. 7B). In CHO cells, MLN64 depletion had no effect on cholesterol content of either mitochondrial or endosomal/lysosomal fractions. For unknown reasons, siRNA-transfected cells had higher mitochondrial cholesterol levels than nontransfected cells regardless of siRNA sequence. Cholesterol content of the endosome-enriched fraction was markedly increased in 4-4-19 cells compared with CHO cells but not affected by MLN64 depletion (Fig. 7C), suggesting that only a small percentage of accumulated cholesterol is transported out of the endosome by MLN64. This observation also speaks for the purity of the isolated mitochondrial fraction, since a contamination with lysosomal cholesterol would have masked the small reduction in the absolute amount of cholesterol in the mitochondrial fraction of MLN64-depleted 4-4-19 cells.
Fig. 7.
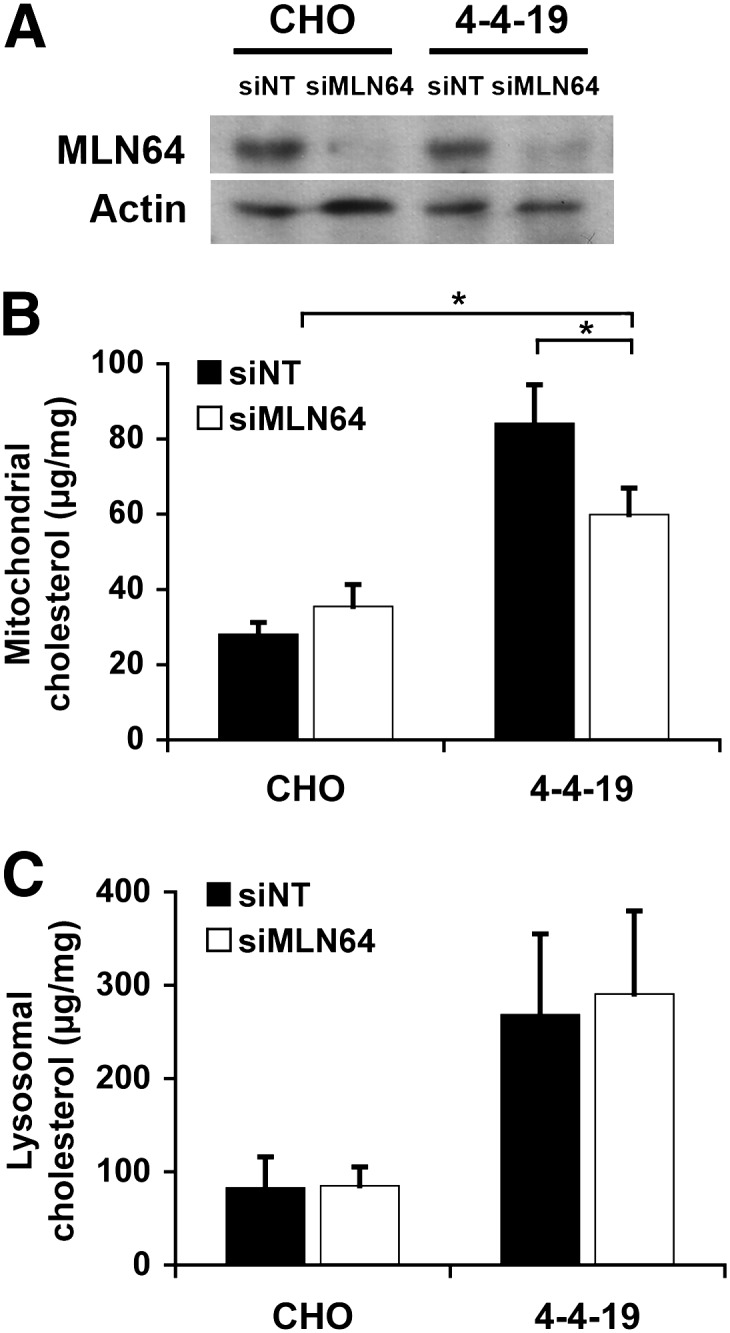
Mitochondrial cholesterol content is decreased following downregulation of MLN64 in 4-4-19 cells. Mitochondria were isolated from CHO and 4-4-19 cells transfected with nontargeting siRNA (siNT) or siRNA directed against MLN64 (siMLN64) and grown in serum-containing medium for 3 days. A: Immunoblot of cell lysates using anti-MLN64 antibodies. Actin was used as loading control on the same membrane. Shown is one immunoblot representative of three independent experiments. B, C: Unesterified cholesterol determined with the Amplex Red assay in mitochondria (B) and lysosomes/endosomes (C) isolated by Percoll gradient centrifugation. Data are standardized to mitochondrial protein and represent the means ± SEM of three independent experiments. * P < 0.05.
DISCUSSION
Alterations in cholesterol metabolism have been observed in several neurodegenerative disorders, but the mechanisms connecting defects of cholesterol metabolism to cellular dysfunction are not clear. One hypothesis is that alterations in mitochondrial cholesterol homeostasis could negatively affect mitochondrial and cellular function. In NPC disease, loss of NPC1 function leads to accumulation of cholesterol in late endosomes/lysosomes and an impaired cholesterol homeostatic response, which could affect mitochondrial cholesterol trafficking. Here, we show that cholesterol is transported to the mitochondrial inner membrane in the absence of functional NPC1 and provide evidence that MLN64 contributes to cholesterol transport to mitochondria, likely using endosomal cholesterol. Our data suggest that in NPC1-deficient cells, cholesterol builds up in mitochondria due to increased cholesterol transfer to the mitochondrial outer membrane by MLN64, while transfer of cholesterol from the mitochondrial outer to inner membrane becomes rate limiting.
Using an assay measuring cholesterol transport to the mitochondrial inner membrane by following its conversion to pregnenolone, we found similar levels of cholesterol transported to the mitochondrial inner membrane in NPC1-defective F2-4-4-19 cells and in F2-CHO cells with siRNA-mediated depletion of NPC1, compared with control F2-CHO cells (Figs. 2 and 3). These data show that NPC1 was not required for basal cholesterol import into mitochondria or that alternative pathways were upregulated in the absence of NPC1. Pregnenolone formation was normal in F2-4-4-19 cells even at the 6 h time point, suggesting that cholesterol movement to the mitochondrial inner membrane was not, or was only slightly, delayed in the absence of NPC1 (Fig. 2D). Cholesterol transport to the mitochondrial inner membrane remained relatively constant in both F2-CHO and F2-4-4-19 cells under conditions of different availability of cholesterol from synthesis and endocytosis (Fig. 4A, C), indicating that both endosomal and endogenously synthesized cholesterol can enter mitochondria. Thus, during prolonged cholesterol deprivation, endogenous synthesis could provide mitochondria with the necessary cholesterol, whereas additional cholesterol reaching the mitochondrial inner membrane during refeeding LDL to cholesterol-deprived cells most likely represented transport of LDL-derived, endosomal cholesterol. The observed increase in cholesterol import into mitochondria in response to LDL is in line with the known stimulation of oxysterol production by lipoproteins (45). Oxysterols play an important role in the regulation of sterol metabolism and are formed in part by 27-cholesterol hydroxylase at the mitochondrial inner membrane (46, 47). In NPC1-deficient fibroblasts and macrophages, oxysterol production during a 24 h time period in the presence of LDL following a 48 h cholesterol deprivation was lower than in wild-type cells (22, 45). Under the same conditions, pregnenolone formation by F2-4-4-19 cells appeared lower than in F2-CHO cells. However, it became evident that while the basal pregnenolone production in LPDS-containing medium without lipoproteins was lower in F2-4-4-19 cells than in F2-CHO cells, the increase in pregnenolone formation due to addition of LDL was similar, suggesting that transport of LDL-derived endosomal cholesterol to the mitochondrial inner membrane was not impaired in the absence of NPC1 function. The decrease in pregnenolone formation in serum-starved F2-4-4-19 compared with serum-starved F2-CHO cells might be due to comparatively lower rates of cholesterol biosynthesis in F2-4-4 19 cells under these conditions or to a defect in another cholesterol trafficking pathway that mediates mitochondrial cholesterol import when only little endosomal cholesterol is available.
These experiments clearly indicated that there are several pathways transporting cholesterol to the mitochondrial membranes, one of them using endosomal cholesterol. A likely candidate for cholesterol transport from endosomes to mitochondria is MLN64, a late endosomal transmembrane protein with a cholesterol-binding START domain on the cytosolic side (28). Expression of N-218 MLN64, an N-truncated, soluble construct of MLN64 containing the START domain, increased cholesterol delivery for mitochondrial steroidogenesis in a mechanism similar to StAR (29, 48, 30, 49, 50). However, it had not been clear whether full-length MLN64 contributed to basal mitochondrial cholesterol import. Our observation that depletion of MLN64 by RNA interference reduces cholesterol transport to the mitochondrial inner membrane by 30–40% depending on culture conditions (Fig. 5) clearly demonstrates that MLN64 plays a significant role in mitochondrial cholesterol homeostasis. The extent to which LDL refeeding increased pregnenolone production in F2-CHO and F2-4-4-19 cells (Fig. 4) agreed well with the extent of inhibition of pregnenolone formation caused by MLN64 depletion under refeeding conditions in F2-CHO cells and in F2-CHO cells transfected with siRNA against NPC1 (Fig. 5C). The comparison of these data suggests that most LDL-derived cholesterol reaching the mitochondrial inner membrane was transported via MLN64. On the other hand, RNA interference against MLN64 also revealed a remarkable redundancy of cholesterol trafficking pathways into mitochondria, in line with the relatively constant pregnenolone formation under different states of cholesterol homeostasis (Fig. 4). A large part of cholesterol transported independently of MLN64 is likely to be endogenously synthesized cholesterol. In addition, a recent study described cholesterol transport from plasma membrane to mitochondria (26). This latter pathway would require cytosolic sterol carrier proteins (51), such as sterol carrier protein 2, or other START proteins such as StARD4 or StARD5, which have both been shown to bind cholesterol (52, 53). Overexpression of StARD4 (but not StARD5) in hepatocytes increased cholesterol ester synthesis in the endoplasmic reticulum as well as bile acid formation in mitochondria (53). The redundancy of pathways to maintain mitochondrial cholesterol homeostasis could explain reports that mice with a targeted deletion of the MLN64 START domain developed only minor phenotypic defects (54).
The relatively constant rates of pregnenolone formation under conditions of widely different cholesterol homeostasis not only suggest the presence of several pathways for cholesterol transport into mitochondria, but also that these pathways might be largely dependent on the availability of cholesterol from different sources. If MLN64-dependent transport was mainly regulated by the amount of endosomal cholesterol, this would be in line with the buildup of cholesterol in mitochondrial fractions from NPC1-deficient cells and tissues that have been reported previously (16, 23) and that we observed in 4-4-19 cells (Fig. 6A). However, mitochondria isolated by density gradient centrifugation may be contaminated by small amounts of endosomes, in particular when endosomal density is altered by NPC1 deficiency. Moreover, since MLN64, in analogy to StAR, likely needs direct contact with mitochondria for cholesterol transfer, endosome-mitochondria contact sites would be required unless MLN64 is cleaved. We therefore took several different approaches to purify mitochondria, including immunoisolation, which does not rely on organellar density, and trypsinization of crude mitochondria to disrupt potential contact sites prior to density gradient centrifugation. Under all conditions, mitochondrial fractions from NPC1-deficient cells had higher levels of cholesterol per protein than the wild type (Fig. 6). Moreover, in imipramine-treated cells, endosomal but not mitochondrial cholesterol was increased compared with control, underlining the purity of the mitochondrial fraction (Fig. 6E). Together, these data strongly suggest that cholesterol builds up in mitochondrial membranes of NPC1-deficient cells, but direct proof would require cholesterol quantification on the ultrastructural level.
Since we did not observe an increase in cholesterol reaching CYP11A1 at the inner membrane in NPC1-deficient 4-4-19 cells (Figs. 2 and 3), our data suggest a model whereby MLN64 transfers endosomal cholesterol to the mitochondrial outer membrane and where cholesterol transfer from mitochondrial outer to inner membrane may become rate limiting. This model is supported by our finding of a significant reduction of mitochondrial cholesterol accumulation in 4-4-19 cells after siRNA-mediated depletion of MLN64. Mitochondrial cholesterol was not completely normalized by MLN64 depletion, which could be due to incomplete turnover of mitochondrial cholesterol during siRNA transfection but would also be in line with increased cholesterol transport by additional proteins. Further investigations are required to determine the relative contributions of different transport mechanisms for different conditions and cell types.
In summary, this study defines a transport pathway from endosomes to mitochondria that requires MLN64 but not NPC1. Endosomal cholesterol accumulation in NPC1 deficiency leads to increased MLN64-mediated transport of cholesterol to mitochondria and a buildup of cholesterol in the mitochondrial outer membrane. Since increased mitochondrial cholesterol is potentially detrimental for mitochondrial function, MLN64-mediated cholesterol transport might present a target for future attempts to correct mitochondrial cholesterol homeostasis and mitochondrial function.
Acknowledgments
The authors thank Laura Liscum (Tufts University, Boston, MA) for the generous gift of CHO and 4-4-19 cells and Walter Miller (University of California, San Francisco, CA) for kindly providing the F2-pcDNA3 vector. The technical help of Debra Fice is gratefully acknowledged, as is the assistance of Mary-Ann Trevors with sample preparation for electron microscopy. We also thank Neale Ridgway and Roger McLeod for the critical reading of the manuscript.
Footnotes
Abbreviations:
- CHO
- Chinese hamster ovary
- LAMP1
- lysosome associated membrane protein 1
- LPDS
- lipoprotein-deficient serum
- MI
- mitochondria isolation
- MLN64
- metastatic lymph node protein 64
- NPC
- Niemann-Pick Type C
- 22-OH-Chol
- 22R-hydroxycholesterol
- RIA
- radioimmunoassay
- siRNA
- short interfering RNA
- StAR
- steroidogenic acute regulatory protein
- START
- STAR-related lipid transfer domain
- VDAC
- voltage-dependent anion channel
This study was supported by an operating grant from the Canadian Institutes of Health Research (MOP 81087) and a New Investigator Award by the Dalhousie Medical Research Foundation. Salary support is provided by the Regional Partnership Program of the Nova Scotia Health Research Foundation to B.E.K.
REFERENCES
- 1.Carstea E. D., Morris J. A., Coleman K. G., Loftus S. K., Zhang D., Cummings C., Gu J., Rosenfeld M. A., Pavan W. J., Krizman D. B., et al. 1997. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 277: 228–231. [DOI] [PubMed] [Google Scholar]
- 2.Naureckiene S., Sleat D. E., Lackland H., Fensom A., Vanier M. T., Wattiaux R., Jadot M., Lobel P. 2000. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 290: 2298–2301. [DOI] [PubMed] [Google Scholar]
- 3.Davies J. P., Ioannou Y. A. 2000. Topological analysis of Niemann-Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. J. Biol. Chem. 275: 24367–24374. [DOI] [PubMed] [Google Scholar]
- 4.Millard E. E., Gale S. E., Dudley N., Zhang J., Schaffer J. E., Ory D. S. 2005. The sterol-sensing domain of the Niemann-Pick C1 (NPC1) protein regulates trafficking of low density lipoprotein cholesterol. J. Biol. Chem. 280: 28581–28590. [DOI] [PubMed] [Google Scholar]
- 5.Friedland N., Liou H. L., Lobel P., Stock A. M. 2003. Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proc. Natl. Acad. Sci. USA. 100: 2512–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ko D. C., Binkley J., Sidow A., Scott M. P. 2003. The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc. Natl. Acad. Sci. USA. 100: 2518–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Infante R. E., Wang M. L., Radhakrishnan A., Kwon H. J., Brown M. S., Goldstein J. L. 2008. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA. 105: 15287–15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohgami N., Ko D. C., Thomas M., Scott M. P., Chang C. C., Chang T. Y. 2004. Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc. Natl. Acad. Sci. USA. 101: 12473–12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Infante R. E., Abi-Mosleh L., Radhakrishnan A., Dale J. D., Brown M. S., Goldstein J. L. 2008. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 283: 1052–1063. [DOI] [PubMed] [Google Scholar]
- 10.Liscum L. 2000. Niemann-Pick type C mutations cause lipid traffic jam. Traffic. 1: 218–225. [DOI] [PubMed] [Google Scholar]
- 11.Walkley S. U., Suzuki K. 2004. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta. 1685: 48–62. [DOI] [PubMed] [Google Scholar]
- 12.Lloyd-Evans E., Morgan A. J., He X., Smith D. A., Elliot-Smith E., Sillence D. J., Churchill G. C., Schuchman E. H., Galione A., Platt F. M. 2008. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14: 1247–1255. [DOI] [PubMed] [Google Scholar]
- 13.Schneiter R. 2007. Intracellular sterol transport in eukaryotes, a connection to mitochondrial function? Biochimie. 89: 255–259. [DOI] [PubMed] [Google Scholar]
- 14.Colell A., Garcia-Ruiz C., Lluis J. M., Coll O., Mari M., Fernandez-Checa J. C. 2003. Cholesterol impairs the adenine nucleotide translocator-mediated mitochondrial permeability transition through altered membrane fluidity. J. Biol. Chem. 278: 33928–33935. [DOI] [PubMed] [Google Scholar]
- 15.Mari M., Caballero F., Colell A., Morales A., Caballeria J., Fernandez A., Enrich C., Fernandez-Checa J. C., Garcia-Ruiz C. 2006. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 4: 185–198. [DOI] [PubMed] [Google Scholar]
- 16.Yu W., Gong J. S., Ko M., Garver W. S., Yanagisawa K., Michikawa M. 2005. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 280: 11731–11739. [DOI] [PubMed] [Google Scholar]
- 17.Griffin L. D., Gong W., Verot L., Mellon S. H. 2004. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med. 10: 704–711. [DOI] [PubMed] [Google Scholar]
- 18.Mellon S., Gong W., Griffin L. D. 2004. Niemann pick type C disease as a model for defects in neurosteroidogenesis. Endocr. Res. 30: 727–735. [DOI] [PubMed] [Google Scholar]
- 19.Chen G., Li H. M., Chen Y. R., Gu X. S., Duan S. 2007. Decreased estradiol release from astrocytes contributes to the neurodegeneration in a mouse model of Niemann-Pick disease type C. Glia. 55: 1509–1518. [DOI] [PubMed] [Google Scholar]
- 20.Fluegel M. L., Parker T. J., Pallanck L. J. 2006. Mutations of a Drosophila NPC1 gene confer sterol and ecdysone metabolic defects. Genetics. 172: 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roff C. F., Strauss J. F., 3rd, Goldin E., Jaffe H., Patterson M. C., Agritellis G. C., Hibbs A. M., Garfield M., Brady R. O., Pentchev P. G. 1993. The murine Niemann-Pick type C lesion affects testosterone production. Endocrinology. 133: 2913–2923. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J. R., Coleman T., Langmade S. J., Scherrer D. E., Lane L., Lanier M. H., Feng C., Sands M. S., Schaffer J. E., Semenkovich C. F., et al. 2008. Niemann-Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J. Clin. Invest. 118: 2281–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez A., Llacuna L., Fernandez-Checa J. C., Colell A. 2009. Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J. Neurosci. 29: 6394–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller W. L. 2007. Mechanism of StAR's regulation of mitochondrial cholesterol import. Mol. Cell. Endocrinol. 265–266: 46–50. [DOI] [PubMed] [Google Scholar]
- 25.Papadopoulos V., Liu J., Culty M. 2007. Is there a mitochondrial signaling complex facilitating cholesterol import? Mol. Cell. Endocrinol. 265–266: 59–64. [DOI] [PubMed] [Google Scholar]
- 26.Lange Y., Steck T. L., Ye J., Lanier M. H., Molugu V., Ory D. S. 2009. Regulation of fibroblast mitochondrial 27-hydroxycholesterol production by active plasma membrane cholesterol. J Lipid Res. 50: 1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alpy F., Tomasetto C. 2005. Give lipids a START: the StAR-related lipid transfer (START) domain in mammals. J. Cell Sci. 118: 2791–2801. [DOI] [PubMed] [Google Scholar]
- 28.Alpy F., Stoeckel M. E., Dierich A., Escola J. M., Wendling C., Chenard M. P., Vanier M. T., Gruenberg J., Tomasetto C., Rio M. C. 2001. The steroidogenic acute regulatory protein homolog MLN64, a late endosomal cholesterol-binding protein. J. Biol. Chem. 276: 4261–4269. [DOI] [PubMed] [Google Scholar]
- 29.Zhang M., Liu P., Dwyer N. K., Christenson L. K., Fujimoto T., Martinez F., Comly M., Hanover J. A., Blanchette-Mackie E. J., Strauss J. F., 3rd 2002. MLN64 mediates mobilization of lysosomal cholesterol to steroidogenic mitochondria. J. Biol. Chem. 277: 33300–33310. [DOI] [PubMed] [Google Scholar]
- 30.Tuckey R. C., Bose H. S., Czerwionka I., Miller W. L. 2004. Molten globule structure and steroidogenic activity of N-218 MLN64 in human placental mitochondria. Endocrinology. 145: 1700–1707. [DOI] [PubMed] [Google Scholar]
- 31.Beal M. F. 2005. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 58: 495–505. [DOI] [PubMed] [Google Scholar]
- 32.Sattler W., Mohr D., Stocker R. 1994. Rapid isolation of lipoproteins and assessment of their peroxidation by high-performance liquid chromatography postcolumn chemiluminescence. Methods Enzymol. 233: 469–489. [DOI] [PubMed] [Google Scholar]
- 33.Dahl N. K., Reed K. L., Daunais M. A., Faust J. R., Liscum L. 1992. Isolation and characterization of Chinese hamster ovary cells defective in the intracellular metabolism of low density lipoprotein-derived cholesterol. J. Biol. Chem. 267: 4889–4896. [PubMed] [Google Scholar]
- 34.Wojtanik K. M., Liscum L. 2003. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 278: 14850–14856. [DOI] [PubMed] [Google Scholar]
- 35.Harikrishna J. A., Black S. M., Szklarz G. D., Miller W. L. 1993. Construction and function of fusion enzymes of the human cytochrome P450scc system. DNA Cell Biol. 12: 371–379. [DOI] [PubMed] [Google Scholar]
- 36.Huang M. C., Miller W. L. 2001. Creation and activity of COS-1 cells stably expressing the F2 fusion of the human cholesterol side-chain cleavage enzyme system. Endocrinology. 142: 2569–2576. [DOI] [PubMed] [Google Scholar]
- 37.Papadopoulos V., Guarneri P., Kreuger K. E., Guidotti A., Costa E. 1992. Pregnenolone biosynthesis in C6–2B glioma cell mitochondria: regulation by a mitochondrial diazepam binding inhibitor receptor. Proc. Natl. Acad. Sci. USA. 89: 5113–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Potts G. O., Creange J. E., Hardomg H. R., Schane H. P. 1978. Trilostane, an orally active inhibitor of steroid biosynthesis. Steroids. 32: 257–267. [DOI] [PubMed] [Google Scholar]
- 39.Karten B., Vance D. E., Campenot R. B., Vance J. E. 2002. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J. Neurochem. 83: 1154–1163. [DOI] [PubMed] [Google Scholar]
- 40.Kristian T., Hopkins I. B., McKenna M. C., Fiskum G. 2006. Isolation of mitochondria with high respiratory control from primary cultures of neurons and astrocytes using nitrogen cavitation. J. Neurosci. Methods. 152: 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loftus S. K., Morris J. A., Carstea E. D., Gu J. Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M. A., Tagle D. A., et al. 1997. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 277: 232–235. [DOI] [PubMed] [Google Scholar]
- 42.Stocco D. M. 2000. Intramitochondrial cholesterol transfer. Biochim. Biophys. Acta. 1486: 184–197. [DOI] [PubMed] [Google Scholar]
- 43.Miller W. L. 1995. Mitochondrial specificity of the early steps in steroidogenesis. J. Steroid Biochem. Mol. Biol. 55: 607–616. [DOI] [PubMed] [Google Scholar]
- 44.Bose H. S., Sugawara T., Strauss J. F., 3rd, Miller W. L. 1996. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. International Congenital Lipoid Adrenal Hyperplasia Consortium. N. Engl. J. Med. 335: 1870–1878. [DOI] [PubMed] [Google Scholar]
- 45.Frolov A., Zielinski S. E., Crowley J. R., Dudley-Rucker N., Schaffer J. E., Ory D. S. 2003. NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J. Biol. Chem. 278: 25517–25525. [DOI] [PubMed] [Google Scholar]
- 46.Bjorkhem I. 2009. Are side-chain oxidized oxysterols regulators also in vivo? J Lipid Res. 50 (Suppl): S213–S218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gill S., Chow R., Brown A. J. 2008. Sterol regulators of cholesterol homeostasis and beyond: the oxysterol hypothesis revisited and revised. Prog. Lipid Res. 47: 391–404. [DOI] [PubMed] [Google Scholar]
- 48.Watari H., Arakane F., Moog-Lutz C., Kallen C. B., Tomasetto C., Gerton G. L., Rio M. C., Baker M. E., Strauss J. F., 3rd 1997. MLN64 contains a domain with homology to the steroidogenic acute regulatory protein (StAR) that stimulates steroidogenesis. Proc. Natl. Acad. Sci. USA. 94: 8462–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bose H. S., Whittal R. M., Huang M. C., Baldwin M. A., Miller W. L. 2000. N-218 MLN64, a protein with StAR-like steroidogenic activity, is folded and cleaved similarly to StAR. Biochemistry. 39: 11722–11731. [DOI] [PubMed] [Google Scholar]
- 50.Bose H. S., Baldwin M. A., Miller W. L. 2000. Evidence that StAR and MLN64 act on the outer mitochondrial membrane as molten globules. Endocr. Res. 26: 629–637. [DOI] [PubMed] [Google Scholar]
- 51.Prinz W. A. 2007. Non-vesicular sterol transport in cells. Prog. Lipid Res. 46: 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez-Agudo D., Ren S., Hylemon P. B., Redford K., Natarajan R., Del Castillo A., Gil G., Pandak W. M. 2005. Human StarD5, a cytosolic StAR-related lipid binding protein. J. Lipid Res. 46: 1615–1623. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez-Agudo D., Ren S., Wong E., Marques D., Redford K., Gil G., Hylemon P., Pandak W. M. 2008. Intracellular cholesterol transporter StarD4 binds free cholesterol and increases cholesteryl ester formation. J. Lipid Res. 49: 1409–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kishida T., Kostetskii I., Zhang Z., Martinez F., Liu P., Walkley S. U., Dwyer N. K., Blanchette-Mackie E. J., Radice G. L., Strauss J. F., 3rd 2004. Targeted mutation of the MLN64 START domain causes only modest alterations in cellular sterol metabolism. J. Biol. Chem. 279: 19276–19285. [DOI] [PubMed] [Google Scholar]