

Abstract
A rare mutation in the RSPH9 gene leading to Primary Ciliary Dyskinesia was previously identified in two Bedouin families, one from Israel and one from the United Arab Emirates (UAE). Herein we analyze mutation segregation in the Israeli family, present the clinical disease spectrum, and estimate mutation age in the two families. Mutation segregation was studied by restriction fragment length analysis. Mutation ages were estimated using a model of the decrease in the length of ancestral haplotypes. The mutations in each of the two families had a common ancestor less than 95 and 17 generations in the past. If the mutations in the two families are descended from a common ancestor, that mutation would have to have arisen at least 150 generations ago. If the Bedouin population has been roughly constant in size for at least 6000 years, it is possible that the mutations in the two families are identical by descent. If there were substantial fluctuations in the size of the Bedouin population, it is more likely that there were two independent mutations. Based on the available data, the population genetic analysis does not strongly favor one conclusion over the other.
Keywords: primary ciliary dyskinesia, Bedouin, founder mutation
INTRODUCTION
Primary ciliary dyskinesia (PCD, MIM 242650) refers to a heterogeneous group of genetic ciliopathies characterized by ultrastructural defects in the axoneme, the microtubule-based core of ‘9+2’ motile cilia, and sperm flagella (Afzelius , 1976; Bush et al., 2007). The incidence is estimated at 1:15,000-1:30,000 (Noone et al., 2004; Bush et al., 2007), with higher incidence in certain consanguineous and isolated populations (Jeganathan et al., 2004; Kennedy et al., 2007; Chilvers et al., 2003). Eight PCD-causing disease genes have been reported and these account for an estimated 17-38% of cases (Zariwala et al., 2006; Hornef et al., 2006; Failly et al., 2008; Failly et al., 2009). With the exception of the KTU gene, these genes all encode structural components of the force-generating axonemal outer dynein arm that is responsible for ciliary beat generation: DNAH5 (Olbrich et al, 2002) and DNAH11 (Bartoloni et al., 2002) (heavy chain dyneins); DNAI1 (Pennarun et al., 1999) and DNAI2 (Loges et al., 2008) (intermediate chain dyneins); TXNDC3 (Duriez et al., 2007) (light chain dynein); RSPH4A and RSPH9 (Castleman et al., 2009) (radial spoke head proteins). KTU encodes a cytoplasmic protein required for preassembly of the dynein arm, prior to its transport into the axoneme (Omran et al., 2008). No work has been published on common ancestors of the mutations identified in these genes.
Different ciliary ultrastructural defects have been described, each of which results in ineffective ciliary function. The most common are lack of inner and /or outer dynein arms, and more rarely ciliary disorientation, ciliary transposition or defective radial spokes (Afzelius et al., 2001; Chilvers et al., 2003). Clinical features include chronic respiratory infections leading to lung damage and sub fertility. About half of patients manifest laterality defects due to a randomization of left-right body axis determination, proposed to result from defective function of embryonic ‘9+0’ ultrastructure nodal cilia (Nonaka et al., 1998; Stannard et al., 2004; Kennedy 2007; Fliegauf et al., 2007). PCD heterozygotes have normal ciliary function and no clinical features of this disease.
A common mutation in the radial spoke head protein-encoding gene RSPH9 was previously identified in two Bedouin families, one from Israel and one from the United Arab Emirates (UAE) (Castleman et al., 2009). Affected individuals in both families were homozygous for the RSPH9 mutation c.801_803delGAA (p.Lys268del) that gives rise to cilia dysmotility associated with central-microtubular-pair abnormalities. We examined here whether the mutation arose in one distant ancestor in the families’ history and subsequently spread into two families, or whether it occurred independently in each of the two families. Herein we focused on the extended 5 generation Israeli Bedouin family, in which we analyzed mutation segregation, and calculated the age of the mutated allele in both families based on haplotypes and haplotype+microsatellite in an attempt to define the source of the mutation.
METHODS
Subjects
Two Bedouin families were studied, one from Israel and one from the UAE. Both were partially described in Castleman et al. (2009) and correlate to UCL152 and UCL146, respectively. The UAE family is detailed in Stannard, et al. (2004); the extended Israeli Bedouin family is detailed here.
Haplotype analysis
Haplotypes were constructed from microsatellite and single nucleotide polymorphism (SNP) genetic marker information using Haplopainter (Thiele et al., 2005). The genotyping data was generated as described previously (Castleman et al., 2009) and presented here. A merged marker map was created using the Illumina Linkage IVb SNP and deCODE Genetics microsatellite maps, and in-house microsatellites were positioned with reference to the University of California Santa Cruz genome browser (NCBI Build 36.1).
Mutation screening by restriction fragment length analysis
In this study, DNA was prepared using the salting out method (Miller & Polesky , 1988) from blood samples obtained from 18 members of the family. Each individual completed an informed consent form.
MboII restriction digestion was used to detect the c.801_803delGAA mutation as described previously (Castleman et al., 2009). Briefly, DNA samples were PCR amplified using primers 5′-CCAGTGGAACCATAGCACCT and 5′-AACAGGCAGGCCAAGTTCAC-3′ and PCR conditions of 5 mins at 94°C, 30 cycles of 1 min each at 94/ 62 /72°C, and a final 10 mins at 72°C, using a prepared 2xReddyMix PCR master mix (1.5mM MgCl2) (Thermo Scientific). PCR products were cleaved by MboII yielding two fragments of 260 and 96 bp for normal alleles and a single fragment of 356 bp for mutant alleles.
Fifty healthy controls of Israeli Arab origin were screened for the mutation. The research protocol was reviewed and approved by the Israeli National Ethics Committee
Calculation of allele age
The time since different copies of an allele are descended from a common ancestor can be estimated from the length of the haplotype shared by all copies. The basic idea is that recombination steadily erodes the ancestral haplotype in each allelic lineage at a known rate. If initially the ancestral haplotype on one side of an allele is of length i (measured in nucleotides), then t generations later the probability that it is of length j
| (1) |
is where c is the recombination rate per nucleotide (Slatkin, 2008). If the rate per nucleotide varies, then the distance is measured in map units instead of nucleotides.
From equation (1), the distribution of lengths of a haplotype shared by two independent lineages can be calculated. If the length of the ancestral haplotype in one lineage is of length j and in the other of length k, then the shared haplotype will be the smaller of j and k. The standard theory of order statistics Kendall and Stuart, 1977) states that the distribution of l=min(j,k) is
| (2) |
The first term represents the probability that both ancestral haplotypes are of length l and the second represents the probability that one is longer.
The data for a given pair of chromosomes is the pair of lengths of shared haplotypes on either side of the mutation, l1 and l2. Because the distributions of those lengths are independent, the probability of observing the data given t is
| (3) |
which is the likelihood of t as a function of the data. We can then estimate the age by finding the value of t that maximizes this likelihood. The curves in Fig. 3 labeled “Haplotype only” are graphs of Equation (3).
Figure 3.

Log-likelihood of the age of the common ancestor within the. UAE family (A) and the Israeli Bedouin family (B). The curves labeled ‘Haplotype only’ were computed from Equations (2) and (3) in the text. The curves labeled ‘Haplotype+microsatellite’ were computed by subtracting −2nμt from the log-likelihood computed for haplotype only, where n=20 for the UAE Bedouin family and n=11 for the Israeli Bedouin family and μ=1.2×10−3. The log-likelihoods were computed for integer values of t only.
Further information about age comes from the fact that none of the microsatellites within the conserved haplotype in each family have mutated. We can account for the observation that no mutations at several microsatellite loci are detected in the data. We can incorporate that information into the likelihood by computing the probability of observing no mutations during the time since the two haplotypes were descended from a common ancestor. The probability of no mutations in two lineages in t generations is exp(−2μnt), where n is the number of loci (20 or 11) and μ is the mutation rate per locus. Weber and Wong (1993) estimated μ to be 1.2×10−3. Multiplying this exponential by the likelihood computed from the length of the shared haplotype gives a new likelihood, shown in Fig. 3 as Haplotype+microsatellite.
RESULTS
Two Bedouin families were previously analyzed, one from Israel and one from the UAE, that link to the RSPH9 locus on chromosome 6p21.1 and carry the RSPH9 c.801_803delGAA mutation (Castleman et al., 2009). The Israeli Bedouin pedigree presented here in detail consisted of 5 generations (Fig. 1; Table 1): 83.3% of affected individuals had neonatal respiratory distress and /or ear disease and/or sinus disease. Respiratory infections in this family were due to Sreptococcus pneumomia (66.6%) and Hemophilus influenzae (50%). Lung functions deteriorated with age, from mildly impaired in infancy and childhood to severely impaired in all 5 adults, who had bronchiectatic changes, particularly in the lung bases, with one of them listed for lung transplant. Infertility or hypo fertility necessitated in vitro fertilization in 100% of affected adults. Patient IV-11 had died due to septic shock complications arising from immunosuppressive therapy for B-Cell lymphoma of the adrenal. Nasal brush biopsies in 2 patients were reported as normal or suboptimal and the ciliary beat frequency was not assessed. Clinical characterization of the UAE Bedouin family was described previously (Stannard et al., 2004).
Figure 1.
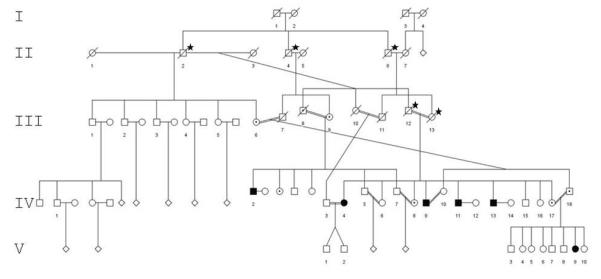
Mutation segregation in the Israeli Bedouin family. Affected: filled circles and squares; dot within circle or square: carrier; asterisk: inferred carrier.
Table 1.
Clinical characteristics of affected individuals in the Israeli Bedouin family.
| Affected individual | IV-2 | IV-4 | IV-8 | IV-11 | IV-13 | V-9 |
|---|---|---|---|---|---|---|
| Age (years) | 45 | 43 | 50 | 42 | 48 | 9 |
| Gender | M | F | M | M | M | F |
| Neonatal respiratory distress |
− | + | + | + | + | + |
| Bilateral bronchiectasis |
+ | + | + | + | + | + |
| FEV1% predicteda | 45 | 51 | 35.9 | NA | 63.8 | 70 |
| Ear disease | + | AOMb | SOMc | + | AOM | − |
| Sinus disease | + | + | + | + | + | − |
| Infertilityd | + | + | + | + | + | − |
| ENOe (ppb) | NA | NA | NA | NA | NA | 7 |
| EMf | NA | Normal | Normal | NA | NA | NA |
| Other | Lung transplantation pending |
Died at 42 Y |
FEV1 predicted - Volume Expired in 1st second, reflecting airflow obstruction degree: mild = 70-80%; moderate = 50-70%; severe <50%.
acute otitis media
serous otitis media
females required IVF; males had abnormal sperm motility/azoospermia
exhaled nitric oxide test - normal >2.4 ppb
electron microscopy for nasal brush biopsy
NA - not assessed
Haplotypes in the two Bedouin families across the RSPH9 locus on chromosome 6p21.1, based on genotyping data from the Castleman et al. (2009), are presented in Fig. 2. This defines a 10.4 Mb region of homozygosity identical-by-descent (IBD) in the UAE Bedouin family between D6S291 and D6S1638, which overlaps with a smaller 1.9 Mb region in the Israeli Bedouin family between D6S400 and rs3734693. Thus, in the two Bedouin families there is only a small shared 1.9 Mb of homozygosity that is IBD. Across this D6S400-rs3736493 critical region, alleles are shared at only two in-house microsatellite markers, UAEPal5 and UAEPal3, located on either side of RSPH9. These are present at 66.5 and 66.6 centiMorgan (cM) from the telomere of the p arm of chromosome 6 (Fig. 1). RFLP performed in the current study showed mutation segregation within the Israeli Bedouin family (shown in part in Fig. 1).
Figure 2.

High density haplotypes across the RSPH9 locus on chromosome 6p21.1. SNP markers, indicated by the prefix ‘rs’, and microsatellite markers are shown against their genetic distance in cM from the p arm telomere of chromosome 6. Boxing indicates the regions of homozygosity across the RSPH9 gene, with flanking markers for the RSPH9 locus defined by loss of homozygosity in affected individuals. The RSPH9 gene is located between markers UAEPal3 and UAEPal5. Arrows indicate the Israeli Bedouin family critical region against that of UAE Bedouin family, which was not genotyped for as many SNP markers. Note that SNP information is inferred in family UCL152 for individuals III:12, V:3-8 and V:10. No microsatellite data is inferred. Three healthy distant members in the Israeli family are not shown.
We used the haplotype information to investigate the history of this mutation in the Bedouin families according to the formulae presented in the Method section. Analyzing each family separately, we assumed that all copies of the mutation are identical by descent within each family and that the source of the mutation in each family was an individual t generations in the past. In the UAE Bedouin family, the shared haplotype extends from 56.86 to 70.14 cM, and there is no heterozygosity of any locus within that range. The mutation is at map position 66.55, and hence l1=70.14–66.55=3.59 and l2=66.55–56.86=9.69, where all distances are measured in cM. We used Equations (2) and (3) to compute the likelihood of t for the UAE Bedouin family (Fig 3A, ‘Haplotype only’ curve). The maximum likelihood estimate (MLE) of t is 7 generations. The support interval, within which the log-likelihood is less than 2 units from the maximum, is (1, 36). Incorporating the information about the lack of mutation in the microsatellite loci in the conserved haplotype (‘Haplotype+microsatellite’), the MLE of t is reduced to 5 generations and the support interval is (1, 17).
The shared haplotype is shorter for the Israeli Bedouin family, extending from 64.36 to 66.90 (l1=66.90–66.55=0.45 and l2=66.55–64.36=2.19). The likelihood curve is shown in Fig. 3B (‘Haplotype only’). The MLE of t based on the shared haplotype alone is i38 generations with a support interval of (6, 119). Within that shared haplotype, 11 microsatellite loci have not mutated. The log-likelihood curve that accounts for these microsatellites is also shown in Fig. 3B (‘Haplotype+microsatellite’). The MLE of t is 30 and the support interval is (5, 95).
The mutation was not found in any of the 100 chromosomes screened from matched Israeli Arabs in this study
DISCUSSION
Bedouin are traditionally pastoral semi-nomadic Arab tribes that are spread out in North Africa and the Middle East. Historically, the Bedouin engaged primarily in nomadic herding, agriculture, raiding, and sometimes fishing. They also generated income by transporting goods and people across the desert. Scarcity of water and permanent pastoral land necessitated their constant movement. The Israeli Bedouin family described here is from a tribe that arrived from Libya at the end of the 19th century and settled near Gadera in the south of Israel. With the establishment of the State of Israel in 1948, they fled to Hebron, and several years later returned to Ramleh, a city in Israel where they were settled in housing provided by the government (Farag & Teebi, 1977; Kressel, 1974). This community is unaware of the other PCD family from the UAE and no historical connections could be gleaned from the known family history.
According to the genetic analysis in this study that demonstrated the mutation segregation among the individuals within the Israeli Bedouin family (Fig. 1), the mutation was introduced 5 generations ago by individual I-1 or I-2, the ancestors of the family, and subsequently spread to their 3 sons in generation II. Inter-familial marriages between first degree cousins in generations III and IV led to a total of 6 affected individuals in generations IV and V, respectively. These patients are the oldest known to carry the RSPH9 defect and display the full spectrum of the disease (Table 1). Their signs relate predominantly to infertility and the pulmonary system, which, when compared with the patients related to the siblings of the UAE family – ages 8 months and 4 and 5 years – showed marked impairment compatible with aggravation of pulmonary functions throughout lifetime in these patients. Bacterial infections were caused by pathogens similar to those reported for other PCD patients (Santamaria et al., 2008), but exhaled nitric oxide (ENO) test was not predictive Horváth et al., 2003).
One of the 5 adults in the Israeli Bedouin family had B cell adrenal lymphoma. Malignancy has not been typically correlated with PCD, but different tumors have been reported in a few cases of Kartagener’s syndrome (Verdejo et al., 2000; Yoshida et al., 1986; Barselo et al., 2008; Nukina et al., 1989).
A larger screen for RSPH9 mutations and homozygosity of the c.801_803delGAA RSPH9 defect may require further study in light of evidence supporting the role of cilia in cell cycle regulation and downstream signaling (Fliegauf et al., 2007; Gerdes et al., 2009). Clinical investigations of nasal brush biopsies could result in under-diagnosis of PCD patients with the central microtubule defect associated with the c.801_803delGAA mutation, as occurred with 2 samples derived from the Israeli Bedouin family (Castleman et al., 2009), and consistent with the UAE Bedouin samples that demonstrated absent central microtubules in only 12.5% to 17% of the cilia on cross-sectional analysis (Stannard et al., 2004). The ultrastructural change involves an intermittent absence of the central microtubular pair, which could remain undetected since only a certain percentage of each epithelial cilia section would show the defect, leading to the detection of the defect in the remainder of the cilia above the plane examined (Stannard et al., 2004). Thus, a nasal biopsy from patients with this specific defect should be interpreted with extra caution in the context of this pitfall.
Although rare diseases are more common in inbred families, we were intrigued that two families of Bedouin origin harbor an identical rare mutation. Given that the incidence of PCD, which is a heterogenic disorder, is estimated at 1;15,000-1;30,000, the gene frequency defect should be 0.008-0.005, and the frequency of the c.801_803delGAA mutation markedly lower. Supporting this is the lack of this mutation in 200 Bedouin and Arab controls’ chromosomes (Castleman et al., 2009), and an additional 100 chromosomes from matched Israeli Arabs analyzed in this study. These findings suggest that the mutation did not occur independently in each family.
If the mutations in the two families are descended from a common ancestor, we can use the same theory employed to estimate the age within each family to estimate the time since that common ancestor. In this case the shared haplotype is much shorter than within each family, only from 66.60 to 66.50. Therefore, l1=l2=0.05, and the likelihood of t for this case is shown in Fig. 4. The MLE is roughly 900 generations with support interval (150, 2900).
Figure 4.

Log-likelihood of the age of the mutant in the two families under the assumption that they are descended from a common ancestor. The results were obtained by evaluating Equations (2) and (3) in the text for t in multiples of 50 generations.
The support intervals for the estimate ages are broad, as is typical of estimates of allele age based on haplotype data. Although some studies claim much narrower confidence intervals for estimates of allele age, such narrow confidence intervals are based on simplifying assumptions that ignore one or more sources of uncertainty. (Slatkin and Rannala 2000) Although we cannot estimate the age of mutations within each family with great precision, we can conclude that the most recent common ancestor carrying the mutation in each family is recent, less than 17 generations ago in the UAE Bedouin family and less than 95 generations in the Israeli Bedouin family, probably much less in both families. Our estimates of age are conservative because the theory used assumes that the mutation is neutral. Any selection against heterozygous carriers would result in lower estimates of age both within and between families (Slatkin 2008). Our estimates are not affected by under-diagnosis because the method used does not assume every case is identified. We assume only that probability that an individual is diagnosed is not dependent on the length of the ancestral haplotype. In other words, we assume that the individuals analyzed represent a random sample of individuals with PCD. Of course, if genotypes of other individuals not included in our study because of under-diagnosis had ancestral haplotypes of different lengths, our analysis would obtain different estimates of allele ages.
We can also conclude that if the mutations in the two families are identical by descent, the original mutation is quite old, at least 150 generations, and probably much older. Whether such a large estimated age for a rare, deleterious mutation is plausible depends on the history of the population. In populations of constant size, low frequency alleles may well be old; the average age in this case is approximately (Kimura & Ohta, 1973) t̄ = −4Nx ln( x) /(1– x)(Kimura & Ohta, 1973); if x=10−3 and N=5000, t̄ =138 generations. But, if the population has grown substantially in the recent past, the average age will be much younger (Slatkin 2002). With a minimum age of 150 generations and 25 years per generation, the population size would have to have been roughly at its current level for about 6000 years. Otherwise, it is less likely that the mutation in the two families is descended from a common ancestor and more likely that there were two independent mutations. Based on the currently available data, the population genetic analysis does not strongly favor one conclusion over the other.
The assessment strategy presented here to resolve allele age and origin may have implications for population genetics, public health considerations, and understanding the dynamics of mutation evolution in other diseases as well.
Acknowledgements
We are grateful to the families for their involvement in this study. We thank Dr. Eddie Chung and Professor Mark Gardiner for helpful discussions. We thank Dr. Aharon Zohar for helpful information on the Bedouin population and their lifestyle. M.S. was supported in part by a grant from the National Institutes of Health (NIH), R01-GM40282. The genotyping work was supported in part by the Medical Research Council (UK), the Wellcome Trust (UK), the Milena Carvajal–Prokartagener Foundation (Switzerland), and the PCD Family Support Group (UK).
Abbreviations
- IBD
identical-by-descent
- MLE
maximum likelihood estimate
- PCD
primary ciliary dyskinesia
- PCR
polymerase chain reaction
- SNP
single nucleotide polymorphism
- UAE
United Arab Emirates
References
- Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- Bartoloni L, Blouin JL, Pan Y, Gehrig C, Maiti AK, Scamuffa N, Rossier C, Jorissen M, Armengot M, Meeks M, Mitchison HM, Chung EM, Delozier-Blanchet CD, Craigen WJ, Antonarakis SE. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci USA. 2002;99:10282–10286. doi: 10.1073/pnas.152337699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush A, Chodhari R, Collins N, Copeland F, Hall P, Harcourt J, Hariri M, Hogg C, Lucas J, Mitchison HM, O’Callaghan C, Phillips G. Primary ciliary dyskinesia: current state of the art. Arch Dis Child. 2007;92:1136–1140. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castleman VH, Romio L, Chodhari R, Hirst RA, de Castro SC, Parker KA, Ybot-Gonzalez P, Eme,s RD, Wilson SW, Wallis C, Johnson CA, Herrera RJ, Rutman A, Dixon M, Shoemark A, Bush A, Hogg C, Gardiner RM, Reish O, Greene ND, O’Callaghan C, Purton S, Chung EM, Mitchison HM. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am J Hum Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilvers MA, Rutman A, O’Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J Allergy Clin Immunol. 2003;112:518–524. doi: 10.1016/S0091-6749(03)01799-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duriez B, Duquesnoy P, Escudier E, Bridoux AM, Escalier D, Rayet I, Marcos E, Vojtek AM, Bercher JF, Amselem S. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc Natl Acad Sci USA. 2007;104:3336–3341. doi: 10.1073/pnas.0611405104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failly M, Bartoloni L, Letourneau A, Munoz A, Falconnet E, Rossier C, de Santi MM, Santamaria F, Sacco O, DeLozier-Blanchet CD, Lazor R, Blouin JL. Mutations in DNAH5 account for only 15% of a non-preselected cohort of patients with primary ciliary dyskinesia. J Med Genet. 2009;46:281–286. doi: 10.1136/jmg.2008.061176. [DOI] [PubMed] [Google Scholar]
- Failly M, Saitta A, Munoz A, Falconnet E, Rossier C, Santamaria F, de Santi MM, Lazo,r R, Delozier-Blanchet CD, Bartoloni L, Blouin JL. DNAI1 mutations explain only 2% of primary ciliary dykinesia. Respiration. 2008;76:198–204. doi: 10.1159/000128567. [DOI] [PubMed] [Google Scholar]
- Farag TI, Teebi AS. Genetic disorders among Bedouins. In: Teebi AS, Farag TI TI, editors. Genetic disorders among Arab populations. Oxford University Press; New York: 1997. pp. 375–410. [Google Scholar]
- Fliegauf M, Benzing T, Omran H. Mechanism disease. When cilia go bad: cilia defects and ciliopathy. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornef N, Olbrich H, Horvath J, Zariwala MA, Fliegauf M, Loges NT, Wildhaber J, Noone PG, Kennedy M, Antonarakis SE, Blouin JL, Bartoloni L, Nusslein T, Ahrens P, Griese M, Kuhl H, Sudbrak R, Knowles MR, Reinhardt R, Omran H. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am J Respir Crit Care Med. 2006;174:120–126. doi: 10.1164/rccm.200601-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth I, Loukides S, Wodehouse T, Csiszér E, Cole PJ, Kharitonov SA, Barnes PJ. Comparison of exhaled and nasal nitric oxide and exhaled carbon monoxide levels in bronchiectatic patients with and without primary ciliary dyskinesia. Thorax. 2003;58:68–72. doi: 10.1136/thorax.58.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan D, Chodhari R, Meeks M, Faeroe O, Smyth D, Nielsen K, Amirav I, Luder AS, Bisgaard H, Gardiner RM, Chung EM, Mitchison HM. Loci for primary ciliary dyskinesia map to chromosome 16p12.1-12.2 and 15q13.1-15.1 in Faroe Islands and Israeli Druze genetic isolates. J Med Genet. 2004;41:233–240. doi: 10.1136/jmg.2003.014084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdejo A. Jiménez, Torres J.L. Martínez, Yago F. Palao, Ranera J. Tinaut, Martín M. Arrabal, Ortiz J.L Miján, Gómez A. Zuluaga. Renal cell carcinoma in patient with situs inversus and kartagener syndrome (Spanish) Actas Urol Espa. 2000;24:169–172. doi: 10.1016/s0210-4806(00)72424-7. [DOI] [PubMed] [Google Scholar]
- Kendall M, Stuart A. The advanced theory of statistics. 4th Vol. 1. MacMillan; New York: 1977. [Google Scholar]
- Kennedy MP, Omran H, Leigh MW, Dell S, Morgan L, Molina PL, Robinson BV, Minnix SL, Olbrich H, Severin T, Ahrens P, Lange L, Morillas HN, Noone PG, Zariwala MA, Knowles MR. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- Kimura M, Ohta T. The age of a neutral mutant persisting in a finite population. Genetics. 1973;75:99–212. doi: 10.1093/genetics/75.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kressel GM. Individuality against tribality. Hakkibutz Hameuchad. Hebrew; Tel Aviv, Israel: 1974. Chapter 1; pp. 24–100. 1974. [Google Scholar]
- Loges NT, Olbric H, Fenske L, Mussaffi H, Horvath J, Fliegauf M, Kuhl H, Baktai G, Peterffy E, Chodhari R, Chung EMK, Rutman A, O’Callaghan C, Blau H, Tiszlavicz L, Voelkel K, Witt M, Ziętkiewicz E, Neesen J, Reinhardt R, Mitchison HM, Omran H. DNAI2 mutations cause primary ciliary dyskinesia with outer dynein arm defects. Am J Hum Genet. 2008;83:547–58. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;18:613–617. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, Zariwala MA, Knowles MR. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med. 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- Nukina S, Fusaoka T, Fukumochi Y, Hihara T, Ikushima S, Fujiwara F, Morioka Y, Todo S, Imashuku S. Malignant lymphoma of the central nervous system in a boy with immotile cilia syndrome (Japanese) Rinsho Ketsueki. 1989;30:568–572. [PubMed] [Google Scholar]
- Olbrich H, Haffner K, Kispert A, Volkel A, Volz A, Sasmaz G, Reinhardt R, Hennig S, Lehrach H, Konietzko N, Zariwala M, Noone PG, Knowles M, Mitchison HM, Meeks M, Chung EM, Hildebrandt F, Sudbrak R, Omran H. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- Omran H, Kobayashi D, Olbrich H, Tsukahara T, Loges NT, Hagiwara H, Zhang Q, Leblond G, O’Toole E, Hara C, Mizuno H, Kawano H, Fliegauf M, Yagi T, Koshida S, Miyawaki A, Zentgraf H, Seithe H, Reinhardt R, Watanabe Y, Kamiya R, Mitchel,l DR, Takeda H. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennarun G, Escudier E, Chapelin C, Bridoux AM, Cacheux V, Roger G, Clement A, Goossens M, Amselem S, Duriez B. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am J Hum Genet. 1999;65:1508–1519. doi: 10.1086/302683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barselo E. Ramos, Martín J.A. Portillo, Gómez M.A. Correas, del Valle Schaan JI, Baños J.I. Gutiérrez, Peña A. Villanueva, Edreira A. Roca, Tubet C. Aguilera, Diego R. Ballestero, Guerrero S. Zubillaga, Sañudo J.A. Campos. Testicular seminoma in a patient with kartagener’s syndrome. Arch Esp Urol. 2008;61:431–434. doi: 10.4321/s0004-06142008000300011. [DOI] [PubMed] [Google Scholar]
- Santamaria F, Montella S, Tiddens HA, Guidi G, Casotti V, Maglione M, de Jong PA. Structural and functional lung disease in primary ciliary dyskinesia. Chest. 2008;134:351–357. doi: 10.1378/chest.07-2812. [DOI] [PubMed] [Google Scholar]
- Slatkin M. The age of alleles. In: Slatkin M, Michel VM, editors. Modern developments in theoretical population genetics. Oxford University Press; Oxford: 2002. pp. 233–259. [Google Scholar]
- Slatkin M. Bayesian method for jointly estimating allele age and selection intensity. Genetics Research. 2008;90:129–137. doi: 10.1017/S0016672307008944. [DOI] [PubMed] [Google Scholar]
- Slatkin M, Rannala B. Estimating allele age. Ann Rev Genomics and Human Genetics. 2000;1:225–249. doi: 10.1146/annurev.genom.1.1.225. [DOI] [PubMed] [Google Scholar]
- Stannard W, Rutman A, Wallis C, O’Callaghan C. Central microtubular agenesis causing primary ciliary dyskinesia. Am J Respir Crit Care Med. 2004;169:634–637. doi: 10.1164/rccm.200306-782OC. [DOI] [PubMed] [Google Scholar]
- Thiele H, Nürnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- Weber JL, Wong C. Mutation of human short tandem repeats. Human Molecular Genetics. 1993;2:1123–1128. doi: 10.1093/hmg/2.8.1123. [DOI] [PubMed] [Google Scholar]
- Yoshida J, Tsuneyoshi M, Nakamura K, Murakami T, Akamine Y. Primary ciliary dyskinesia with transverse colon carcinoma. Am J Clin Pathol. 1986;85:101–104. doi: 10.1093/ajcp/85.1.101. [DOI] [PubMed] [Google Scholar]
- Zariwala MA, Leigh MW, Ceppa F, Kennedy MP, Noon,e PG, Carson JL, Hazucha MJ, Lori A, Horvath J, Olbrich H, Loges NT, Bridoux AM, Pennarun G, Duriez B, Escudier E, Mitchison HM, Chodhari R, Chung EM, Morgan LC, de Iongh RU, Rutland J, Prada,l U, Omran H, Amselem S, Knowles MR. Mutations of DNAI1 in primary ciliary dyskinesia: evidence of founder effect in a common mutation. Am J Respir Crit Care Med. 2006;174:858–866. doi: 10.1164/rccm.200603-370OC. [DOI] [PMC free article] [PubMed] [Google Scholar]