

Abstract
In neurons L-type calcium currents contribute to synaptic plasticity and to activity-dependent gene regulation. The subcellular localization of CaV1.2 and its association with upstream and downstream signaling proteins is important for efficient and specific signal transduction. Here we tested the hypothesis that A-kinase anchoring proteins (AKAPs) or PDZ-proteins are responsible for the targeting and anchoring of CaV1.2 in the postsynaptic compartment of glutamatergic neurons. Double-immunofluorescence labeling of hippocampal neurons transfected with external HA epitope-tagged CaV1.2 demonstrated that clusters of membrane-incorporated CaV1.2-HA were colocalized with AKAP79/150 but not with PSD-95 in the spines and shafts of dendrites. To disrupt the interactions with these scaffold proteins, we mutated known binding sequences for AKAP79/150 and PDZ proteins in the C terminus of CaV1.2-HA. Unexpectedly, the distribution pattern, the density, and the fluorescence intensity of clusters were similar for wild-type and mutant CaV1.2-HA, indicating that interactions with AKAP and PDZ proteins are not essential for the correct targeting of CaV1.2. In agreement, brief treatment with NMDA (a chemical LTD paradigm) caused the degradation of PSD-95 and the redistribution of AKAP79/150 and α-actinin from dendritic spines into the shaft, without a concurrent loss or redistribution of CaV1.2-HA clusters. Thus, in the postsynaptic compartment of hippocampal neurons CaV1.2 calcium channels form signaling complexes apart from those of glutamate receptors and PSD-95. Their number and distribution in dendritic spines is not altered upon NMDA-induced disruption of the glutamate receptor signaling complex, and targeting and anchoring of CaV1.2 is independent of its interactions with AKAP79/150 and PDZ proteins.
Keywords: L-type Ca2+ channels, hippocampal neurons, dendritic spines, PSD-95, α-actinin, chemical LTD, immunofluorescence microscopy
Introduction
In neurons calcium influx through ligand- and voltage-gated channels causes long-lasting changes of synaptic strength, which underlie learning and memory. NMDA receptor-mediated calcium influx into dendritic spines leads to the rearrangement of the postsynaptic signaling complex and ultimately to the altered surface expression of AMPA receptors. Activation of L-type calcium channels is required to transmit synaptic activity to the regulation of gene expression (Bading et al., 1993; Impey et al., 1996; Graef et al., 1999; Mermelstein et al., 2000; Dolmetsch et al., 2001). Knock-out of the L-type calcium channel CaV1.2 isoform in the neocortex and hippocampus impairs NMDA receptor-independent long-term potentiation (LTP) and spatial memory (Moosmang et al., 2005). Thus, postsynaptic CaV1.2 calcium channels are critical in initiating long-lasting changes in synaptic strength that are dependent on protein synthesis but independent of NMDA receptor activation.
The postsynaptic compartment of excitatory synapses contains two prominent classes of scaffold proteins: PDZ proteins and AKAPs. The postsynaptic density protein-95 (PSD-95) and AKAP79/150 recruit protein kinases and phosphatases to glutamate receptors and are important for the modulation of synaptic strength (Lüscher et al., 1999). L-type calcium channels also functionally interact with PDZ proteins and AKAPs. The C-terminal PDZ-binding sequence (VSNL) of CaV1.2 is essential for L-type calcium current-dependent activation of CREB-mediated gene-expression (Weick et al., 2003). A leucine zipper in the C terminus of CaV1.2 binds AKAP79/150. This interaction is important for the reversible phosphorylation of CaV1.2 by PKA and calcineurin, and for the signaling to the nucleus through NFATc4 (Oliveria et al., 2007). Thus, in dendritic spines of hippocampal neurons AKAP79/150 integrates CaV1.2 in a signaling complex with the upstream β2 adrenergic receptor (Hoogland and Saggau, 2004) as well as with downstream signaling proteins (Davare et al., 2001).
Furthermore, PDZ proteins control the targeting of voltage-gated calcium channels into presynaptic and postsynaptic neuronal compartments (Maximov and Bezprozvanny, 2002; Zhang et al., 2005), and coexpression of AKAP79/150 promotes membrane expression of CaV1.2 in Xenopus oocytes (Altier et al., 2002). Moreover, strong stimulation of cortical neurons has recently been shown to induce endocytosis of CaV1.2 (Green et al., 2007). Therefore we examined whether interactions with AKAPs and PDZ proteins are necessary for the targeting and stabilization of CaV1.2 calcium channels in the postsynaptic compartments of hippocampal neurons, and whether disruption of these scaffolds in dendritic spines affects the distribution or membrane expression of CaV1.2.
Three lines of evidence indicate that CaV1.2 signaling complexes in dendritic spines of hippocampal neurons are structurally and functionally independent of the glutamate receptor signaling complexes. First, CaV1.2 clusters were not colocalized with PSD-95. Second, deletion of known interaction sequences for PDZ proteins and AKAPs did not reduce the density or size of CaV1.2 clusters. Third, NMDA-induced disruption of the PSD-95/AKAP scaffold in dendritic spines was not accompanied by the loss of CaV1.2 clusters. Together, these results suggest that glutamate receptors and CaV1.2 coexist in dendritic spines and interact with common scaffold proteins, but are constituents of separate signaling complexes and their membrane expression is regulated by independent mechanisms.
Materials and Methods
Primary culture of hippocampal neurons.
Low-density cultures of hippocampal neurons were prepared from 16.5-d-old embryonic BALB/c mice as described previously (Goslin et al., 1998; Obermair et al., 2003, 2004). Briefly, dissected hippocampi were dissociated by trypsin treatment and trituration. Neurons were plated on poly-l-lysine-coated glass coverslips in 60 mm culture dishes at a density of 3500 cells/cm2. After plating, cells were allowed to attach for 3–4 h before transferring the coverslips neuron-side-down into a 60 mm culture dish with a glial feeder layer. Neurons and glial feeder layer were cultured in serum-free Neurobasal medium (Invitrogen) supplemented with Glutamax and B27 supplements (Invitrogen).
Transfection of hippocampal neurons.
Plasmids were introduced into neurons on day 6 using Lipofectamine 2000-mediated transfection (Invitrogen) as previously described (Obermair et al., 2004). For single transfection (pβA-eGFP) 1 μg of DNA, for cotransfection experiments (pβA-eGFP or eCFP plus HA-tagged constructs) 2 μg of total DNA at a molar ratio of 1:2 was used. Cells were stained and analyzed 11–14 d after transfection.
Plasmids.
The generation of pβA-eGFP and pβA-CaV1.2-HA (CaV1.2: GenBank accession no. M67515) has been described earlier (Obermair et al., 2004). pECFP-C1 (BD Biosciences Clontech) was a gift from D. Garczarczyk (Innsbruck Medical University, Innsbruck, Austria). The CaV1.2 C-terminal mutants were generated by standard overlapping PCR protocols with specific oligonucleotides carrying a premature stop codon for ΔVSNL, or three alanine substitutions (I2046A, L2053A, I2060A) for ΔLZ. These PCRs were performed using pcDNA3-CaV1.2 as template. C-terminal cassettes (2.2 kb Sbfi1-NotI fragment) carrying the mutations were sequenced to verify the presence of the desired mutations and then cloned into the pβA-CaV1.2-HA plasmid backbone to generate pβA-CaV1.2-HA(ΔVSNL) and pβA-CaV1.2-HA(ΔLZ). The functional expression of all CaV1.2-HA constructs was confirmed using patch-clamp recordings in HEK cells and in dysgenic myotubes (data not shown).
NMDA treatment of hippocampal neurons.
NMDA (Tocris) was applied to 18- to 20-d-old neurons in neuronal medium at a final concentration of 25 μm for 3 min. Subsequently the cultures were washed with fresh medium and incubated in a 1:1 mix of glia-conditioned and fresh medium for 30 min at 37°C, before processing for immunocytochemistry or labeling with DiIC16 (Invitrogen). Controls were handled identically but without NMDA.
DiIC16 labeling.
Living neurons were incubated for 1 min in DiIC16 diluted ∼5 μg/ml in conditioned medium, washed, mounted in PBS, and immediately observed.
Fixation and immunocytochemistry.
Neurons were either fixed in 4% paraformaldehyde, 4% sucrose in PBS (pF) at room temperature or in methanol at −20°C for experiments involving PSD-95 (Rao and Craig, 1997). Fixed neurons were incubated in 5% normal goat serum (NGS) in PBS containing 0.2% bovine serum albumin (BSA) and 0.2% Triton X-100 (PBS/BSA/Triton) for 30 min. Primary antibodies were applied in PBS/BSA/Triton at 4°C overnight and detected by fluorochrome-conjugated secondary antibodies (Obermair et al., 2004).
Live cell surface labeling.
For staining of surface-expressed HA-tagged CaV1.2 constructs living neurons were incubated with the rat anti-HA antibody for 30 min at 37°C (Watschinger et al., 2008). In NMDA experiments this was done during the 30 min post NMDA incubation. Then the cultures were rinsed in HBSS, pF fixed for 10 min, blocked with NGS, and incubated with the secondary antibody for 1 h (Obermair et al., 2004).
Live cell surface labeling combined with regular immunocytochemistry.
For colocalization of surface-expressed CaV1.2-HA constructs and cytoplasmic scaffold proteins, live cell-stained neurons were postfixed for 5 min in pF (for AKAP79/150 and α-actinin) or in methanol (for PSD-95). Then neurons were rinsed in PBS, permeabilized, and blocked again with 5% NGS in PBS/BSA/Triton and subsequently incubated with the second primary antibody overnight at 4°C. After washing, the Alexa 488-conjugated secondary antibody was applied for 1 h at room temperature. Coverslips were then washed and mounted in p-phenylene-diamine-glycerol to retard photobleaching (Flucher et al., 1993). Preparations were analyzed on an Axiophot or an Axiovert 200M microscope (Carl Zeiss) using a 63×, 1.4 NA objective. Images were recorded with a cooled CCD camera (SPOT; Diagnostic Instruments) and Metavue image processing software (Universal Imaging).
Antibodies.
Primary antibodies: rabbit anti-AKAP79/150 (4361J, polyclonal, generously supplied by Dr. Yvonne Lai, ICOS, Bothell, WA, 1:8000); mouse anti-α-actinin (monoclonal, clone EA-53, Sigma; 1:10,000); rat anti-HA (monoclonal, clone 3F10, Roche Diagnostics, 1:100); mouse anti-PSD-95 (monoclonal, clone 6G6–1C9, Affinity Bioreagents, 1:1000). Secondary antibodies: goat anti-mouse Alexa 488 (1:2000), goat anti-rabbit Alexa 488, and goat anti-rat Alexa 594 (Invitrogen, 1:4000).
Quantification of the density and fluorescent intensity of CaV1.2-HA clusters.
Fourteen-bit gray-scale images of the red (HA) and green (eGFP) channel were acquired. Corresponding images were aligned in an image stack and one dendritic segment of 10–15 μm length was selected for analysis. Using the eGFP image as reference, ROIs were drawn around the shaft and the adjacent regions containing the spines. The shaft area was measured and the number of spines was counted. The CaV1.2-HA image was background-flattened (MetaMorph software, Universal Imaging) and thresholded to trace the fluorescent clusters as accurately as possible. Using the integrated morphometric analysis option of MetaMorph, the number of clusters was counted and their average gray values were determined and corrected for background fluorescence.
Quantification of colocalization of CaV1.2-HA clusters with α-actinin, AKAP79/150, and PSD-95 in dendritic spines.
Fourteen-bit gray-scale images of the red (HA) and green (α-actinin, AKAP79/150, and PSD-95) channel were acquired. Corresponding images were 2D-deconvolved (MetaMorph), aligned, and background subtracted. A ROI was drawn around the region containing the dendritic spines along a dendritic segment of 20–50 μm length. Colocalization was analyzed using the distance based colocalization method, which is part of the JACoP plugin (Bolte and Cordelières, 2006) in ImageJ (W. S. Rasband, ImageJ, National Institutes of Health, Bethesda, MD, http://rsb.info.nih.gov/ij/, 1997–2007). Results are expressed as percentage of CaV1.2-HA objects colocalizing with α-actinin, AKAP79/150, or PSD-95, respectively.
Quantification of fluorescent intensity in dendritic spines and shafts.
To calculate the spine/shaft ratio, a line was placed through the spine and the adjacent shaft region in the eGFP image, along which background-subtracted intensities were recorded in the eGFP and the corresponding immunofluorescence image (cf. Fig. 1). The threshold for determining average intensities of the spine and shaft was set to 68% of eGFP fluorescence between the minimum intensity value of the line scan and the maximum of each peak.
Figure 1.
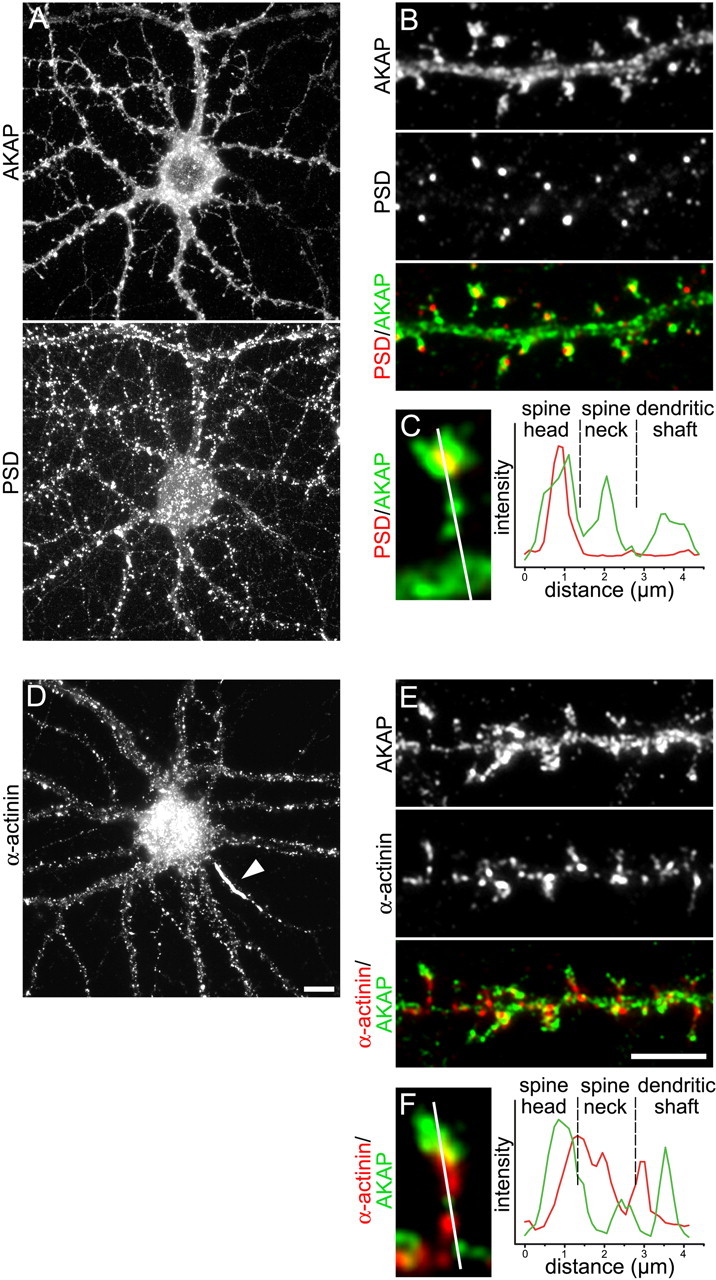
Localization of AKAP79/150, PSD-95, and α-actinin in dendrites of cultured hippocampal neurons. A, A representative glutamatergic neuron (20 DIV) labeled with anti-AKAP79/150 and anti-PSD-95 shows the distribution of both proteins in the soma and dendrites. B, AKAP79/150 staining is found throughout the dendritic shaft and is concentrated in the heads of dendritic spines (green in merged color image). Clusters of PSD-95, a marker of the excitatory postsynaptic compartment, are localized exclusively in the spine heads (red/yellow). C, A line scan (∼4 μm) across a typical spine demonstrates overlapping peaks of AKAP79/150 (green line) and PSD-95 (red line) labeling intensity in the spine head. D, A hippocampal neuron (20 DIV) labeled for α-actinin shows clusters in the somatodendritic compartment and a strong accumulation in the axon hillock (arrowhead). E, The dendritic segment shows that the α-actinin clusters (red) are concentrated in the proximal parts of the spine and in the dendritic shaft with little overlap with AKAP79/150 (green/yellow). F, A line scan across a double-labeled spine shows the maximum intensity of α-actinin (red line) in the spine neck alternating with AKAP79/150 peaks (green line) in the heads and the shaft. Scale bars: A, D, 10 μm; B, E, 5 μm.
Statistical analysis.
Absolute values of all the measurements were expressed as percentage of control. Statistical significance was calculated using the t test (Microsoft Excel) on normalized data. Analyses were performed on 15–25 neurons from at least two different experiments and at least two separate culture preparations. All data are reported as mean ± 95% confidence interval. Graphs and figures were generated using Origin 7 and Adobe Photoshop 8.0 software.
Results
The distribution of CaV1.2 clusters in the membrane of hippocampal neurons overlaps with AKAP79/150 but is distinct from that of PSD-95 and α-actinin
AKAP79/150, PSD-95, and α-actinin are all constituents of dendritic spines. AKAP79/150 and α-actinin were reported to interact with the CaV1.2 calcium channel (Hall et al., 2007; Oliveria et al., 2007) (J. W. Hell, personal communication). PSD-95 is a marker of the postsynaptic signaling complex of glutamatergic synapses and recruits AKAP79/150 and the associated protein kinases and phosphatases to the postsynaptic density. If any one of these scaffold proteins participates in the CaV1.2 signaling complex in hippocampal neurons it is expected to be colocalized with this channel. We used double-immunofluorescence labeling to determine the distribution patterns of AKAP79/150, PSD-95, and α-actinin in the somatodendritic compartment in differentiated cultured mouse hippocampal neurons. Figure 1A shows that AKAP79/150 is widely distributed throughout the neurons, whereas PSD-95 is confined to discrete puncta along the dendrites and on the soma. A magnified dendritic segment reveals that AKAP79/150 and PSD-95 colocalize in the spine heads, where PSD-95 marks the site of the glutamatergic synapse (Fig. 1B). In the dendritic spines AKAP79/150 reaches its highest concentrations and its distribution usually exceeds that of PSD-95 clusters in that it fills the entire spine. In addition AKAP79/150 but not PSD-95 is found in the spine neck and in dendritic shafts (Fig. 1C). α-Actinin also shows a clustered distribution pattern in many neurons (Fig. 1D). However, in contrast to AKAP79/150 and PSD-95, α-actinin clusters are typically excluded from the spine head but enriched in the spine necks and along the shafts of the dendrites (Fig. 1E,F). This distribution is consistent with dense α-actinin labeling of the spine apparatus in the necks of spines demonstrated by immunogold staining in electron microscopic sections by Wyszynski et al. (1998) (see Fig. 6C); although in addition these authors also find α-actinin in the postsynaptic density. Whereas AKAP79/150 and PSD-95 were restricted to the somatodendritic compartment, α-actinin was also found in the axon, where it often accumulated in the axon hillock (Fig. 1D, arrowhead).
Figure 6.

Distribution, density, and fluorescence intensity of CaV1.2-HA clusters are not reduced by NMDA-induced redistribution of AKAP79/150. A, Neurons transfected with CaV1.2-HA and CFP were treated with 25 μm NMDA and live-cell labeled with anti-HA. Following fixation and permeabilization they were labeled with anti-AKAP79/150. In dendritic segments of control cells AKAP79/150 (green) is abundant in dendritic spines, some of which also contain CaV1.2-HA clusters (red/yellow, arrowheads). In NMDA-treated neurons AKAP79/150 is concentrated in the dendritic shaft (turquoise) and absent from the spines. However CaV1.2-HA clusters (red, arrowheads) still persist in the spines. The magnified spines (right) show CaV1.2-HA clusters (red) in the control containing AKAP79/150 (green), and in a spine of an NMDA-treated neuron that lacks AKAP79/150 immunolabeling (the blue CFP outlines the spine). For better orientation the contours of the example spines and the CaV1.2-HA clusters are outlined in the sketches at right. Scale bars: 5 μm (A); 0.5 μm (enlargement). B, C, Quantification of the density and fluorescence intensity of CaV1.2-HA clusters; expressed as % of control and analyzed per length of dendrites (B, control n = 15, NMDA n = 17; N = 4; *p < 0.015), and separately as clusters per spine and clusters per area of shafts (C, control n = 13, NMDA n = 14; N = 3). NMDA-induced removal of AKAP79/150 and PSD-95 from dendritic spines (see also Fig. 5) did not cause a reduction in the number of CaV1.2-HA clusters, nor a reduction of their fluorescence intensity. Bar graphs: mean ± 95% C.I.
Previously we demonstrated that CaV1.2 exists in small clusters that are distributed along the dendritic shafts and in the heads and necks of dendritic spines (Obermair et al., 2004; Szabo et al., 2006). To determine which of the three scaffold proteins actually coexist with CaV1.2 in differentiated hippocampal neurons, we compared the distribution patterns of AKAP79/150, PSD-95, and α-actinin directly with that of CaV1.2 membrane clusters. For this purpose hippocampal neurons were transfected with a CaV1.2 construct containing an HA-tag in the extracellular loop immediately after the transmembrane helix IIS5. Before fixation and permeabilization the living cells were incubated with an HA antibody, which under these conditions exclusively stained CaV1.2-HA expressed in the plasma membrane (see Obermair et al., 2004). Subsequently the neurons were fixed, permeabilized, and labeled with the antibodies against AKAP79/150, PSD-95, and α-actinin. Figure 2A shows the typical clustered distribution pattern of CaV1.2-HA in hippocampal neurons. We previously demonstrated that this pattern closely resembles that of endogenous CaV1.2 (Obermair et al., 2004). Double labeling with AKAP79/150 indicated that most of the CaV1.2-HA clusters coincided with this scaffold protein both in the dendritic shafts and in spines (Fig. 2B). This was to be expected because of the high abundance of AKAP79/150 label throughout the dendritic arbor. Accordingly, we also observed spines containing AKAP79/150 but no CaV1.2-HA clusters, and rarely spines in which the CaV1.2-HA clusters did not overlap with AKAP79/150 (Fig. 2B). Double labeling with PSD-95 demonstrated that CaV1.2-HA clusters are not localized in the postsynaptic densities (Fig. 2C). Whereas PSD-95 clusters were located along both sides of the dendrite, presumably at the tips of the spines, the majority of CaV1.2-HA clusters was located in between. Some spines that were identified by their PSD-95 clusters did not contain calcium channel clusters. However, even when CaV1.2-HA and PSD-95 coexisted on a spine their clusters did not coincide, rather were located adjacent to each other with different degrees of overlap (Fig. 2C, high-magnification examples). Double labeling of CaV1.2-HA with α-actinin revealed that these two proteins were clearly separate from each other (Fig. 2D). Whereas the CaV1.2-HA clusters were found on dendritic shafts and spines, α-actinin was mostly concentrated in the neck of the spines and only rarely coincided with CaV1.2-HA clusters (Fig. 2D). These observations were further supported using a distance-based colocalization analysis. This method indicates the percentage at which the centroids of CaV1.2-HA clusters coincide with clusters of the corresponding protein. Consistent with our qualitative observation, the highest value was obtained with AKAP79/150 (68 ± 3% SEM, n = 11), and the lowest degree of overlap with α-actinin (53 ± 4%, n = 6). The value for PSD-95 (61 ± 6%, n = 7) was not significantly different from the other two, but fell in been those for AKAP79/150 and α-actinin. Together this immunofluorescence analysis indicates that CaV1.2-HA calcium channel clusters overlap with the widely distributed AKAP79/150. In contrast, CaV1.2-HA clusters do not colocalize with the discrete clusters of PSD-95 (see also Obermair et al., 2004 and Tippens et al., 2008), nor with those of α-actinin.
Figure 2.

Differential distribution of CaV1.2-HA relative to AKAP79/150, PSD-95, and α-actinin in dendritic shaft and spines. A, A hippocampal neuron (18 DIV) transfected with CaV1.2-HA and labeled with an antibody against the extracellular HA epitope before fixation reveals clusters of surface-expressed CaV1.2-HA on the soma and dendrites, and at lower density on the proximal axon (arrowheads). B–D, After live cell labeling of CaV1.2-HA, neurons were fixed and stained for AKAP79/150 (B), PSD-95 (C), or α-actinin (D). B, Membrane expressed CaV1.2-HA clusters (red) are found on the shafts and spines of dendrites. Due to the extent of the AKAP79/150 distribution (green), CaV1.2-HA clusters frequently colocalize with AKAP79/150 (yellow, see example of enlarged spine). The line scan across a representative spine shows that the peaks of CaV1.2-HA (red) and AKAP79/150 (green) intensity coincide. C, Whereas CaV1.2-HA clusters are abundant on the dendritic shafts and spines, PSD-95 clusters (green) are restricted to the spines; therefore they show little colocalization. When clusters of CaV1.2-HA (red) and PSD-95 (green) are found together in spines they are adjacent to each other (see enlarged example). The line scan shows that CaV1.2-HA (red) and PSD-95 (green) fluorescence overlap but that their peaks do not coincide. D, Clusters of α-actinin (green) are larger and less abundant than those of CaV1.2-HA (red). α-Actinin clusters are found in necks of spines and typically do not colocalize with CaV1.2-HA clusters (see magnified example and line scan). B–D, Note that not all spines labeled with AKAP79/150, PSD-95, or α-actinin also contain CaV1.2-HA clusters. Scale bars: A, 10 μm; B–D, 5 μm.
Mutation of C-terminal interaction domains for AKAP79/150 and PDZ-proteins, respectively, does not alter the distribution and size of CaV1.2-HA clusters in hippocampal neurons
The C terminus of CaV1.2 contains binding sites for AKAP79/150 and for PDZ proteins (Weick et al., 2003; Oliveria et al., 2007). Based on the colocalization (see above) and previous functional studies (Oliveria et al., 2007), AKAP79/150 is a potential interaction partner of CaV1.2 in hippocampal neurons. Furthermore, coexpression of AKAP79/150 in oocytes promoted functional membrane expression of CaV1.2-HA (Altier et al., 2002). Therefore we hypothesized that AKAP79/150 may be involved in the targeting and/or anchoring of CaV1.2 in dendrites of hippocampal neurons. To test this we mutated the three nonpolar amino acids of the modified leucine zipper in the C terminus of CaV1.2-HA into alanines (Fig. 3B). This mutation has previously been demonstrated to abolish AKAP79/150 binding (Oliveria et al., 2007). When channels with the mutated leucine zipper [CaV1.2-HA(ΔLZ)] were expressed in hippocampal neurons, the expression pattern was indistinguishable from that of wild-type CaV1.2-HA (Fig. 3A). CaV1.2-HA(ΔLZ) clusters were found in dendritic shafts and spines at the same density as those of CaV1.2-HA. Also, the intensity of the clusters, which corresponds to the number of channels per cluster, was not altered by the mutation of the AKAP79/150 binding site (Fig. 3C).
Figure 3.

Distribution and size of membrane expressed clusters of CaV1.2-HA constructs with mutated binding sites for AKAP and PDZ proteins. B, The domain model of CaV1.2-HA indicates the positions of the extracellular HA-tag, the amino acid substitutions in the leucine zipper (AKAP binding site), and the truncation of the C-terminal VSNL sequence (PDZ binding site). A, Immunofluorescence labeling of surface-expressed CaV1.2-HA, CaV1.2-HA(ΔLZ), and CaV1.2-HA(ΔVSNL) in dendritic segments of hippocampal neurons (DIV 17). Soluble eGFP (green) outlines the morphology of the dendritic shafts and spines. Clusters of wild-type and mutant channels (red/yellow) show the same distribution pattern in the dendritic shaft and spines. Scale bar, 5 μm. C, Quantification of the density (number of clusters per spine or per μm2 of dendritic shaft) and the intensity (average gray value) of CaV1.2-HA(ΔLZ) or CaV1.2-HA(ΔVSNL) expressed as a percentage of the corresponding wild-type CaV1.2-HA values (normalized to 100%). Spines and shaft regions were defined in the eGFP image, the corresponding CaV1.2-HA image was thresholded and analyzed as described in the Materials and Methods. For CaV1.2-HA(ΔLZ) both values were indistinguishable from those of the wild-type channel [25 and 24 neurons (n) analyzed for CaV1.2-HA and CaV1.2(ΔLZ), respectively, in 6 independent experiments (N)]. For CaV1.2-HA(ΔVSNL) cluster density and intensity showed a small increase (Cav1.2-HA n = 22, Cav1.2(ΔVSNL) n = 18; N = 4). Bar graphs: mean ± 95% confidence interval (C.I.); *significance, t test, 0.05 > p > 0.03.
The C-terminal PDZ binding sequence is essential for downstream signaling to activate gene expression and it was suggested that these interactions may also be involved in the targeting of L-type calcium channels to distinct intracellular scaffolding and signaling proteins (Weick et al., 2003). However, the PDZ protein interacting with CaV1.2 channels in neurons remains unknown. To examine whether interactions with PDZ proteins are required for the correct targeting and anchoring of CaV1.2-HA in hippocampal neurons, we deleted the VSNL sequence in CaV1.2-HA (Fig. 3B). Similar deletions of the PDZ binding domains of CaV1.3 and CaV2.2 channels had been reported to alter the subcellular distribution of these channels in neurons (Maximov and Bezprozvanny, 2002; Zhang et al., 2005). In contrast, the data presented in Figure 3A demonstrate that expression of CaV1.2-HA(ΔVSNL) in hippocampal neurons showed similar distribution patterns as the wild-type CaV1.2-HA. Unexpectedly, the density of CaV1.2-HA(ΔVSNL) clusters even showed a modest but statistically significant increase in dendritic spines and shafts (Fig. 3C), and, at least in the shafts, also the intensity of the fluorescence clusters increased.
Figure 4 provides a direct comparison of these mutant calcium channels with AKAP79/150 and PSD-95, respectively. Double labeling of membrane incorporated CaV1.2-HA(ΔLZ) and AKAP79/150 revealed the same relationship as shown before for the wild-type CaV1.2-HA (compare Fig. 2B). CaV1.2-HA(ΔLZ) clusters overlap with the AKAP79/150 stain in the shafts and spines of dendrites. Similarly, the double labeling pattern of CaV1.2-HA(ΔVSNL) with PSD-95 closely resembled that shown for wild-type CaV1.2-HA and PSD-95 (compare Fig. 2C). CaV1.2-HA(ΔVSNL) clusters were not colocalized with PSD-95 clusters but were frequently found adjacent to them in dendritic spines. Thus, mutation and deletion of CaV1.2 sequences known to interact with AKAP79/150 and PDZ proteins, respectively, did not abolish the characteristic subcellular distribution of the calcium channel constructs or their spatial relationship with the scaffold proteins in dendrites of hippocampal neurons. On the contrary, deletion of the PDZ binding sequence even increased the density and size of the calcium channel clusters. Moreover, deletion of the binding sites for AKAP79/150 and PDZ proteins in CaV1.2-HA did not affect the distribution of the corresponding scaffold proteins in the dendritic shafts and spines.
Figure 4.

Mutating the binding sites for AKAP79/150 and PSD-95 does not change their distribution and spatial relationship to CaV1.2-HA. A, Hippocampal neurons (DIV 18) transfected with CaV1.2-HA(ΔLZ) were double labeled with anti-HA (live cell labeling) and subsequently with anti-AKAP79/150. A representative dendritic segment shows that AKAP79/150 (green) is distributed throughout the shafts and spines of dendrites and has a large degree of overlap with CaV1.2-HA(ΔLZ) clusters (yellow). B, A neuron transfected with CaV1.2-HA(ΔVSNL) was double labeled with anti-HA (live cell labeling) and subsequently with anti-PSD-95. The clusters of PSD-95 (green) and of CaV1.2-HA(ΔVSNL) (red) showed little overlap. The overall distribution of AKAP79/150 and PSD-95 was indistinguishable from that observed in neurons expressing wild-type CaV1.2-HA (compare Fig. 2). Scale bar, 5 μm.
NMDA-induced removal of AKAP79/150 and PSD-95 from dendritic spines does not disrupt CaV1.2 clusters in dendritic spines
The observation that deletion of the binding sites for either AKAP79/150 or PDZ proteins did not hamper normal targeting and distribution of CaV1.2-HA in hippocampal neurons, did not exclude the possibility that interactions of these scaffold proteins with additional or alternative binding sites are required for the targeting and immobilization of CaV1.2 in dendrites. If this was the case, then disruption of the postsynaptic scaffolds containing AKAP79/150 and PDZ proteins should also disrupt the postsynaptic calcium channel clusters. Brief application of NMDA has been used to induce chemical LTD (Lee et al., 1998). This process involves the disruption of the actin cytoskeleton in dendritic spines, the relocation of AKAP79/150 from the spine into the shafts of dendrites, and the ubiquitin-dependent degradation of PSD-95 in the proteasome. As a result AMPA receptors are removed from the postsynaptic membrane and the efficacy of glutamatergic synaptic transmission is reduced (Colledge et al., 2003). Here we used this experimental paradigm to test for the potential involvement of AKAP79/150, α-actinin, and PSD-95 in the stabilization of CaV1.2-HA in spines. We reasoned that if anyone of these scaffold proteins or their concerted action was necessary for anchoring CaV1.2-HA in dendritic spines, their NMDA-induced removal or degradation should be accompanied by a disruption of the CaV1.2-HA clusters.
First we analyzed the effect of NMDA on AKAP79/150, α-actinin, and PSD-95 in our culture system. Figure 5 compares the distribution of these scaffold proteins in hippocampal neurons 30 min after NMDA treatment with their distribution in mock-treated control neurons. The double-immunofluorescence images of the control neurons show the same distribution patterns as seen in the untreated cultures (Fig. 5A,B; compare Fig. 1). Most importantly, AKAP79/150, α-actinin, and PSD-95 were all found in the dendritic spines. In contrast, after NMDA treatment the labeling patterns of all three scaffold proteins had undergone striking and characteristic changes. Consistent with previous reports (Smith et al., 2006), AKAP79/150 and α-actinin became concentrated in the shafts of the dendrites (Fig. 5A,B). In many neurons spines labeled with AKAP79/150 or α-actinin were no longer visible. In parallel, PSD-95 clusters disappeared from the dendritic spines (Fig. 5A) (Colledge et al., 2003; Smith et al., 2006). Nevertheless, labeling of the membrane with the fluorescent lipid marker DiIC16 demonstrated that spines were still present after NMDA treatment (Fig. 5C). Furthermore, NMDA-induced removal of AKAP79/150 and PSD-95 from dendritic spines was directly visualized in neurons transfected with soluble eGFP. Whereas in control cells clusters of the scaffold proteins coexisted with eGFP in the spine heads, in NMDA-treated neurons eGFP labeled spines were devoid of AKAP79/150 and PSD-95 (Fig. 5D). We quantified the NMDA induced disruption of AKAP79/150 and PSD-95 clusters in the dendritic spines by measuring the ratio of the fluorescence intensity in the spines and the adjacent segment of the shaft of the dendrite. The spine/shaft ratio of eGFP showed little change upon NMDA treatment, indicating that the spines themselves were still intact. However, as expected from the qualitative analysis the spine/shaft ratios for AKAP79/150 and PSD-95 were dramatically reduced to 31.6% and 17.2% of control values, respectively. Thus, the applied NMDA protocol caused the removal of AKAP79/150, α-actinin, and PSD-95 from dendritic spines without inducing a retraction of the spines.
Figure 5.

NMDA treatment causes the loss of PSD-95 clusters and the redistribution of AKAP79/150 and α-actinin from spines to dendritic shaft. Hippocampal neurons (19 DIV) were treated for 3 min with 25 μm NMDA, fixed after 30 min, and subsequently immunolabeled. A, Double immunolabeling of AKAP79/150 and PSD-95 shows that, compared with mock-treated neurons (control), the number and intensity of PSD-95 clusters (red/yellow) was greatly reduced. AKAP79/150 (green) was absent from the spines and concentrated in the dendritic shafts. B, Double immunolabeling of AKAP79/150 and α-actinin shows that also α-actinin (red/yellow) was absent from the spines and concentrated in large patches in the dendritic shafts. C, Control and NMDA-treated neurons stained with the fluorescent lipid probe DiIC16 reveal that 30 min after NMDA treatment the dendritic spines were still present. D, The extent of the NMDA-induced rearrangement of AKAP79/150 and the loss of PSD-95 was quantified in eGFP-transfected neurons. NMDA treatment reduced the ratios of the fluorescence intensities in spines and shafts to 31.6 ± 4.7% for AKAP79/150 (control n = 20, NMDA n = 21; N = 2) and to 17.2 ± 11.9% for PSD-95 of controls (control n = 18, NMDA n = 15; N = 2). In the same dendrites the spine/shaft ratios for eGFP were not dramatically reduced. Bar graphs: mean ± 95% C.I., *p = 0.011, **p < 0.0005. Scale bars: A–C, 5 μm; D, 0.5 μm.
To examine potential effects of NMDA-induced disruption of scaffold proteins on the CaV1.2 calcium channels in the spines, hippocampal neurons were transfected with CaV1.2-HA plus CFP, and then exposed to NMDA as described above. To specifically label the CaV1.2-HA inserted into the plasma membrane the living neurons were incubated with anti-HA before fixation. Figure 6A shows dendritic segments of triple labeled neurons. In the controls the CFP-labeled shaft (blue) is flanked by AKAP79/150-containing spines (green), some of which contain clusters of CaV1.2-HA (red/yellow, arrowheads). In NMDA-treated neurons the AKAP79/150 label was concentrated in the shaft (resulting in a reduction of green spines and in a turquoise color in the shaft), whereas the CaV1.2-HA clusters (red) persisted in the spines. Quantification of the triple-labeled neurons confirmed that CaV1.2-HA clusters withstood NMDA treatment (Fig. 6B). Neither the overall number of CaV1.2-HA clusters, expressed as the mean density of clusters, nor the number of channels per cluster, expressed as the mean fluorescent intensity of the CaV1.2-HA clusters, decreased on NMDA treatment. On the contrary, both values actually showed a slight increase. This finding was further substantiated by separately analyzing the density and intensity of clusters in the spines and the shafts of neurons labeled with eGFP and anti-HA (Fig. 6C). The number and size of CaV1.2-HA clusters was not reduced in the spines, which lost all three analyzed scaffold proteins, nor were they changed in the shafts, where AKAP79/150 and α-actinin were now concentrated. Together the data presented in Figures 5 and 6 demonstrate that NMDA-induced disruption of postsynaptic scaffolds in spines is not accompanied by a loss or shrinking of CaV1.2-HA clusters.
Discussion
The results described above clearly demonstrate that neither an interaction with AKAP79/150 nor interactions of the C-terminal VSNL sequence with PDZ proteins are required for the targeting and localization of CaV1.2 calcium channels in dendrites of hippocampal neurons. The distribution, density, and the fluorescence intensity of CaV1.2-HA clusters in dendritic spines and shafts remained unaltered when the known interaction domains for these scaffold proteins—the C-terminal leucine zipper and VSNL, respectively—were mutated, or when the AKAP79/150 and PSD-95 scaffolds in spines were disrupted in response to pharmacological NMDA receptor activation.
AKAP79/150 is the major AKAP protein in neurons, where it is widely distributed and has been shown to anchor protein kinases and other signaling proteins to multiple receptors and ion channels (for review, see Bauman et al., 2004). It is concentrated in the dendritic spines and recruits the protein kinase A (PKA) and the protein phosphatase 2B (calcineurin) to the AMPA receptor (Colledge et al., 2000). In neurons AKAP79/150 also associates with CaV1.2 (Hall et al., 2007). It recruits PKA and calcineurin to the channel and is necessary for the β-adrenergic stimulation of L-type calcium currents (Hall et al., 2007) as well as for the L-type calcium current-mediated activation of the transcriptional regulator NFATc4 (Oliveria et al., 2007). This dual role of AKAP79/150 is consistent with its colocalization with both CaV1.2 and PSD-95 in dendritic spines of hippocampal neurons. Three different binding sites for AKAP79/150 were described in the N terminus, the cytoplasmic loop connecting repeats I and II, and in the C terminus of CaV1.2 (Hall et al., 2007). The C-terminal leucine zipper was shown to be essential for AKAP binding and for β-adrenergic stimulation and reversible phosphorylation of CaV1.2 in heart muscle and in neurons (Hulme et al., 2003; Oliveria et al., 2007). Mutation of the three basic residues of this motif blocked AKAP79/150 and PKA binding and phosphorylation of the Ca2+ channel in response to β-adrenergic stimulation (Oliveria et al., 2007). Therefore it was reasonable to expect that the same mutations in the C-terminal leucine zipper would also inhibit any other AKAP79/150-dependent effects on the channel, including those on membrane expression or targeting of CaV1.2. Altier et al. (2002) demonstrated that in oocytes AKAP79 directly regulated the surface expression of CaV1.2. Our present findings, showing that mutation of the leucine zipper or NMDA-induced removal of AKAP79/150 from dendritic spines did not alter the distribution pattern or the size of the CaV1.2-HA clusters, indicate that in neurons the interactions of AKAP79/150 with the C-terminal leucine zipper or with any other sequences are not essential for anchoring CaV1.2-HA at postsynaptic sites. This conclusion is consistent with functional data from AKAP79/150 siRNA experiments (Oliveria et al., 2007). Knock-down of AKAP79/150 blocked the effects on channel phosphorylation and current modulation, but did not result in decreased basal L-type Ca2+ currents, suggesting that the total number of functional channel in the membrane was not altered. Apparently the role of AKAP79/150 with respect to the L-type calcium channel is to recruit upstream and downstream signaling proteins to the channel, but AKAP79/150 itself is not important for targeting CaV1.2 into the signaling complex. Interestingly, the ties of AKAP79/150 with the CaV1.2 channel are not strong enough to retain detectable amounts of AKAP79/150 at the channel clusters upon strong NMDA receptor activation. Hell et al. (1996) demonstrated that NMDA treatment causes proteolytic cleavage of the C terminus of CaV1.2. One possibility is that cleavage of the C terminus directly leads to the redistribution of AKAP79/150 together with the CaV1.2 C terminus into the dendritic shafts, while the distribution of the truncated calcium channels in the spines remains unaltered. However, at least in cardiac myocytes the cleaved C terminus appears to remain associated with the channel complex in the membrane (Gao et al., 1997).
Another candidate for targeting and anchoring of the CaV1.2 calcium channel in the postsynaptic compartment is the interaction of its C-terminal VSNL sequence with PDZ proteins. When expressed in hippocampal neurons an eGFP-VSNL fusion protein showed a distribution pattern similar to that of CaV1.2, and it specifically attenuated L-type calcium channel-induced CREB phosphorylation and CRE-dependent transcription (Weick et al., 2003). A truncated CaV1.2 construct in which VSNL was lacking had the same effect, although its overall distribution in the somatodendritic compartment was the same as that of the full-length CaV1.2. Thus, the authors speculated that differences in their localization may exist at the subcellular level (Weick et al., 2003). Here we analyzed the subcellular distribution specifically of membrane incorporated CaV1.2-HA with and without the VSNL sequence. Unexpectedly, the deletion of VSNL did not result in an altered distribution or a reduction of membrane expression. On the contrary, the density of CaV1.2-HA(ΔVSNL) clusters in the shafts and spines of dendrites as well as their fluorescence intensity, which is a measure of channel number per cluster, were slightly increased compared with the control. Therefore, interactions of VSNL with PDZ-proteins may be important for L-type calcium current-dependent signaling to CREB, but are not necessary for the correct targeting of CaV1.2-HA in dendrites of hippocampal neurons. In this regard the role of the PDZ ligand of CaV1.2 differs from that of CaV1.3, which has been reported to be important for the insertion of this L-type channel into postsynaptic sites (Zhang et al., 2005), and from that of CaV2.2, which has been reported to be necessary and sufficient for the targeting of N-type calcium channels into presynaptic terminals (Maximov and Bezprozvanny, 2002). If interactions of the VSNL sequence with PDZ-proteins are at all involved in the regulation of CaV1.2 expression in the postsynaptic compartment, then the increased density and size of CaV1.2-HA(ΔVSNL) clusters observed here would indicate a limiting role in channel targeting or stabilization. Interactions between VSNL with a PDZ protein could either reduce the insertion rate of CaV1.2 into dendritic clusters or increase its turnover and internalization.
By themselves, the mutagenesis experiments do not exclude the possibility that interactions of the leucine zipper and VSNL cooperate in anchoring of CaV1.2. However, CaV1.2-HA clusters also remain stable when AKAP79/150, PSD-95, and other scaffolding proteins are removed from dendritic spines upon NMDA application. Therefore it is unlikely that interactions of AKAP79/150 and PDZ-proteins with these or any other binding sites in CaV1.2 (Hall et al., 2007) are required for stabilizing the channel in dendritic spines. This raises the question which other proteins might be responsible for anchoring CaV1.2 clusters in the postsynaptic compartment. One possible candidate is the auxiliary calcium channel β subunit, which is essential for membrane expression of CaV channels (Leroy et al., 2005; Obermair and Flucher, unpublished results), and may serve as adaptor for other scaffolds or signaling proteins. However, due to the high sequence homology between the β isoforms and the great promiscuity of their interactions with all high-voltage-activated calcium channels it is unlikely that β subunits determine the specific localization of CaV channels in distinct neuronal compartments.
Moreover, our experiments indicate that CaV1.2 clusters are stable regardless of the activity-induced changes in the postsynaptic compartment of glutamatergic synapses. NMDA-induced remodeling of the postsynaptic density ultimately results in the endocytosis of AMPA receptors (Colledge et al., 2003), and is therefore used as an experimental paradigm to induce LTD (also termed chemical LTD; Lee et al., 1998). Interestingly, the CaV1.2 calcium channels, which are involved in the signaling of long-term potentiation of synaptic strength (Impey et al., 1996), are not subject to downregulation during chemical LTD. This conclusion is in apparent conflict with the results of a recent study reporting an activity-induced downregulation of calcium currents in cortical neurons that was accompanied by an increased turnover rate and an internalization of CaV1.2 channels (Green et al., 2007). However, that study was performed on immature cortical neurons and Neuro2A neuroblastoma cells, and the effects on channel expression were not specific for CaV1.2. Therefore, it is likely that the effects observed in that study resemble a developmental change in channel expression patterns, possibly induced by the onset of electrical activity. In contrast, here we studied the membrane expression of CaV1.2-HA in the context of mature glutamatergic synapses, and find that during NMDA-induced synaptic remodeling of the postsynaptic compartment clusters of CaV1.2 calcium channels in dendritic spines remain stable.
Together with recent findings on the signaling pathways of L-type calcium channels in neurons our current results allow drawing the following picture of the CaV1.2 signaling complex in the postsynaptic compartments of excitatory hippocampal neurons (Fig. 7). Clusters of a small number of CaV1.2 channels are found along the soma and dendrites of differentiated hippocampal neurons, including dendritic spines (Obermair et al., 2004; Tippens et al., 2008). However within the spines CaV1.2 clusters are not colocalized with PSD-95, indicating that they are not part of the glutamate receptor signaling complex. A distinct localization of CaV1.2 and glutamate receptors is consistent with their specific roles in signaling to the nucleus (Graef et al., 1999; Weick et al., 2003; Oliveria et al., 2007), as well as with the finding that activation of L-type calcium channels, but not strong activation of NMDA receptors, leads to the depression of R-type calcium channels in dendritic spines (Yasuda et al., 2003). Only if CaV1.2 and NMDA receptors are spatially separated, it is conceivable that calcium nanodomains in the immediate vicinity of these channels can activate distinct signal transduction pathways. This may in part be achieved by their association with distinct PDZ-proteins (Colledge et al., 2000; Weick et al., 2003). Nevertheless, CaV1.2 and glutamate receptors use the same AKAP to recruit protein kinases and phosphatases to either signaling complex. AKAP79/150 is colocalized and functionally interacts with both CaV1.2 and the AMPA receptor (Bauman et al., 2004; Hall et al., 2007; Oliveria et al., 2007). In the case of CaV1.2 this signaling complex is involved in the phosphorylation and modulation of the calcium channel in response to β-adrenergic stimulation (Davare et al., 2001; Oliveria et al., 2007), as well as in the activation of the transcriptional regulator NFATc4 (Graef et al., 1999; Oliveria et al., 2007). Stimuli that induce chemical LTD not only lead to the degradation of PSD-95 (Colledge et al., 2003), but also remove AKAP79/150 from the spine (Fig. 7B). Therefore it is expected that this process disrupts PKA and calcineurin-dependent signaling in both the glutamate receptor as well as in the CaV1.2 signaling complex. In the glutamate receptor complex this causes the removal of AMPA receptors from the postsynaptic membrane. However targeting and membrane expression of the CaV1.2 channel remain unaffected by this form of NMDA-induced synaptic plasticity. The stability of CaV1.2 clusters in dendritic spines may be important for synapses weakened by LTD, to be strengthened again during subsequent reinforcing stimuli.
Figure 7.

Differential effects of NMDA-induced synaptic remodeling on CaV1.2 and glutamate receptor signaling complexes in dendritic spines. A, CaV1.2 and glutamate receptors form separate signaling complexes in dendritic spines. Both are associated with PDZ-proteins and an AKAP79/150/PKA/calcineurin complex. In CaV1.2 a PDZ protein interacting sequence is necessary for activating the transcriptional regulator CREB, and AKAP79/150 interactions are necessary for activating NFATc4. B, Strong stimulation of NMDA receptors activates CREB as well as the disassembly of the glutamate receptor signaling complex. AKAP79/150 is relocated from the spine into the dendritic shaft, PSD-95 is degraded, which in turn causes the internalization of AMPA receptors and long-term depression of synaptic strength. The calcium channels clusters in the spines are no longer colocalized with AKAP79/150; however, their size and localization within the spine remains unaltered.
Footnotes
This work was supported by grants from the Austrian Science Fund and the Austrian National Bank (P17806-B05 and P20059-B05 to B.E.F. and P17807-B05 to G.J.O.) and from the Tyrolean Science Fund (to G.J.O.). This work is part of the thesis of V.D.B. We thank S. Baumgartner for her excellent technical assistance and P. Tuluc for functional analysis of calcium channel constructs.
References
- Altier C, Dubel SJ, Barrère C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW, Nargeot J, Bourinet E. Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem. 2002;277:33598–33603. doi: 10.1074/jbc.M202476200. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Bauman AL, Goehring AS, Scott JD. Orchestration of synaptic plasticity through AKAP signaling complexes. Neuropharmacology. 2004;46:299–310. doi: 10.1016/j.neuropharm.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel CaV1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Flucher BE, Andrews SB, Fleischer S, Marks AR, Caswell A, Powell JA. Triad formation: organization and function of the sarcoplasmic reticulum calcium release channel and triadin in normal and dysgenic muscle in vitro. J Cell Biol. 1993;123:1161–1174. doi: 10.1083/jcb.123.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Puri TS, Gerhardstein BL, Chien AJ, Green RD, Hosey MM. Identification and subcellular localization of the subunits of L-type calcium channels and adenylyl cyclase in cardiac myocytes. J Biol Chem. 1997;272:19401–19407. doi: 10.1074/jbc.272.31.19401. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. Cambridge, MA: MIT; 1998. pp. 339–370. [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Green EM, Barrett CF, Bultynck G, Shamah SM, Dolmetsch RE. The tumor suppressor eIF3e mediates calcium-dependent internalization of the L-type calcium channel CaV1.2. Neuron. 2007;55:615–632. doi: 10.1016/j.neuron.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel CaV1.2 via A-kinase anchor protein 150 in neurons. Biochemistry. 2007;46:1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Breeze LJ, Wang KK, Chavkin C, Catterall WA. N-methyl-d-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc Natl Acad Sci U S A. 1996;93:3362–3367. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland TM, Saggau P. Facilitation of L-type Ca2+ channels in dendritic spines by activation of β2 adrenergic receptors. J Neurosci. 2004;24:8416–8427. doi: 10.1523/JNEUROSCI.1677-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003;100:13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Leroy J, Richards MW, Butcher AJ, Nieto-Rostro M, Pratt WS, Davies A, Dolphin AC. Interaction via a key tryptophan in the I-II linker of N-type calcium channels is required for β1 but not for palmitoylated β2, implicating an additional binding site in the regulation of channel voltage-dependent properties. J Neurosci. 2005;25:6984–6996. doi: 10.1523/JNEUROSCI.1137-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Maximov A, Bezprozvanny I. Synaptic targeting of N-type calcium channels in hippocampal neurons. J Neurosci. 2002;22:6939–6952. doi: 10.1523/JNEUROSCI.22-16-06939.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein PG, Bito H, Deisseroth K, Tsien RW. Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J Neurosci. 2000;20:266–273. doi: 10.1523/JNEUROSCI.20-01-00266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T. Role of hippocampal CaV1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermair GJ, Kaufmann WA, Knaus HG, Flucher BE. The small conductance Ca2+-activated K+ channel SK3 is localized in nerve terminals of excitatory synapses of cultured mouse hippocampal neurons. Eur J Neurosci. 2003;17:721–731. doi: 10.1046/j.1460-9568.2003.02488.x. [DOI] [PubMed] [Google Scholar]
- Obermair GJ, Szabo Z, Bourinet E, Flucher BE. Differential targeting of the L-type Ca channel α1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci. 2004;19:2109–2122. doi: 10.1111/j.0953-816X.2004.03272.x. [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML. cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci. 2006;26:2391–2402. doi: 10.1523/JNEUROSCI.3092-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo Z, Obermair GJ, Cooper CB, Zamponi GW, Flucher BE. Role of the synprint site in presynaptic targeting of the calcium channel CaV2.2 in hippocampal neurons. Eur J Neurosci. 2006;24:709–718. doi: 10.1111/j.1460-9568.2006.04947.x. [DOI] [PubMed] [Google Scholar]
- Tippens AL, Pare JF, Langwieser N, Moosmang S, Milner TA, Smith Y, Lee A. Ultrastructural evidence for pre- and postsynaptic localization of CaV1.2 L-type Ca2+ channels in the rat hippocampus. J Comp Neurol. 2008;506:569–583. doi: 10.1002/cne.21567. [DOI] [PubMed] [Google Scholar]
- Watschinger K, Horak SB, Schulze K, Obermair GJ, Wild C, Koschak A, Sinnegger-Brauns MJ, Tampe R, Striessnig J. Functional properties and modulation of extracellular epitope-tagged Ca(V)2.1 voltage-gated calcium channels. Channels (Austin) Advance online publication. 2008 doi: 10.4161/chan.2.6.6793. Retrieved November 24, 2008. Online ISSN:1933-6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weick JP, Groth RD, Isaksen AL, Mermelstein PG. Interactions with PDZ proteins are required for L-type calcium channels to activate cAMP response element-binding protein-dependent gene expression. J Neurosci. 2003;23:3446–3456. doi: 10.1523/JNEUROSCI.23-08-03446.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Kharazia V, Shanghvi R, Rao A, Beggs AH, Craig AM, Weinberg R, Sheng M. Differential regional expression and ultrastructural localization of α-actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J Neurosci. 1998;18:1383–1392. doi: 10.1523/JNEUROSCI.18-04-01383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K. Plasticity of calcium channels in dendritic spines. Nat Neurosci. 2003;6:948–955. doi: 10.1038/nn1112. [DOI] [PubMed] [Google Scholar]
- Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I. Association of CaV1.3 L-type calcium channels with Shank. J Neurosci. 2005;25:1037–1049. doi: 10.1523/JNEUROSCI.4554-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]