

Abstract
Thrombospondin-1 is the first and most studied naturally occurring protein inhibitor of angiogenesis. Its characteristic multi-domain structure determines thrombospondin-1 divergent functions, which include but are not limited to the regulation of angiogenesis. Below we overview the structural determinants and receptors expressed on the endothelial and other cell types, that are at the root of thrombospondin-1 striking ability to block neovascularization. We specifically emphasize thrombospondin-1 direct apoptotic action on the remodeling vascular endothelium and summarize current knowledge of its pro-apoptotic signaling and transcriptional networks. Further, we provide comprehensive survey of the thrombospondin-based anti-angiogenic strategies with special focus on the combination treatments. We convincingly illustrate how precise knowledge of the pro-apoptotic events and intermediates elicited by thrombospondin in the vascular endothelial cells facilitates the design of the most effective treatment combinations, where the efficacy of thrombospondin-derived compounds is maximized by the partner drug(s) (“complementation” strategies) and provide examples of such fine-tuning of the thrombospondin-based anti-angiogenic treatments.
Keywords: Thrombospondin, angiogenesis, apoptosis, complementation treatment
TSP1 structure and function
The members of the large family of matricellular proteins including thrombospondin-1 (TSP1) play important roles in genesis and remodeling of multiple tissues including cartilage and vasculature. TSP1 is one of the important pivots that regulate vascular tissue homeostasis whereas its key function is the negative control of angiogenesis. TSP1 was the first naturally occurring protein inhibitor of angiogenesis [1] to be identified; its anti-angiogenic effects have since been established in a multitude of experimental models and linked to specific epitopes in the multi-domain, multi-functional TSP1 molecule. TSP1 is the first identified, and therefore best studied thrombospondin family representative, its structure is thus considered as prototype for the other family members. In the thrombospondin family, another member, TSP2 has a similar domain structure and, non-surprisingly, its functions significantly overlap with those of TSP1. Specifically, both TSP1 and TSP2 potently inhibit angiogenesis [2].
In adult organisms, TSP1 expression is limited to the sites of tissue remodeling where it resides in the extracellular space and determines cell phenotype and extracellular matrix structure and composition [3]. Although not all TSP1 domains have been ascribed cognate cell surface receptors, the assumption that each domain has a corresponding receptor(s) is not far from the truth: the array of TSP1 receptors expressed by a given cell determines the repertoire for cellular responses, which are extremely diverse. TSP1 promotes the migration of vascular smooth muscle cells but suppresses chemotaxis and motility of the endothelial cells with equal potency. TSP1 stimulates matrix assembly by binding other matrix proteins, such as fibronectin and collagen and regulates matrix digestion by matrix metalloproteinases and plasmin. Finally, TSP1 stimulates apoptosis of the endothelial cells and T cells but promotes survival of the vascular smooth muscle cells. This diversity of function marks TSP1 and its close homolog TSP2 as critical players in physiological angiogenesis, development, wound healing, synaptogenesis, and the corresponding tissue remodeling but also in pathological processes including atherosclerosis, neoplasia and tumor angiogenesis.
The current state of the structure-function analysis of TSP1 and 2, which is pertinent to angiogenesis is summarized in Figure 1a. TSP1 functional epitopes probably originate from exon shuffling in the course of evolution. The N-terminal (N-ter) domain contains high-affinity binding site for heparin and heparan sulfate proteoglycans; N-ter, but not the heparin-binding site also mediates VEGF uptake and clearance via low-density lipoprotein receptor-related protein (LRP) [4]. The core of the TSP1 contains three thrombospondin type 1 repeats (TSRs), also called malarial (Mal) or properdin repeats and three epidermal growth factor or thrombospondin type 2 repeats. TSRs are found in more than a hundred human proteins [5] and represent the most studied TSP1 anti-angiogenic epitope. TSRs and their peptide mimetics are successfully used to block angiogenesis and tumor growth in preclinical models [6] and the mimetics have been evaluated in early stage clinical trials [7]. TSP1 is the only member of the family that has the ability to activate TGF-β: this function is mapped to the critical RFK sequence between TSR1 and TSR2 [8, 9] (Figure 1). This ability indirectly impinges on angiogenesis, since TGF-β is the major modifier of the vascular smooth muscle cells (VSMC) proliferation and motility (recruitment to the vasculature), the two factors critical for the stability of the microvasculature [10]. TSP1 C-terminal portion (from the last type 2 repeat to the C-terminus, C-ter) contains over 30 calcium-binding sites wrapped around a sandwich structure formed by the last 200 amino acids of the TSP1 [3]. This is the most conserved of all TSP domains, the signature domain for the entire family. The presence of the calcium-binding domain suggests TSP1 functions in the calcium homeostasis, a role that only begins to unravel: recent study demonstrates the loss of another calcium-binding protein, calbindin, and compensatory repression of the transient potential receptor channel 4, lowers calcium intake in the renal carcinoma cells. The resulting decrease of intracellular calcium, in turn, causes TSP1 misfolding and impaired secretion due to the retrograde transport of misfolded protein from the Golgi complex to the endoplasmic reticulum [11]. Another study ascribes the Type 3 TSP1 repeats the ability to block angiogenesis by quenching bFGF [12].
Figure 1.

TSP1 domain structure. Fragments relevant for anti-angiogenesis are indicated. Modified from review by Zhang and Lawler, 2007 [99].
TSP1 anti-angiogenic activity is not limited to the TSRs: the procollagen homology domain proximal to the TSRs also interferes with angiogenesis, however its anti-angiogenic function is little explored [13]. Another peptide at the C-ter region of TSP1, 4NIK may either inhibit or induce angiogenesis, depending on the cell and tissue context [14, 15].
TSP1 angioinhibitory epitopes
TSP1 effects on angiogenesis can be classified as direct and indirect. The direct ones target and modify the behavior of endothelial cells proper, while the indirect effects are mediated by the endothelial cell environment, where the changes occur to the complex milieu of growth factors and cytokines, angiogenesis inhibitors, proteases and adhesion factors produced by other cell types. Indirect effects of TSP1 include the recruitment of the vascular smooth muscle cells thereby stabilizing the remodeling capillaries and precluding further angiogenesis [16]; the binding and sequestration of the pro-angiogenic growth factors and cytokines secreted by non-endothelial cells, such as bFGF and VEGF [17, 18] thus lowering endothelial cell survival, migration and vascular sprouting. In addition, TSP1 promotes VEGF internalization by the non-endothelial cells via low density lipoprotein receptor-related protein (LRP) and partially suppresses VEGF expression by the same cells [4]. Finally, TSP1 pro-apoptotic effect on activated macrophages reduces the levels of inflammatory cytokines, which are important players in the pathological angiogenesis [19, 20]. TSP1 direct effects on the endothelial cells encompass the repression of motility and chemotaxis [21], cell cycle arrest [22] and apoptosis [23, 24].
The domain(s) responsible for the sequestration of the growth factors reside at the N-terminus of the TSP1 molecule, in the heparin-binding domain [17]; TGF-β activating sequence is positioned at the proximal end of the TSRs [5] and the peptide sequences required for the killing of T cells, macrophages and dendritic cells has been mapped to the C-terminus [20].
TSP1 direct anti-angiogenic activities map largely to TSRs (Figure 1a). The anti-angiogenic activity has been ascribed to the NGVQYRN sequence in the procollagen homology domain [13] and to the KRFKQDGGWSHWSPWSSC in TSR2 [25]. Unexpectedly, although the KRFKQDGGWSHWSPWSSC sequence encompasses TGF-β activating motif RFK, its ability to suppress proliferation of the endothelial and tumor cells is independent of the TGF-β activation. The anti-angiogenic fragment from the procollagen domain inhibits endothelial cell chemotaxis in vitro and angiogenesis in vivo; however, its function has not been further explored. Three independent studies by Tolsma, Iruela-Arispe and Dawson using synthetic peptides identified the sequence SPWSSCSVTCGDGVITRIR, or MalII peptide within TSR2 [13, 25, 26] and a similar sequence in the TSR3 (SPWDIASVTAGGVQKRSK). Two L- to D-isoleucine substitution further stabilized SPWSSCSVTCGDGVITRIR peptide and increased its specific activity [26]. Minimal version of the D-Ile MalII peptide yielding DI-TSP, a heptapeptide containing D-isoleucyl substitution for the first L-isoleucine29 and the internal Arg residue replaced by Nva showed activity at low nanomolar concentrations [26] (Figure 2). Further optimized TSP1 peptide mimetics ABT-510 and ABT-526 (Figure 2) have improved stability and pharmacokinetics [27] and have been showing promise in early-phase clinical trials [7, 28].
Figure 2.

The evolution of CD36-binding peptides.
TSP1 induces endothelial cell apoptosis to block angiogenesis
TSP1 was the first anti-angiogenic agent, for which apoptosis was shown to be the main way of exerting anti-angiogenic activity [23]. Later this observation was generalized for the majority of natural angiogenesis inhibitors including angiostatin, endostatin, tumstatin, SPARC and many others [29]. The apoptosis is induced predominantly in the endothelial cells by intact thrombospondin and by the KRFKQDGGWSHWSPWSSC peptide (see above), while the peptide from procollagen domain, NGVQYRNC lacked the pro-apoptotic capacity [24]. The apoptotic effect of KRFKQDGGWSHWSPWSSC peptide is independent of TGF-β activation but requires the WSXW motif. Interestingly, the pro-apoptotic action of TSP1 and its peptides is independent of their anti-proliferative activity. Later studies have demonstrated that TSP1 and 2 anti-proliferative activities are due to the inhibition of G1/S phase transition and are mediated by p21 accumulation and Rb dephosphorylation, and are presumably triggered at CD36 cell surface receptor [22]. Alternatively, TSP can induce CD36-independent growth arrest in HUVEC via p53-dependent p21 induction [30]. Similar pathway have been shown for another TSR containing protein, brain-specific angiogenesis inhibitor I (BAI1), where p53 induction is mediated via αvβ5 integrin [31]. Another study directly linked apoptosis and anti-angiogenesis by demonstrating that pan-caspase inhibitor, z-VAD relieved angiogenesis blockade by TSP1 in vivo, in the mouse cornea model [23].
CD36 is the endothelial anti-angiogenic and pro-apoptotic receptor for TSP1
In the endothelial cells, TSP1 induces caspase-dependent apoptosis [23]. Several studies identified CD36 as essential receptor for the TSP1 and 2 anti-angiogenic and pro-apoptotic activity [23, 32]. CD36 is an 88-kDa cell-surface transmembrane glycoprotein of the scavenger receptor B family, which is key for multiple cell functions, including platelet aggregation, anti-angiogenesis, uptake of the long chain fatty acids and oxidized LDL (oxLDL), cell adhesion and phagocytosis [33]. TSP1 binding to CD36 is inhibited by the CSVTCG peptide [34, 35] (Figure 1). Large vessel endothelial cells lacking CD36 fail to respond to TSP1 in the in vitro assays for angiogenesis, unless transfected with exogenous CD36 [32]. CD36 neutralizing antibodies or CD36 soluble fragments, which disrupt CD36-TSP binding, also hamper TSP1 inhibition of the endothelial cell migration, morphogenesis and apoptosis [23, 32]. Moreover, TSP in vivo anti-angiogenic activity critically depends on CD36: neither TSP-1 nor TSP-2 effectively block bFGF or VEGF induced neovascularization in CD36 null mice in the matrigel or corneal assays [23, 36]. Moreover, CD36 null mice mount a more vigorous response to the angiogenic growth factors than their wild type counterparts [36], suggesting the importance of CD36-TSP axis in the maintenance of angiostasis in adult organisms. The important structural features of CD36 are shown in Figure 3a. The cationic face of the TSRs interacts with the CD36 amino acids 93–100, a charged domain conserved in all CD36 family members and between species, termed CLESH (CD36, LIMP2, EMP, SRB-1) [37]. Interestingly, LDL, which binds another CD36 epitope upstream of CLESH (aa139-155) can also block angiogenesis and causes endothelial cell apoptosis via mechanism similar to that of TSP1 [38]. TSP1 binding to CD36 and anti-angiogenic activity is regulated on two levels: by competitive binding between TSP1 and Histidine-Rich Glycoprotein (HGRP), which also contains a CLESH domain and thus acts as a soluble decoy receptor for TSR-containing proteins [36] (Figure 3b), and possibly by CD36 phosphorylation at the consensus site between aa87-99 by the protein kinase C, which also suppresses TSP1 binding [39]. Thus the CD36-TSP-HGRP (Figure 3c) is an important module that determines angiostasis vs. angiogenesis.
Figure 3. CD36-TSP-HRGP axis: structure and function.
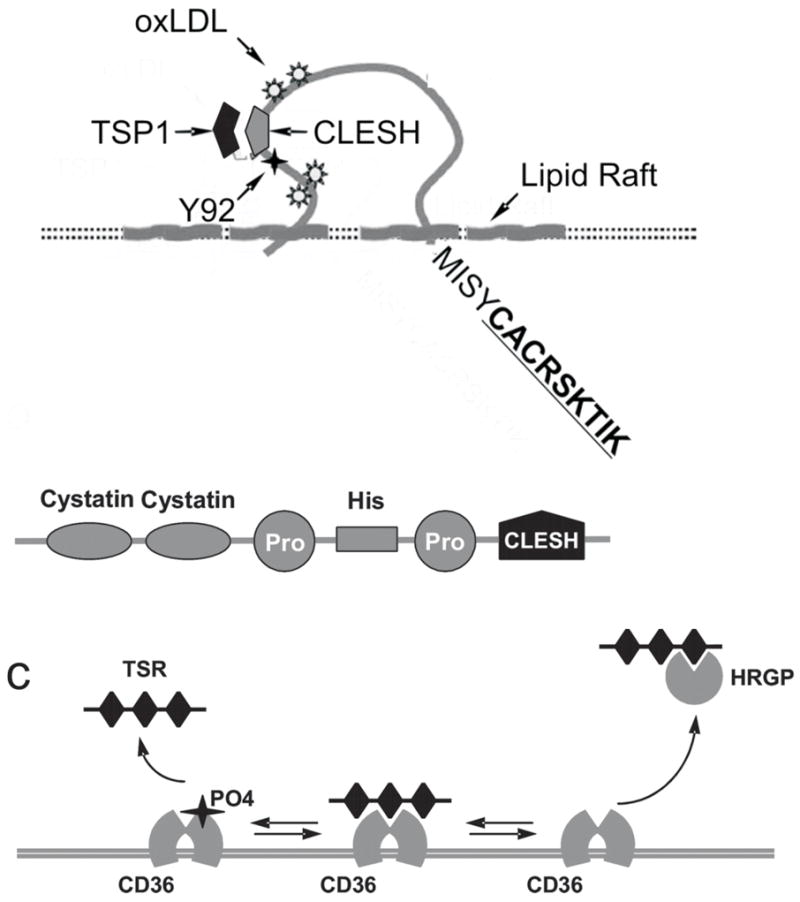
(a) CD36 features important for ant-angiogenesis: CLESH domain, PKC phosphorylation site (Y92), intracellular signaling sequence (CACRSKTIK).
TSP1 apoptotic pathways
Multiple groups have demonstrated that TSP-induced apoptosis of the microvascular endothelial cells is caspase-dependent and indicated caspases 3 and 8 ([23, 40] and Roya Khrosavi-Far, personal communication). This process is mediated by CD36, which is clustered in cholesterol-rich lipid rafts in association with the src family kinases (SFK), Fyn, Lyn and Yes [41]. TSR binding to CD36 increases CD36-Fyn association, and leads to Fyn activation, which is critical for TSP-dependent apoptosis and anti-angiogenesis: microinjections of Fyn neutralizing antibodies abolish subsequent signaling events leading to apoptosis; moreover, mice null for Fyn mount robust response to bFGF in the corneal assay despite the presence of TSP1 in slow-release implants [23]. TSP1 interaction with CD36 results in the activation of p38 kinase and c-Jun N-terminal kinase (JNK). JNK induction is critical for the TSP-1 dependent apoptosis and angiogenesis since TSP1 anti-angiogenic activity is severely compromised in JNK-1 and to some degree in JNK-2 null mice [42]. On the other hand, p38 inhibitor SB203580 blocks TSP1-induced apoptosis in vitro and restores corneal angiogenesis in vivo [23].
TSP1 causes apoptosis selectively in the remodeling endothelium in vivo (e.g. tumor vasculature or growth factor-induced neovessels), which is best represented in vitro by the endothelial cells treated with angiogenic stimuli, including VEGF, bFGF and IL-8 [43]. This selectivity is attributed to the induction of the intrinsic apoptotic cascade triggered by the interaction between the CD95 (Fas) receptor and its cognate ligand, CD95L (FasL). CD95 is extremely low on the surface of resting endothelial cells and in quiescent microvessels: it becomes prominent in the presence of angiogenic stimuli, both in vitro and in vivo due to redistribution of the intracellular pool of CD95 from the Golgi complex to the cell membrane. In contrast, TSP1 has no effect on CD95 expression or presentation, but strongly increases p38-dependent expression of CD95L mRNA and protein. The CD95L induction initiates the secondary receptor-ligand interaction, which triggers apoptotic events including the activation of initiator caspase-8 and executioner caspase-3. These in vitro findings are supported by the loss of TSP1 inhibitory activity in mice deficient for CD95 or CD95L and by the increased vascularization in the tissues of mice deficient for both CD95 and its ligand [43].
At the transcription level, pro-angiogenic signaling events by both VEGF and bFGF include the induction of the Nuclear Factor of the Activated T-cells (NFATc2) transcription factor, which contributes to angiogenesis by driving the expression of COX-2 and the endogenous caspase-8 inhibitor, c-FLIP [44, 45]. NFATc2 is activated and undergoes nuclear import upon dephosphorylation by its associated phosphatase, calcineurin [46]. TSP-1 activation of JNK-1 and 2 leads to re-phosphorylation and subsequent nuclear export and cytoplasmic retention of NFATc2. Interestingly, NFAT phosphorylation, TSP1-dependent apoptosis and anti-angiogenic activity in the mouse corneal assay were significantly inhibited by the specific JNK inhibitor, SP-600125 ([44] and Volpert and Zaichuk, unpublished observations). These findings are summarized in Figure 4.
Figure 4. Summary of the TSP1 signaling pathways causing apoptosis and anti-angiogenesis.

Angiogenic stimuli (right) activate survival kinases and pro-angiogenic transcription factor NFAT leading to the transcriptional activation of cFLIP and Bcl-2 and concomitantly increases surface CD95. Anti-angiogenic TSP1 activates Fyn, leading to sustained actitivation of p38. It concomitantly activates JNK1-2 and NFκB. Active p38 and NFκB (p65/p50) cause transcriptional activation of CD95L, its interaction with CD95, activation of caspases 8 and 3 and apoptosis. JNK1 and 2 directly phosphorylate NFAT and facilitate its displacement from cFLIP promoter by NFκB (p50/p50 dimers), causing transcriptional repression and increased susceptibility to apoptotic signals. Cumulative result of these events is endothelial cell apoptotsis and angiogenesis blockade.
Recent studies reveal that the regulation of angiogenesis and apoptosis by TSP1 involves another transcription factor, nuclear factor kappa beta (NFκB). Previous studies have identified the induction of NFκB by VEGF as necessary part of angiogenesis [47]. Surprisingly, TSP1 further enhanced NFκB activation. A specific inhibitor of NFκB, which blocks upstream IKKα and IKKβ kinases, abolished apoptosis and reversed anti-angiogenesis by TSP1 in vitro and in vivo, in matrigel plug assay, suggesting critical role of NFκB in TSP1 anti-angiogenic, pro-apoptotic function i. Timed measurements of NFκB activity in VEGF-stimulated endothelial cells and in the absence or in the presence of TSP1 reveal major distinctions in the activation kinetics: while VEGF causes short and transient NFκB activation, which repeats with 3–4 hour intervals, the effect of TSP1 was sustained for at least 6 hours and persisted for 12 hours (Figure 5) i. In the case of JNK and p38 kinases, short and transient activation leads to survival, while long and sustained activation causes apoptosis [48]: it is not unlikely that similar differences in NFκB activation kinetics also produce opposing biological effects. NFκB plays a pivotal role in the regulation of both survival and apoptotic molecules: chromatin immunoprecipitation studies have shown that TSP1-driven NFκB acts as a transcriptional activator for CD95L but represses c-FLIP transcription. These contrasting activities depend on the composition of NFκB complex, promoter-specific context and chromatin reorganization. At the CD95L promoter, NFκB p65/p50 dimers recruit p300 histone acetylase (HAT) and subsequently decrease the DNA association with histones H3 and H4. In contrast, at the c-FLIP promoter, NFκB is present in the form of p50/p50 transcriptionally inactive dimers, which recruit histone deacetylase HDAC1 causing deacetylation and increased DNA binding of the H3 histone and restricting the presence of p300. Moreover, NFκB appears to be responsible for the displacement of NFATc2 from c-FLIP promoter: the dissociation of NFAT from the cFLIP promoter is abolished by NFκB inhibitor, BMS-345541, suggesting that NFAT and NFκB compete for similar or adjacent binding sites, an event that has been speculated upon but never actually demonstrated [49]. The effects of TSP1 on NFκB/NFAT driven transcription are summarized in Figure 6 i.
Figure 5.

The comparison of the NFκB activation kinetics by VEGF and VEGF+TSP1.
Figure 6.

Detailed events at the CD5L and cFLIP promoters due to NFkB induction by TSP1.
Polverini and co-workers noted the decreased Bcl-2 expression in the endothelial cells exposed to TSP1 [50]. Although the mechanism has not been explored, it is feasible that Bcl-2 is also downregulated via NFAT-dependent mechanism. In fact, Endothelin-1 drives Bcl-2 expression via NFAT and therefore promotes endothelial cell survival [51]. The decrease of the two important survival factors, like cFLIP and Bcl-2 make the endothelial cells highly susceptible to the TSP1 apoptotic molecular events.
TSP1-induced killing of non-endothelial cells
Interestingly, TSP1 can induce cell death in non-endothelial cells. In particular, TSP1 type 3 repeats and a minimal active peptide for the C-terminal region, 4N1K cause the ligation of the CD47 receptor/integrin-associated protein (IAP), which results in the death of T-cells, macrophages and dendritic cells [20, 52]. This is thought to limit the number of mature T cells and macrophages, a process necessary for healthy immunostasis [53]. The cell death caused by CD47 ligation is resistant to the exogenous rescuing cytokines, such as interleukin-4 or interferon-γ. Importantly, it occurs independent of caspase activation and is not associated with DNA fragmentation, although phosphatidylserine exposure is symptomatic of apoptosis [54]. The CD47-dependent, caspase-independent T cell apoptosis is attributed to ROS production, which occurs independently of the release of cytochrome C and apoptosis inhibitory factor (AIF) by the mitochondria [55]. ROS accumulation, in turn, drives the increased production of the dynamin-related protein Drp-1, which causes dramatic and rapid actin reorganization and subsequent membrane rupture [56]. In fibroblasts subjected to mechanical stress, increased TSP1 production and subsequent CD47 ligation also cause cell death [57]. In contrast, in the vascular smooth muscle cells and endothelial cells CD47/IAP ligation stimulates focal adhesion kinase and cyclic GMP production and results in the survival and increased angiogenesis [24]. Thus it appears that TSP1 regulates cell survival in multiple ways and across multiple tissues and cell types. The physiological meaning of TSP1 killing of monocytes is yet unclear, as well as the meaning of the TSP1 and CD36-dependent suppression of the megacaryocytopoiesis [58]. However, this is a feasible mechanism of TSP1-dependent suppression of the circulating endothelial precursor cells, which, are critical components of tumor angiogenesis and metastases [59, 60]. Finally, studies with synthetic peptides indicated two heparin-binding peptides, Hep-I at the NH2-terminus and GGWSHW within the TSRs potently induced differentiation and apoptosis of HL-60 and NB4 leukemia cells, suggesting that cell surface heparan sulfates may be involved in the TSP1 apoptotic effect on promyelocytic cells [61].
TSP1 based therapies and combinations
There are several excellent reviews on TSP-based cancer therapies including those by the groups of Lawler, Mosher and Khrosavi-Far [3, 6, 21, 62], which give detailed and comprehensive overview of existing preclinical models using TSP1 and TSP2. Here we will briefly recapitulate this information, with specific emphasis on the TSP1 pro-apoptotic properties and discuss the combinatorial strategies whose design was based on the knowledge gained from the studies of TSP1 signaling pathways.
a. Rationale for the use of TSP1
TSP1 is frequently decreased in angiogenesis-dependent diseases, especially in cancer, an event that is followed by the shift of angiogenic equilibrium from angiosuppression to angiogenesis (angiogenic switch) in the environment permissive for angiogenesis and therefore free nutrient delivery/waste removal critical to the unlimited tumor expansion. The decrease of TSP1 expression in the tumors is typically driven by the main genetic relays of tumor progression, by oncogenes and tumor suppressors [63]. As is the case with many angiogenesis inhibitors, TSP1 expression is sustained by the tumor suppressor genes (APC, p53, p73, PTEN, SMAD4 [64–68]) and repressed by the oncogenes (Ras, Myc, Id-1, src, c-Jun, HER2 and others [69–76]). TSP1 can also be inhibited through the promoter hypermethylation as it happens in lymphoma and neuroblastoma [77, 78] and by hypoxia/anoxia, which are common in the exponentially growing tumors [79]. Increased, expression of HRGP, a soluble counterpart of CD36, can sequester TSP1 from the tumor microenvironment and also flip the angiogenic switch [37]. Finally, TSP secretion may be hampered by aberrant folding due to the deficit of intracellular calcium [11]. In the light of early discoveries by Folkman and colleagues [80], restoring the TSP axis and thus flipping back the angiogenic switch appears an attractive alternative to the highly toxic conventional treatments or a useful adjuvant approach that may allow lowering the doses of toxic compounds, reduce the emergence of resistant tumor and increase the duration of treatment. TSP1-based strategies are especially attractive because they target exclusively remodeling endothelium and are harmless for the quiescent vasculature.
b. Means of restoring TSP1 expression
The team of Robert Kerbel and other groups found that TSP1 levels are restored by the continuous and frequent administration of low-dose chemotherapy (metronomic dosing), particularly cyclophosphamide in both endothelial and tumor cells leading to decreased tumor growth and apoptosis in the non-endothelial and endothelial compartments [81, 82]. However, this approach did not receive clinical verification: in children with recurring refractory solid tumors metronomic therapy with cyclophosphamide or vinblastin produced highly variable levels of circulating TSP1, which did not correlate with the disease progression [83]. However, in the combination therapy with bevacizumab and metronomic cyclophosphamide, which proved effective in blocking tumor growth and angiogenesis, neither VEGF nor TSP1 levels in patients’ serum reflected the outcome [84] suggesting that the circulating levels of the target protein are not reflective of the treatment efficacy. The hypermethylation of TSP1 promoter in some cancer types suggested the use of methylation inhibitor, 5-azacytidine in preclinical animal studies, the treatment significantly lowered the methylation of TSP1 promoter on chromosome 15, restored TSP1 expression and effectively decreased angiogenesis and tumor growth [77]. A cell-based delivery approach to reconstitute TSP-2 levels in squamous cell carcinomas, melanomas and Lewis lung carcinoma was used in the study by Detmar and colleagues [85]. Biodegradable polymer containing TSP-2 expressing fibroblasts was implanted into ovarian pedicle and the decreased tumor growth was accompanied by reduced angiogenesis and increased apoptosis. Finally, an elegant approach by Silverstein and co-workers employs inactivation of HRP expression in tissues and tumors expressing TSP1 and HRGP simultaneously [37], however this technique needs further validation.
b. Rationale for the use of TSP1 peptide fragments
Overexpression of the 180 kDa TSP1 molecule in most cases delays tumor growth, and causes significant reduction in mean vessel density (MVD), supporting the clinical utility of TSP1. This proved true for the breast and cutaneous carcinomas, fibrosarcomas, glioblastoma and more [86–90]. The injections of the TSP-1 expressing plasmid into the tumor site effectively suppressed the growth of the prostate cancer xenografts (DU-145) [91] and systemic injections of purified TSP1 from platelets inhibited melanoma growth [92]. However, the high molecular weight, the difficulties and costs of the large-scale production, possible immunogenicity, and especially the multiplicity of the TSP1 receptors, functions and target cells all present serious obstacles to the development of the intact TSP1 molecule for human use. Some studies demonstrate increased angiogenesis associated with TSP-1 overexpression (reviewed in [21]), which could be due to the TSP1-dependent CD47 ligation, which may increase the motility and invasiveness of the tumor cells [93]. Another possibility is that selective pressure posed by continuous TSP1 application leads to the selection and overgrowth of the pre-existing tumor cells expressing higher levels or broader spectra of angiogenic stimuli and are therefore resistant to the anti-angiogenic effect of TSP1 [94, 95]. Although the nascent molecule may not be suitable for clinical use, it is feasible to use the shorter fragments (peptides) that bind a single receptor on the endothelial cells and produce exclusively anti-angiogenic effect: they are easier to generate on a large scale and less immunogenic. Interestingly, recent studies report the cleavage of TSP-1 and TSP2 by ADAMTS-1 metalloproteinase, which releases shorter peptides with enhanced anti-angiogenic activity, suggesting that in vivo TSP1 and 2 in their matrix-bound intact form act as a reservoir of anti-angiogenic activity, which can be released by proteolytic degradation [96]. Consistent with that notion, we observed TSP1 fragments of varying length in the plasma of mice bearing TSP1 producing tumors (Volpert and Veliceasa, unpublished observations). Therefore, TSP1 peptides present a more viable therapeutic alternative for the full-length parental molecule, and a better replication of the in vivo mode of action.
c. Ectopic delivery of the TSR-based fragments/peptides
The list of the TSP1 and 2 peptides that were successfully tested in vivo for the inhibition of angiogenesis and in vitro, in preclinical models is given in Table 1. Although the N-terminal 25 kDa TSP1 fragment has not been tested in tumor models, it blocks angiogenesis by internalizing VEGF via LRP-1 receptor on the surface of cultured ovarian granulosa cells, concomitantly decreases VEGFR2 and induces apoptosis in vitro and in vivo [4]. The synthetic peptide derived from the TSR1 containing TGFβ activation and heparin binding sites (KRFKQDGGWSHWSPWSSC) effectively inhibited the growth of breast carcinoma in mouse xenograft model [25] and rodent C6 glioma in the orthotopic model [97]. The latter study reported impressive decrease in tumor size in the peptide-treated tumors, but no changes in the average vessel counts per mm2. However, the average vessel area was significantly larger in control group. A direct effect of the peptides on tumor cells was not ruled out in this study. Another angiostatic peptide, TSP1ang, was designed to retain proper folding required for the biological activity [98]. For that purpose the two cisteine residues and procollagen domain were retained along with the TSRs (aa 167–569). In contrast to the KRFKQDGGWSHWSPWSSC treatment, tumors formed by the C6 glioma cells expressing TSP1ang showed potent inhibition of angiogenesis manifested by decreased MVD and vessel branching in both hetero- and orthotropic models. Unfortunately, resultant tumors became more aggressive and invasive, possibly due to the exposure of the heparin-binding module. Systemic injections of synthetic peptide TSP1 3TSR (aa361-530) effectively blocked the growth of B16 mouse melanoma and Lewis lung carcinoma [92]. Recombinant protein composed of all three TSRs (also 3TSR) was tested in an orthotopic model of human pancreatic cancer where it significantly reduced angiogenesis and tumor growth due to massive apoptosis in tumor-associated endothelium, but showed no direct effect on the tumor cells proper [99]. TSP-2/NTF (80 kDa recombinant N-terminal fragment) inhibited the growth and angiogenesis of squamous cell carcinoma in a mouse model and its angioinhibitory effect is attributed to the direct action on the endothelial rather then tumor cells [100].
Table 1.
TSP1 active anti-angiogenic peptides
| Source | Domain | Sequence | Receptor | Mechanism |
|---|---|---|---|---|
| TSP1 | N-ter, heparin binding (25K) | Heparin | Internalizes VEGF, decreases VEGFR2, induces apoptosis [1] | |
| TSP1 | TSR2 | SPWSSCSVTCGDGVITRIR | CD36 | Apoptosis [2–4] |
| TSP1 | TSR2 | KRFKQDGGWSHWSPWSSC | Heparin | activates TGFβ [5, 6] quenches VEGF [7] |
| TSP1 | Procollagen + 3 TSR | CD36 | Apoptosis [8, 9] | |
| TSP2 | Procollagen + 3 TSR | CD36? | Apoptosis [10] | |
| TSP1 | 3 TSR | CD36 β1 integrin | Apoptosis [11–13] | |
| TSP1 | TSR2 | SPWSSCSVTCGDGVdITRIR (DI-TSP) | CD36 | Apoptosis [14] |
| TSP1 | TSR2 | Ac-Sar-GVdITNvdIR-NHEt (ABT-510) | CD36 | Apoptosis [15] |
| TSP1 | TSR2 | Ac-Sar-GVd-allo-ITNvdIR- NHEt (ABT-526) | CD36 | Apoptosis [15] |
Greenaway J, Lawler J, Moorehead R, Bornstein P, Lamarre J, Petrik J. Thrombospondin-1 inhibits VEGF levels in the ovary directly by binding and internalization via the low density lipoprotein receptor-related protein-1 (LRP-1). Journal of cellular physiology. Mar 2007;210(3):807–818.
Tolsma SS, Volpert OV, Good DJ, Frazier WA, Polverini PJ, Bouck N. Peptides derived from two separate domains of the matrix protein thrombospondin-1 have anti-angiogenic activity. The Journal of cell biology. Jul 1993;122(2):497–511.
Guo N, Krutzsch HC, Inman JK, Roberts DD. Thrombospondin 1 and type I repeat peptides of thrombospondin 1 specifically induce apoptosis of endothelial cells. Cancer research. May 1 1997;57(9):1735–1742.
Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. Sep 28 1999;100(13):1423–1431.
Ribeiro SM, Poczatek M, Schultz-Cherry S, Villain M, Murphy-Ullrich JE. The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-beta. The Journal of biological chemistry. May 7 1999;274(19):13586–13593.
Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. Jun 26 1998;93(7):1159–1170.
Margosio B, Marchetti D, Vergani V, Giavazzi R, Rusnati M, Presta M, Taraboletti G. Thrombospondin 1 as a scavenger for matrix-associated fibroblast growth factor 2. Blood. Dec 15 2003;102(13):4399–4406.
Streit M, Velasco P, Brown LF, Skobe M, Richard L, Riccardi L, Lawler J, Detmar M. Overexpression of thrombospondin-1 decreases angiogenesis and inhibits the growth of human cutaneous squamous cell carcinomas. The American journal of pathology. Aug 1999;155(2):441–452.
Yee KO, Streit M, Hawighorst T, Detmar M, Lawler J. Expression of the type-1 repeats of thrombospondin-1 inhibits tumor growth through activation of transforming growth factor-beta. The American journal of pathology. Aug 2004;165(2):541–552.
Lawler J, Detmar M. Tumor progression: the effects of thrombospondin-1 and -2. The international journal of biochemistry & cell biology. Jun 2004;36(6):1038–1045.
Zhang X, Galardi E, Duquette M, Lawler J, Parangi S. Antiangiogenic treatment with three thrombospondin-1 type 1 repeats versus gemcitabine in an orthotopic human pancreatic cancer model. Clin Cancer Res. Aug 1 2005;11(15):5622–5630.
Zhang X, Galardi E, Duquette M, Delic M, Lawler J, Parangi S. Antiangiogenic treatment with the three thrombospondin-1 type 1 repeats recombinant protein in an orthotopic human pancreatic cancer model. Clin Cancer Res. Mar 15 2005;11(6):2337–2344.
Short SM, Derrien A, Narsimhan RP, Lawler J, Ingber DE, Zetter BR. Inhibition of endothelial cell migration by thrombospondin-1 type-1 repeats is mediated by beta1 integrins. The Journal of cell biology. Feb 14 2005;168(4):643–653.
Dawson DW, Volpert OV, Pearce SF, Schneider AJ, Silverstein RL, Henkin J, Bouck NP. Three distinct D-amino acid substitutions confer potent antiangiogenic activity on an inactive peptide derived from a thrombospondin-1 type 1 repeat. Molecular pharmacology. Feb 1999;55(2):332–338.
Haviv F, Bradley MF, Kalvin DM, Schneider AJ, Davidson DJ, Majest SM, McKay LM, Haskell CJ, Bell RL, Nguyen B, Marsh KC, Surber BW, Uchic JT, Ferrero J, Wang YC, Leal J, Record RD, Hodde J, Badylak SF, Lesniewski RR, Henkin J. Thrombospondin-1 mimetic peptide inhibitors of angiogenesis and tumor growth: design, synthesis, and optimization of pharmacokinetics and biological activities. Journal of medicinal chemistry. Apr 21 2005;48(8):2838–2846.
The minimal TSP1 peptide mimetics based on the anti-angiogenic sequence GVITRIR in the TSR2 of TSP1 were designed by Abbott in a study initiated by Bouck and colleagues. The peptides are designated ABT-510 and ABT-526 [27] and are the most advanced of the TSP-based anti-angiogenic peptides, with specific activity in a low nanomolar range. Both are accepted and used in domestic and international research as the standard anti-angiogenic compounds. ABT-526 is highly effective in mouse cancer model and in dogs with naturally occurring cancers. Treatment with ABT-510 or ABT-526 lead to significant stabilization of disease in companion dogs with spontaneous tumors including head and neck carcinoma, mammary carcinoma, non-Hodgkin’s lymphoma and others. Part of the effect has been attributed to the reduction of the circulating endothelial cells or of the endothelial progenitor cells, with increased numbers of the circulating apoptotic endothelial cells [101]. ABT-510 also inhibits the orthotopic growth of malignant rodent glioma in rats [102]. In Phase I clinical trial for the treatment of cancer ABT-510 peptide was well tolerated and it is now in phase II clinical trial in various cancers including sarcoma and lymphoma. The statistical data so far are not overwhelming, although some patients undergo delayed recurrence, stabilization of disease and even tumor shrinkage. Most of the studies indicate the need for testing in combinations with cytotoxic therapies [7, 103, 104].
Interestingly, when ABT-510, which lacks direct immunosuppresive properties, was tested in the rat model of graft arteriosclerosis, it reduced adventitial angiogenesis and decreased the entry of inflammatory cells approximately twofold. This decrease resulted in dramatic reduction of collagen deposition and thereby prevented subsequent constrictive remodeling of the adventitia and reduction of lumen area, indicating that ABT-510 may be synergistic with conventional immunosuppressive therapy in preventing graft arteriosclerosis, a crucial feature of chronic graft rejection [105].
d. TSP1-based combination treatments: intelligent design
Knowledge gained in the studies of the TSP1 signaling pathways could become valuable aid in optimizing of the TSP-based anti-angiogenic therapies. Once the rate-limiting mediators of TSP1 dependent apoptosis are identified, strategies can be devised to maximize their levels on endothelial or non-endothelial cells and thus the anti-angiogenic action of TSP1. Previous studies of the TSP1 apoptotic signaling revealed two such intermediates: CD36 and CD95 [23, 43]. Below we will discuss the examples of the fine-tuning of the TSP-based anti-angiogenic compounds, mainly ABT-510.
e. Enhancing CD36 expression
CD36 is positively regulated by PPARγ (Peroxisome Proliferator activated receptor γ) [106, 107]. Peroxisome proliferator activated receptors (PPARs), are ligand-activated transcription factors with pleiotropic effects on cell fate. Due to its anti-proliferative, pro-apoptotic and differentiation activities, PPARγ has been intensively studied as potential anti-cancer target and found to harbor capabilities to both promote and suppress neoplastic growth. Thus the therapeutic value of PPARγ agonists remains controversial. Prompted by the fact that PPARγ ligands thiozolinediones (TZDs) block the growth effects in PPARγ-deficient embryonic stem cell tumors [108], Panigrahy and colleagues showed potent inhibition of angiogenesis and tumor growth by the PPARγ ligand rosiglitazone and thus extended the target repertoire of PPARγ ligands from cancer cells proper to the tumor endothelium [109]. In this study anti-angiogenic effect of PPARγ ligands was attributed to their direct action on the endothelial cells and reduced VEGF production by the tumor cells. Subsequent study by Huang and co-authors used TZDs troglitazone and rosiglitazone to augment CD36 expression by the endothelial cells in order to enhance anti-angiogenic and anti-tumor effects of ABT-510 [110]. PPARγ natural agonist 15-deoxy-delta(12,14)-prostaglandin J2, troglitazone, and rosiglitazone increased PPARgamma and CD36 expression by the endothelial cells and improved the efficacy of TSP1 and ABT510 in a CD36-dependent manner. PPARγ ligands increased endothelial CD36 protein by 20–40%. The ABT-510 and 15-deoxy-delta(12,14)-prostaglandin J2, troglitazone and reosiglitazone cooperatively blocked endothelial cell chemotaxis and tubulogenesis and induced apoptosis, These cooperative effects were CD36-dependent and completely abolished by the CD36 neutralizing antibody, FA6-152. In vivo 15-deoxy-delta(12,14)-prostaglandin J2 and troglitazone synergistically improved antiangiogenic and antitumor effects of ABT510 in the mouse corneal model, in Matrigel plugs and in tumor xenografts. In the tumors treated with the combination of troglitazone and ABT-510, the apoptosis was significantly increased and the MVD proportionally decreased, compared to the control tumors treated with the single compounds. Moreover, the larger, productive vessels in the tumors treated with ABT-510 alone were weakly positive for CD36 and appeared resistant to apoptosis. Conversely, in tumors subjected to the combination treatment CD36 presentation and apoptosis in the larger vessels were significantly higher. This was the first demonstration of the fine-tuning of antiangiogenic efficacy via targeted up-regulation of the limiting factors such as surface receptors.
f. Enhancing Fas levels
Redondo and co-workers discovered that in the endothelial cells treated with sub-lethal doses of doxorubicin CD95 mRNA and surface protein were upregulated in a p53-dependent manner (the induction was abolished by the inhibitor of p53-dependent transcription pifithrin-α [111]. Using this result as starting point, Nelius and Quesada combined ABT-510 with low dose doxorubicin in order to augment the expression of the rate-limiting CD95 on remodeling endothelium [112]. Doxorubicin titrated to a dose that produced CD95 increase, but failed to induce endothelial cells apoptosis, was combined with sub-optimal dosing of ABT-510. Low-dose doxorubicin strongly enhanced endothelial cell apoptosis by ABT-510, anti-angiogenesis in the subcutaneous Matrigel plugs, and the inhibition of angiogenesis and tumor growth in prostate cancer xenografts. Predictably, concomitant in vivo increases of the vascular endothelial CD95 and its cognate ligand accompanied by massive endothelial cell apoptosis were observed only when ABT-510/doxorubicin combination was used.
The ability to augment CD95 expression by the vascular endothelial cells was not unique to doxorubicin: cyclophosphamide (Cytoxan) also dramatically increased microvascular CD95. Studies by Volpert and Kerbel demonstrated similar interactions between ABT-510 and cytoxan where increased CD95 expression due to cytoxan synergistically increased the anti-angiogenesis and apoptosis by ABT-510 in vitro and in vivo, in Matrigel and tumor models [113]. This approach was termed a complementation antiangiogenic strategy. In the studies above the in vivo dosing of cytotoxic drugs was reduced to 1/20–1/50 of the maximally tolerated dose. Further studies demonstrated the validity of this approach: ABT-526 showed cooperative effect with cytotoxic agent lomustine (CeeNu, Bristol-Meyers-Squibb) in pet dogs with relapsed non-Hodgkin’s lymphoma [114] and with gemcitabine in the mouse model of pancreatic cancer [99, 115].
g. Modulating histone deacetylase (HDAC) activity
Recent study by Aurora and Volpert demonstrated intimate involvement of HDACs in the anti-angiogenic actions of TSP1 and another angiogenesis inhibitor, pigment epithelial-derived factor (PEDF) i. However the effect of TSP1 on HDACs was twofold: the release of HDACs association with the CD95L promoter and the enhancement of HDACs recruitment to the cFLIP promoter. Thus, the effect of HDAC inhibitors (HDACi) on the TSP1 angioinhibitory effect is difficult to predict. Earlier studies showed strong anti-angiogenic action of HDACi in multiple models of tumor growth and angiogenesis. In the tumor microenvironment, HDACi cause EC apoptosis and vascular regression by altering VEGF signaling via VEGFR2 and semaphorin, vascular integrity via Ang2, and permeability via eNOS production [116]. Interestingly, a mild HDAC inhibitor, valproic acid (VA) greatly potentiated the anti-angiogenic and anti-tumor effects of the TSP1 peptide, ABT-510 [117]. Combination therapy inhibited the growth of small neuroblastoma xenografts with better efficacy than single-agent treatments, and in animals with large xenografts, only combination treatment resulted in total cessation of tumor growth. The MVD was significantly reduced in the xenografts treated with combination therapy compared with the single-agent treatments and the structurally abnormal vessels were less abundant suggesting that the ABT-510 combination with VA may normalize tumor vasculature and pointing to the potential utility of this new antiangiogenic complementation treatment in children with high-risk neuroblastoma. In our hands, another HDAC inhibitor, Vorinostat showed a more complex, biphasic effect i. When tested in combination with PEDF, another natural angiogenesis inhibitor that acts via CD95/CD95L secondary death cascade [43, 44]. Vorinostat synergistically increased apoptosis and anti-angiogenesis at low doses, which had no independent effect (20 μM and lower). At 100–200 μM Vorinostat completely abolished the anti-angiogenic activity of PEDF or its active peptides. Further molecular analysis showed that lower doses of Vorinostat were sufficient to augment the expression of CD95L, but not cFLIP, while at higher doses both CD95L and cFLIP were de-repressed. One caveat for the long-term use of HDACi is their non-specific toxic effects produced by the extremely high doses required to reach efficacy in vivo [118]. The use in combination with TSP1 or PEDF derived anti-angiogenic drugs may offer a way to circumvent this problem.
Conclusions
In conclusion the information available to date indicates that TSP1 is a potent angiogenesis inhibitor and good candidate for combination treatments where its anti-angiogenic activity may be strongly augmented by the complementing drug. It also provides an important consideration, that the knowledge of the molecular pathways of a particular angiogenesis inhibitor or its derivatives allows intelligent design of the most successful drug combinations and the possibility of “fine-tuning” angiogenic activity.
Footnotes
Aurora AB, Biyashev D, Mirochnik Y, Zaichuk TA, Renault MA, Losordo D, Kwiatek A and Volpert OV. NFκB AND NFAT: BALANCING VASCULAR REGRESSION AND ANGIOGENESIS. Submitted.
References
- 1.Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, Bouck NP. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci U S A. 1990;87(17):6624–6628. doi: 10.1073/pnas.87.17.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bornstein P, Agah A, Kyriakides TR. The role of thrombospondins 1 and 2 in the regulation of cell-matrix interactions, collagen fibril formation, and the response to injury. Int J Biochem Cell Biol. 2004;36(6):1115–1125. doi: 10.1016/j.biocel.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Carlson CB, Lawler J, Mosher DF. Thrombospondins: from structure to therapeutics: Structures of thrombospondins. Cell Mol Life Sci. 2008;65(5):672–686. doi: 10.1007/s00018-007-7484-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenaway J, Lawler J, Moorehead R, Bornstein P, Lamarre J, Petrik J. Thrombospondin-1 inhibits VEGF levels in the ovary directly by binding and internalization via the low density lipoprotein receptor-related protein-1 (LRP-1) J Cell Physiol. 2007;210(3):807–818. doi: 10.1002/jcp.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36(6):961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang X, Lawler J. Thrombospondin-based antiangiogenic therapy. Microvasc Res. 2007;74(2–3):90–99. doi: 10.1016/j.mvr.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoekstra R, de Vos FY, Eskens FA, de Vries EG, Uges DR, Knight R, Carr RA, Humerickhouse R, Verweij J, Gietema JA. Phase I study of the thrombospondin-1-mimetic angiogenesis inhibitor ABT-510 with 5-fluorouracil and leucovorin: a safe combination. Eur J Cancer. 2006;42(4):467–472. doi: 10.1016/j.ejca.2005.08.040. [DOI] [PubMed] [Google Scholar]
- 8.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93(7):1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 9.Ribeiro SM, Poczatek M, Schultz-Cherry S, Villain M, Murphy-Ullrich JE. The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-beta. J Biol Chem. 1999;274(19):13586–13593. doi: 10.1074/jbc.274.19.13586. [DOI] [PubMed] [Google Scholar]
- 10.Wang XQ, Lindberg FP, Frazier WA. Integrin-associated protein stimulates alpha2beta1-dependent chemotaxis via Gi-mediated inhibition of adenylate cyclase and extracellular-regulated kinases. J Cell Biol. 1999;147(2):389–400. doi: 10.1083/jcb.147.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veliceasa D, Ivanovic M, Hoepfner FT, Thumbikat P, Volpert OV, Smith ND. Transient potential receptor channel 4 controls thrombospondin-1 secretion and angiogenesis in renal cell carcinoma. Febs J. 2007;274(24):6365–6377. doi: 10.1111/j.1742-4658.2007.06159.x. [DOI] [PubMed] [Google Scholar]
- 12.Margosio B, Rusnati M, Bonezzi K, Cordes BL, Annis DS, Urbinati C, Giavazzi R, Presta M, Ribatti D, Mosher DF, Taraboletti G. Fibroblast growth factor-2 binding to the thrombospondin-1 type III repeats, a novel antiangiogenic domain. Int J Biochem Cell Biol. 2008;40(4):700–709. doi: 10.1016/j.biocel.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tolsma SS, Volpert OV, Good DJ, Frazier WA, Polverini PJ, Bouck N. Peptides derived from two separate domains of the matrix protein thrombospondin-1 have anti-angiogenic activity. J Cell Biol. 1993;122(2):497–511. doi: 10.1083/jcb.122.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyata Y, Koga S, Takehara K, Kanetake H, Kanda S. Expression of thrombospondin-derived 4N1K peptide-containing proteins in renal cell carcinoma tissues is associated with a decrease in tumor growth and angiogenesis. Clin Cancer Res. 2003;9(5):1734–1740. [PubMed] [Google Scholar]
- 15.Kanda S, Shono T, Tomasini-Johansson B, Klint P, Saito Y. Role of thrombospondin-1-derived peptide, 4N1K, in FGF-2-induced angiogenesis. Exp Cell Res. 1999;252(2):262–272. doi: 10.1006/excr.1999.4622. [DOI] [PubMed] [Google Scholar]
- 16.Isenberg JS, Calzada MJ, Zhou L, Guo N, Lawler J, Wang XQ, Frazier WA, Roberts DD. Endogenous thrombospondin-1 is not necessary for proliferation but is permissive for vascular smooth muscle cell responses to platelet-derived growth factor. Matrix Biol. 2005;24(2):110–123. doi: 10.1016/j.matbio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Gupta K, Gupta P, Wild R, Ramakrishnan S, Hebbel RP. Binding and displacement of vascular endothelial growth factor (VEGF) by thrombospondin: effect on human microvascular endothelial cell proliferation and angiogenesis. Angiogenesis. 1999;3(2):147–158. doi: 10.1023/a:1009018702832. [DOI] [PubMed] [Google Scholar]
- 18.Margosio B, Marchetti D, Vergani V, Giavazzi R, Rusnati M, Presta M, Taraboletti G. Thrombospondin 1 as a scavenger for matrix-associated fibroblast growth factor 2. Blood. 2003;102(13):4399–4406. doi: 10.1182/blood-2003-03-0893. [DOI] [PubMed] [Google Scholar]
- 19.Johansson U, Londei M. Ligation of CD47 during monocyte differentiation into dendritic cells results in reduced capacity for interleukin-12 production. Scand J Immunol. 2004;59(1):50–57. doi: 10.1111/j.0300-9475.2004.01354.x. [DOI] [PubMed] [Google Scholar]
- 20.Johansson U, Higginbottom K, Londei M. CD47 ligation induces a rapid caspase-independent apoptosis-like cell death in human monocytes and dendritic cells. Scand J Immunol. 2004;59(1):40–49. doi: 10.1111/j.0300-9475.2004.01355.x. [DOI] [PubMed] [Google Scholar]
- 21.Ren B, Yee KO, Lawler J, Khosravi-Far R. Regulation of tumor angiogenesis by thrombospondin-1. Biochim Biophys Acta. 2006;1765(2):178–188. doi: 10.1016/j.bbcan.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Armstrong LC, Bjorkblom B, Hankenson KD, Siadak AW, Stiles CE, Bornstein P. Thrombospondin 2 inhibits microvascular endothelial cell proliferation by a caspase-independent mechanism. Mol Biol Cell. 2002;13(6):1893–1905. doi: 10.1091/mbc.E01-09-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6(1):41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 24.Guo N, Krutzsch HC, Inman JK, Roberts DD. Thrombospondin 1 and type I repeat peptides of thrombospondin 1 specifically induce apoptosis of endothelial cells. Cancer Res. 1997;57(9):1735–1742. [PubMed] [Google Scholar]
- 25.Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100(13):1423–1431. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 26.Dawson DW, Volpert OV, Pearce SF, Schneider AJ, Silverstein RL, Henkin J, Bouck NP. Three distinct D-amino acid substitutions confer potent antiangiogenic activity on an inactive peptide derived from a thrombospondin-1 type 1 repeat. Mol Pharmacol. 1999;55(2):332–338. doi: 10.1124/mol.55.2.332. [DOI] [PubMed] [Google Scholar]
- 27.Haviv F, Bradley MF, Kalvin DM, Schneider AJ, Davidson DJ, Majest SM, McKay LM, Haskell CJ, Bell RL, Nguyen B, Marsh KC, Surber BW, Uchic JT, Ferrero J, Wang YC, Leal J, Record RD, Hodde J, Badylak SF, Lesniewski RR, Henkin J. Thrombospondin-1 mimetic peptide inhibitors of angiogenesis and tumor growth: design, synthesis, and optimization of pharmacokinetics and biological activities. J Med Chem. 2005;48(8):2838–2846. doi: 10.1021/jm0401560. [DOI] [PubMed] [Google Scholar]
- 28.Gietema JA, Hoekstra R, de Vos FY, Uges DR, van der Gaast A, Groen HJ, Loos WJ, Knight RA, Carr RA, Humerickhouse RA, Eskens FA. A phase I study assessing the safety and pharmacokinetics of the thrombospondin-1-mimetic angiogenesis inhibitor ABT-510 with gemcitabine and cisplatin in patients with solid tumors. Ann Oncol. 2006;17(8):1320–1327. doi: 10.1093/annonc/mdl102. [DOI] [PubMed] [Google Scholar]
- 29.Folkman J. Endogenous angiogenesis inhibitors. Apmis. 2004;112(7–8):496–507. doi: 10.1111/j.1600-0463.2004.apm11207-0809.x. [DOI] [PubMed] [Google Scholar]
- 30.Yamauchi M, Imajoh-Ohmi S, Shibuya M. Novel antiangiogenic pathway of thrombospondin-1 mediated by suppression of the cell cycle. Cancer Sci. 2007;98(9):1491–1497. doi: 10.1111/j.1349-7006.2007.00534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koh JT, Kook H, Kee HJ, Seo YW, Jeong BC, Lee JH, Kim MY, Yoon KC, Jung S, Kim KK. Extracellular fragment of brain-specific angiogenesis inhibitor 1 suppresses endothelial cell proliferation by blocking alphavbeta5 integrin. Exp Cell Res. 2004;294(1):172–184. doi: 10.1016/j.yexcr.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Dawson DW, Pearce SF, Zhong R, Silverstein RL, Frazier WA, Bouck NP. CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. J Cell Biol. 1997;138(3):707–717. doi: 10.1083/jcb.138.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108(6):785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li WX, Howard RJ, Leung LL. Identification of SVTCG in thrombospondin as the conformation-dependent, high affinity binding site for its receptor, CD36. J Biol Chem. 1993;268(22):16179–16184. [PubMed] [Google Scholar]
- 35.Asch AS, Silbiger S, Heimer E, Nachman RL. Thrombospondin sequence motif (CSVTCG) is responsible for CD36 binding. Biochem Biophys Res Commun. 1992;182(3):1208–1217. doi: 10.1016/0006-291x(92)91860-s. [DOI] [PubMed] [Google Scholar]
- 36.Simantov R, Febbraio M, Silverstein RL. The antiangiogenic effect of thrombospondin-2 is mediated by CD36 and modulated by histidine-rich glycoprotein. Matrix Biol. 2005;24(1):27–34. doi: 10.1016/j.matbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Silverstein RL, Febbraio M. CD36-TSP-HRGP interactions in the regulation of angiogenesis. Curr Pharm Des. 2007;13(35):3559–3567. doi: 10.2174/138161207782794185. [DOI] [PubMed] [Google Scholar]
- 38.Wintergerst ES, Jelk J, Rahner C, Asmis R. Apoptosis induced by oxidized low density lipoprotein in human monocyte-derived macrophages involves CD36 and activation of caspase-3. Eur J Biochem. 2000;267(19):6050–6059. doi: 10.1046/j.1432-1327.2000.01682.x. [DOI] [PubMed] [Google Scholar]
- 39.Asch AS, Liu I, Briccetti FM, Barnwell JW, Kwakye-Berko F, Dokun A, Goldberger J, Pernambuco M. Analysis of CD36 binding domains: ligand specificity controlled by dephosphorylation of an ectodomain. Science. 1993;262(5138):1436–1440. doi: 10.1126/science.7504322. [DOI] [PubMed] [Google Scholar]
- 40.Nor JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PJ. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J Vasc Res. 2000;37(3):209–218. doi: 10.1159/000025733. [DOI] [PubMed] [Google Scholar]
- 41.Simantov R, Silverstein RL. CD36: a critical anti-angiogenic receptor. Front Biosci. 2003:8s874–882. doi: 10.2741/1168. [DOI] [PubMed] [Google Scholar]
- 42.Jimenez B, Volpert OV, Reiher F, Chang L, Munoz A, Karin M, Bouck N. c-Jun N-terminal kinase activation is required for the inhibition of neovascularization by thrombospondin-1. Oncogene. 2001;20(26):3443–3448. doi: 10.1038/sj.onc.1204464. [DOI] [PubMed] [Google Scholar]
- 43.Volpert OV, Zaichuk T, Zhou W, Reiher F, Ferguson TA, Stuart PM, Amin M, Bouck NP. Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat Med. 2002;8(4):349–357. doi: 10.1038/nm0402-349. [DOI] [PubMed] [Google Scholar]
- 44.Zaichuk TA, Shroff EH, Emmanuel R, Filleur S, Nelius T, Volpert OV. Nuclear factor of activated T cells balances angiogenesis activation and inhibition. J Exp Med. 2004;199(11):1513–1522. doi: 10.1084/jem.20040474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hernandez GL, Volpert OV, Iniguez MA, Lorenzo E, Martinez-Martinez S, Grau R, Fresno M, Redondo JM. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med. 2001;193(5):607–620. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 47.Grosjean J, Kiriakidis S, Reilly K, Feldmann M, Paleolog E. Vascular endothelial growth factor signalling in endothelial cell survival: a role for NFkappaB. Biochem Biophys Res Commun. 2006;340(3):984–994. doi: 10.1016/j.bbrc.2005.12.095. [DOI] [PubMed] [Google Scholar]
- 48.Weitzman JB, Yaniv M. Signal transduction pathways and modulation of gene activity. Clin Chem Lab Med. 1998;36(8):535–539. doi: 10.1515/CCLM.1998.091. [DOI] [PubMed] [Google Scholar]
- 49.Serfling E, Berberich-Siebelt F, Avots A, Chuvpilo S, Klein-Hessling S, Jha MK, Kondo E, Pagel P, Schulze-Luehrmann J, Palmetshofer A. NFAT and NF-kappaB factors-the distant relatives. Int J Biochem Cell Biol. 2004;36(7):1166–1170. doi: 10.1016/j.biocel.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 50.Nor JE, Christensen J, Liu J, Peters M, Mooney DJ, Strieter RM, Polverini PJ. Up-Regulation of Bcl-2 in microvascular endothelial cells enhances intratumoral angiogenesis and accelerates tumor growth. Cancer Res. 2001;61(5):2183–2188. [PubMed] [Google Scholar]
- 51.Kawamura T, Ono K, Morimoto T, Akao M, Iwai-Kanai E, Wada H, Sowa N, Kita T, Hasegawa K. Endothelin-1-dependent nuclear factor of activated T lymphocyte signaling associates with transcriptional coactivator p300 in the activation of the B cell leukemia-2 promoter in cardiac myocytes. Circ Res. 2004;94(11):1492–1499. doi: 10.1161/01.RES.0000129701.14494.52. [DOI] [PubMed] [Google Scholar]
- 52.Mateo V, Lagneaux L, Bron D, Biron G, Armant M, Delespesse G, Sarfati M. CD47 ligation induces caspase-independent cell death in chronic lymphocytic leukemia. Nat Med. 1999;5(11):1277–1284. doi: 10.1038/15233. [DOI] [PubMed] [Google Scholar]
- 53.Pettersen RD. CD47 and death signaling in the immune system. Apoptosis. 2000;5(4):299–306. doi: 10.1023/a:1009612821625. [DOI] [PubMed] [Google Scholar]
- 54.Mateo V, Brown EJ, Biron G, Rubio M, Fischer A, Deist FL, Sarfati M. Mechanisms of CD47-induced caspase-independent cell death in normal and leukemic cells: link between phosphatidylserine exposure and cytoskeleton organization. Blood. 2002;100(8):2882–2890. doi: 10.1182/blood-2001-12-0217. [DOI] [PubMed] [Google Scholar]
- 55.Roue G, Bitton N, Yuste VJ, Montange T, Rubio M, Dessauge F, Delettre C, Merle-Beral H, Sarfati M, Susin SA. Mitochondrial dysfunction in CD47-mediated caspase-independent cell death: ROS production in the absence of cytochrome c and AIF release. Biochimie. 2003;85(8):741–746. doi: 10.1016/s0300-9084(03)00129-9. [DOI] [PubMed] [Google Scholar]
- 56.Bras M, Yuste VJ, Roue G, Barbier S, Sancho P, Virely C, Rubio M, Baudet S, Esquerda JE, Merle-Beral H, Sarfati M, Susin SA. Drp1 mediates caspase-independent type III cell death in normal and leukemic cells. Mol Cell Biol. 2007;27(20):7073–7088. doi: 10.1128/MCB.02116-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Graf R, Freyberg M, Kaiser D, Friedl P. Mechanosensitive induction of apoptosis in fibroblasts is regulated by thrombospondin-1 and integrin associated protein (CD47) Apoptosis. 2002;7(6):493–498. doi: 10.1023/a:1020634924760. [DOI] [PubMed] [Google Scholar]
- 58.Yang M, Li K, Ng MH, Yuen PM, Fok TF, Li CK, Hogg PJ, Chong BH. Thrombospondin-1 inhibits in vitro megakaryocytopoiesis via CD36. Thromb Res. 2003;109(1):47–54. doi: 10.1016/s0049-3848(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 59.Benezra R, Henke E, Ciarrocchi A, Ruzinova M, Solit D, Rosen N, Nolan D, Mittal V, de Candia P. Induction of complete regressions of oncogene-induced breast tumors in mice. Cold Spring Harb Symp Quant Biol. 2005:70375–381. doi: 10.1101/sqb.2005.70.006. [DOI] [PubMed] [Google Scholar]
- 60.Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319(5860):195–198. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 61.Bruel A, Touhami-Carrier M, Thomaidis A, Legrand C. Thrombospondin-1 (TSP-1) and TSP-1-derived heparin-binding peptides induce promyelocytic leukemia cell differentiation and apoptosis. Anticancer Res. 2005;25(2A):757–764. [PubMed] [Google Scholar]
- 62.Kazerounian S, Yee KO, Lawler J. Thrombospondins: from structure to therapeutics: Thrombospondins in cancer. Cell Mol Life Sci. 2008;65(5):700–712. doi: 10.1007/s00018-007-7486-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lawler J, Detmar M. Tumor progression: the effects of thrombospondin-1 and -2. Int J Biochem Cell Biol. 2004;36(6):1038–1045. doi: 10.1016/j.biocel.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 64.Wen S, Stolarov J, Myers MP, Su JD, Wigler MH, Tonks NK, Durden DL. PTEN controls tumor-induced angiogenesis. Proc Natl Acad Sci U S A. 2001;98(8):4622–4627. doi: 10.1073/pnas.081063798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwarte-Waldhoff I, Volpert OV, Bouck NP, Sipos B, Hahn SA, Klein-Scory S, Luttges J, Kloppel G, Graeven U, Eilert-Micus C, Hintelmann A, Schmiegel W. Smad4/DPC4-mediated tumor suppression through suppression of angiogenesis. Proc Natl Acad Sci U S A. 2000;97(17):9624–9629. doi: 10.1073/pnas.97.17.9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265(5178):1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 67.Gutierrez LS, Suckow M, Lawler J, Ploplis VA, Castellino FJ. Thrombospondin 1--a regulator of adenoma growth and carcinoma progression in the APC(Min/+) mouse model. Carcinogenesis. 2003;24(2):199–207. doi: 10.1093/carcin/24.2.199. [DOI] [PubMed] [Google Scholar]
- 68.Vikhanskaya F, Bani MR, Borsotti P, Ghilardi C, Ceruti R, Ghisleni G, Marabese M, Giavazzi R, Broggini M, Taraboletti G. p73 Overexpression increases VEGF and reduces thrombospondin-1 production: implications for tumor angiogenesis. Oncogene. 2001;20(50):7293–7300. doi: 10.1038/sj.onc.1204896. [DOI] [PubMed] [Google Scholar]
- 69.Dejong V, Degeorges A, Filleur S, Ait-Si-Ali S, Mettouchi A, Bornstein P, Binetruy B, Cabon F. The Wilms’ tumor gene product represses the transcription of thrombospondin 1 in response to overexpression of c-Jun. Oncogene. 1999;18(20):3143–3151. doi: 10.1038/sj.onc.1202654. [DOI] [PubMed] [Google Scholar]
- 70.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kalas W, Gilpin S, Yu JL, May L, Krchnakova H, Bornstein P, Rak J. Restoration of thrombospondin 1 expression in tumor cells harbouring mutant ras oncogene by treatment with low doses of doxycycline. Biochem Biophys Res Commun. 2003;310(1):109–114. doi: 10.1016/j.bbrc.2003.08.128. [DOI] [PubMed] [Google Scholar]
- 72.Kalas W, Yu JL, Milsom C, Rosenfeld J, Benezra R, Bornstein P, Rak J. Oncogenes and Angiogenesis: down-regulation of thrombospondin-1 in normal fibroblasts exposed to factors from cancer cells harboring mutant ras. Cancer Res. 2005;65(19):8878–8886. doi: 10.1158/0008-5472.CAN-05-1479. [DOI] [PubMed] [Google Scholar]
- 73.Liu YJ, Xu Y, Yu Q. Full-length ADAMTS-1 and the ADAMTS-1 fragments display pro- and antimetastatic activity, respectively. Oncogene. 2006;25(17):2452–2467. doi: 10.1038/sj.onc.1209287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rak J, Yu JL, Klement G, Kerbel RS. Oncogenes and angiogenesis: signaling three-dimensional tumor growth. J Investig Dermatol Symp Proc. 2000;5(1):24–33. doi: 10.1046/j.1087-0024.2000.00012.x. [DOI] [PubMed] [Google Scholar]
- 75.Slack JL, Bornstein P. Transformation by v-src causes transient induction followed by repression of mouse thrombospondin-1. Cell Growth Differ. 1994;5(12):1373–1380. [PubMed] [Google Scholar]
- 76.Wen XF, Yang G, Mao W, Thornton A, Liu J, Bast RC, Jr, Le XF. HER2 signaling modulates the equilibrium between pro- and antiangiogenic factors via distinct pathways: implications for HER2-targeted antibody therapy. Oncogene. 2006;25(52):6986–6996. doi: 10.1038/sj.onc.1209685. [DOI] [PubMed] [Google Scholar]
- 77.Yang QW, Liu S, Tian Y, Salwen HR, Chlenski A, Weinstein J, Cohn SL. Methylation-associated silencing of the thrombospondin-1 gene in human neuroblastoma. Cancer Res. 2003;63(19):6299–6310. [PubMed] [Google Scholar]
- 78.Gonzalez-Gomez P, Bello MJ, Lomas J, Arjona D, Alonso ME, Aminoso C, Lopez-Marin I, Anselmo NP, Sarasa JL, Gutierrez M, Casartelli C, Rey JA. Aberrant methylation of multiple genes in neuroblastic tumours. relationship with MYCN amplification and allelic status at 1p. Eur J Cancer. 2003;39(10):1478–1485. doi: 10.1016/s0959-8049(03)00312-5. [DOI] [PubMed] [Google Scholar]
- 79.Zhou W, Dasgupta C, Negash S, Raj JU. Modulation of pulmonary vascular smooth muscle cell phenotype in hypoxia: role of cGMP-dependent protein kinase. Am J Physiol Lung Cell Mol Physiol. 2007;292(6):L1459–1466. doi: 10.1152/ajplung.00143.2006. [DOI] [PubMed] [Google Scholar]
- 80.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6(4):273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 81.Bocci G, Francia G, Man S, Lawler J, Kerbel RS. Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc Natl Acad Sci U S A. 2003;100(22):12917–12922. doi: 10.1073/pnas.2135406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hamano Y, Sugimoto H, Soubasakos MA, Kieran M, Olsen BR, Lawler J, Sudhakar A, Kalluri R. Thrombospondin-1 associated with tumor microenvironment contributes to low-dose cyclophosphamide-mediated endothelial cell apoptosis and tumor growth suppression. Cancer Res. 2004;64(5):1570–1574. doi: 10.1158/0008-5472.can-03-3126. [DOI] [PubMed] [Google Scholar]
- 83.Stempak D, Gammon J, Halton J, Moghrabi A, Koren G, Baruchel S. A pilot pharmacokinetic and antiangiogenic biomarker study of celecoxib and low-dose metronomic vinblastine or cyclophosphamide in pediatric recurrent solid tumors. J Pediatr Hematol Oncol. 2006;28(11):720–728. doi: 10.1097/01.mph.0000243657.64056.c3. [DOI] [PubMed] [Google Scholar]
- 84.Garcia AA, Hirte H, Fleming G, Yang D, Tsao-Wei DD, Roman L, Groshen S, Swenson S, Markland F, Gandara D, Scudder S, Morgan R, Chen H, Lenz HJ, Oza AM. Phase II clinical trial of bevacizumab and low-dose metronomic oral cyclophosphamide in recurrent ovarian cancer: a trial of the California, Chicago, and Princess Margaret Hospital phase II consortia. J Clin Oncol. 2008;26(1):76–82. doi: 10.1200/JCO.2007.12.1939. [DOI] [PubMed] [Google Scholar]
- 85.Streit M, Stephen AE, Hawighorst T, Matsuda K, Lange-Asschenfeldt B, Brown LF, Vacanti JP, Detmar M. Systemic inhibition of tumor growth and angiogenesis by thrombospondin-2 using cell-based antiangiogenic gene therapy. Cancer Res. 2002;62(7):2004–2012. [PubMed] [Google Scholar]
- 86.Weinstat-Saslow DL, Zabrenetzky VS, VanHoutte K, Frazier WA, Roberts DD, Steeg PS. Transfection of thrombospondin 1 complementary DNA into a human breast carcinoma cell line reduces primary tumor growth, metastatic potential, and angiogenesis. Cancer Res. 1994;54(24):6504–6511. [PubMed] [Google Scholar]
- 87.Bleuel K, Popp S, Fusenig NE, Stanbridge EJ, Boukamp P. Tumor suppression in human skin carcinoma cells by chromosome 15 transfer or thrombospondin-1 overexpression through halted tumor vascularization. Proc Natl Acad Sci U S A. 1999;96(5):2065–2070. doi: 10.1073/pnas.96.5.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Streit M, Velasco P, Brown LF, Skobe M, Richard L, Riccardi L, Lawler J, Detmar M. Overexpression of thrombospondin-1 decreases angiogenesis and inhibits the growth of human cutaneous squamous cell carcinomas. Am J Pathol. 1999;155(2):441–452. doi: 10.1016/S0002-9440(10)65140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hsu SC, Volpert OV, Steck PA, Mikkelsen T, Polverini PJ, Rao S, Chou P, Bouck NP. Inhibition of angiogenesis in human glioblastomas by chromosome 10 induction of thrombospondin-1. Cancer Res. 1996;56(24):5684–5691. [PubMed] [Google Scholar]
- 90.Tenan M, Fulci G, Albertoni M, Diserens AC, Hamou MF, El Atifi-Borel M, Feige JJ, Pepper MS, Van Meir EG. Thrombospondin-1 is downregulated by anoxia and suppresses tumorigenicity of human glioblastoma cells. J Exp Med. 2000;191(10):1789–1798. doi: 10.1084/jem.191.10.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jin RJ, Kwak C, Lee SG, Lee CH, Soo CG, Park MS, Lee E, Lee SE. The application of an anti-angiogenic gene (thrombospondin-1) in the treatment of human prostate cancer xenografts. Cancer Gene Ther. 2000;7(12):1537–1542. doi: 10.1038/sj.cgt.7700266. [DOI] [PubMed] [Google Scholar]
- 92.Miao WM, Seng WL, Duquette M, Lawler P, Laus C, Lawler J. Thrombospondin-1 type 1 repeat recombinant proteins inhibit tumor growth through transforming growth factor-beta-dependent and -independent mechanisms. Cancer Res. 2001;61(21):7830–7839. [PubMed] [Google Scholar]
- 93.Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57(1):25–40. doi: 10.1007/s000180050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Filleur S, Volpert OV, Degeorges A, Voland C, Reiher F, Clezardin P, Bouck N, Cabon F. In vivo mechanisms by which tumors producing thrombospondin 1 bypass its inhibitory effects. Genes Dev. 2001;15(11):1373–1382. doi: 10.1101/gad.193501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fontana A, Filleur S, Guglielmi J, Frappart L, Bruno-Bossio G, Boissier S, Cabon F, Clezardin P. Human breast tumors override the antiangiogenic effect of stromal thrombospondin-1 in vivo. Int J Cancer. 2005;116(5):686–691. doi: 10.1002/ijc.20584. [DOI] [PubMed] [Google Scholar]
- 96.Lee NV, Sato M, Annis DS, Loo JA, Wu L, Mosher DF, Iruela-Arispe ML. ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. Embo J. 2006;25(22):5270–5283. doi: 10.1038/sj.emboj.7601400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bogdanov A, Jr, Marecos E, Cheng HC, Chandrasekaran L, Krutzsch HC, Roberts DD, Weissleder R. Treatment of experimental brain tumors with trombospondin-1 derived peptides: an in vivo imaging study. Neoplasia. 1999;1(5):438–445. doi: 10.1038/sj.neo.7900044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.de Fraipont F, Keramidas M, El Atifi M, Chambaz EM, Berger F, Feige JJ. Expression of the thrombospondin 1 fragment 167–569 in C6 glioma cells stimulates tumorigenicity despite reduced neovascularization. Oncogene. 2004;23(20):3642–3649. doi: 10.1038/sj.onc.1207438. [DOI] [PubMed] [Google Scholar]
- 99.Zhang X, Galardi E, Duquette M, Delic M, Lawler J, Parangi S. Antiangiogenic treatment with the three thrombospondin-1 type 1 repeats recombinant protein in an orthotopic human pancreatic cancer model. Clin Cancer Res. 2005;11(6):2337–2344. doi: 10.1158/1078-0432.CCR-04-1900. [DOI] [PubMed] [Google Scholar]
- 100.Noh YH, Matsuda K, Hong YK, Kunstfeld R, Riccardi L, Koch M, Oura H, Dadras SS, Streit M, Detmar M. An N-terminal 80 kDa recombinant fragment of human thrombospondin-2 inhibits vascular endothelial growth factor induced endothelial cell migration in vitro and tumor growth and angiogenesis in vivo. J Invest Dermatol. 2003;121(6):1536–1543. doi: 10.1046/j.1523-1747.2003.12643.x. [DOI] [PubMed] [Google Scholar]
- 101.Rusk A, McKeegan E, Haviv F, Majest S, Henkin J, Khanna C. Preclinical evaluation of antiangiogenic thrombospondin-1 peptide mimetics, ABT-526 and ABT-510, in companion dogs with naturally occurring cancers. Clin Cancer Res. 2006;12(24):7444–7455. doi: 10.1158/1078-0432.CCR-06-0109. [DOI] [PubMed] [Google Scholar]
- 102.Anderson JC, Grammer JR, Wang W, Nabors LB, Henkin J, Stewart JE, Jr, Gladson CL. ABT-510, a modified type 1 repeat peptide of thrombospondin, inhibits malignant glioma growth in vivo by inhibiting angiogenesis. Cancer Biol Ther. 2007;6(3):454–462. doi: 10.4161/cbt.6.3.3630. [DOI] [PubMed] [Google Scholar]
- 103.Ebbinghaus S, Hussain M, Tannir N, Gordon M, Desai AA, Knight RA, Humerickhouse RA, Qian J, Gordon GB, Figlin R. Phase 2 study of ABT-510 in patients with previously untreated advanced renal cell carcinoma. Clin Cancer Res. 2007;13(22 Pt 1):6689–6695. doi: 10.1158/1078-0432.CCR-07-1477. [DOI] [PubMed] [Google Scholar]
- 104.Markovic SN, Suman VJ, Rao RA, Ingle JN, Kaur JS, Erickson LA, Pitot HC, Croghan GA, McWilliams RR, Merchan J, Kottschade LA, Nevala WK, Uhl CB, Allred J, Creagan ET. A phase II study of ABT-510 (thrombospondin-1 analog) for the treatment of metastatic melanoma. Am J Clin Oncol. 2007;30(3):303–309. doi: 10.1097/01.coc.0000256104.80089.35. [DOI] [PubMed] [Google Scholar]
- 105.Thaunat O, Louedec L, Graff-Dubois S, Dai J, Groyer E, Yacoub-Youssef H, Mandet C, Bruneval P, Kaveri S, Caligiuri G, Germain S, Michel JB, Nicoletti A. Antiangiogenic treatment prevents adventitial constrictive remodeling in graft arteriosclerosis. Transplantation. 2008;85(2):281–289. doi: 10.1097/TP.0b013e318160500a. [DOI] [PubMed] [Google Scholar]
- 106.Majai G, Sarang Z, Csomos K, Zahuczky G, Fesus L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol. 2007;37(5):1343–1354. doi: 10.1002/eji.200636398. [DOI] [PubMed] [Google Scholar]
- 107.Szanto A, Nagy L. Retinoids potentiate peroxisome proliferator-activated receptor gamma action in differentiation, gene expression, and lipid metabolic processes in developing myeloid cells. Mol Pharmacol. 2005;67(6):1935–1943. doi: 10.1124/mol.104.006445. [DOI] [PubMed] [Google Scholar]
- 108.Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001;61(16):6213–6218. [PubMed] [Google Scholar]
- 109.Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, Moses MA, Kilroy S, Duensing S, Fletcher C, Fletcher JA, Hlatky L, Hahnfeldt P, Folkman J, Kaipainen A. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest. 2002;110(7):923–932. doi: 10.1172/JCI15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huang H, Campbell SC, Bedford DF, Nelius T, Veliceasa D, Shroff EH, Henkin J, Schneider A, Bouck N, Volpert OV. Peroxisome proliferator-activated receptor gamma ligands improve the antitumor efficacy of thrombospondin peptide ABT510. Mol Cancer Res. 2004;2(10):541–550. [PubMed] [Google Scholar]
- 111.Lorenzo E, Ruiz-Ruiz C, Quesada AJ, Hernandez G, Rodriguez A, Lopez-Rivas A, Redondo JM. Doxorubicin induces apoptosis and CD95 gene expression in human primary endothelial cells through a p53-dependent mechanism. J Biol Chem. 2002;277(13):10883–10892. doi: 10.1074/jbc.M107442200. [DOI] [PubMed] [Google Scholar]
- 112.Quesada AJ, Nelius T, Yap R, Zaichuk TA, Alfranca A, Filleur S, Volpert OV, Redondo JM. In vivo upregulation of CD95 and CD95L causes synergistic inhibition of angiogenesis by TSP1 peptide and metronomic doxorubicin treatment. Cell Death Differ. 2005;12(6):649–658. doi: 10.1038/sj.cdd.4401615. [DOI] [PubMed] [Google Scholar]
- 113.Yap R, Veliceasa D, Emmenegger U, Kerbel RS, McKay LM, Henkin J, Volpert OV. Metronomic low-dose chemotherapy boosts CD95-dependent antiangiogenic effect of the thrombospondin peptide ABT-510: a complementation antiangiogenic strategy. Clin Cancer Res. 2005;11(18):6678–6685. doi: 10.1158/1078-0432.CCR-05-0621. [DOI] [PubMed] [Google Scholar]
- 114.Rusk A, Cozzi E, Stebbins M, Vail D, Graham J, Valli V, Henkin J, Sharpee R, Khanna C. Cooperative activity of cytotoxic chemotherapy with antiangiogenic thrombospondin-I peptides, ABT-526 in pet dogs with relapsed lymphoma. Clin Cancer Res. 2006;12(24):7456–7464. doi: 10.1158/1078-0432.CCR-06-0110. [DOI] [PubMed] [Google Scholar]
- 115.Zhang X, Galardi E, Duquette M, Lawler J, Parangi S. Antiangiogenic treatment with three thrombospondin-1 type 1 repeats versus gemcitabine in an orthotopic human pancreatic cancer model. Clin Cancer Res. 2005;11(15):5622–5630. doi: 10.1158/1078-0432.CCR-05-0459. [DOI] [PubMed] [Google Scholar]
- 116.Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32(3):157–165. doi: 10.1016/j.ctrv.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 117.Yang Q, Tian Y, Liu S, Zeine R, Chlenski A, Salwen HR, Henkin J, Cohn SL. Thrombospondin-1 peptide ABT-510 combined with valproic acid is an effective antiangiogenesis strategy in neuroblastoma. Cancer Res. 2007;67(4):1716–1724. doi: 10.1158/0008-5472.CAN-06-2595. [DOI] [PubMed] [Google Scholar]
- 118.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]