

Introduction
Excessive proliferation of vascular smooth muscle cells (VSMC) contributes to the pathogenesis of many cardiovascular diseases, including atherosclerosis and pulmonary arterial hypertension (PAH). VSMC proliferation also underlies the failure of many therapies, notable examples being restenosis following coronary angioplasty, vein graft failure in patients with coronary artery bypass grafts and transplant vasculopathy. Few therapies directly target excess VSMC proliferation, in part because the underlying pathways have been unknown. Recently, several pathways of VSMC proliferation have been defined and new therapeutic targets have emerged.
An example of the power of preventing VSMC proliferation in reducing human cardiovascular disease is the rapamycin (Sirolimus)-coated coronary stent. After dozens of agents failed to prevent the 30% restenosis rate post-angioplasty, this VSMC proliferation inhibitor hit the target and restenosis rates tumbled to ~6%.1 However, rapamycin has toxicities, limiting its systemic use. Moreover, the mechanisms of accelerated VSMC proliferation may vary by disease and thus the efficacy of an anti-proliferative drug will likely be contextual (i.e. disease dependent). Understanding the pathways controlling proliferation offers hope for identifying drugs that selectively target proliferating cells. The contribution by Gizard and colleagues in the current issue of Circulation Research identifies such a pathway and suggests new therapeutic targets.2
Bruemmer’s group expands on their prior work, which showed that peroxisome proliferator-activated receptor-∝ (PPARα) activation suppresses G1→S cell cycle progression by increasing the expression of the cyclin dependent kinase (CDK) inhibitor, p16INK4a.3
Their latest study is built on solid body of knowledge of the cell cycle. While growth factors are necessary to initiate proliferation in normal cells, cells become independent of these external stimuli during the G1 phase. This point of no return is highly regulated by the interaction between cyclins, CDKs and CDK inhibitors4. In nonproliferating cells, the retinoblastoma (Rb) proteins (pRB, p107, and p130) bind to E2F transcription factors and inhibit transcription2. During proliferation, CDK-cyclin complexes phosphorylate Rb proteins, allowing release of E2F and transcription commences. However another group of molecules, the CDK inhibitors (p16INK4a, p21WAF1 and p27KIP1) can block this transcription of S phase genes, offering further opportunity for inhibiting proliferation.
Gizard et al provide a new understanding of the transcriptional regulation underlying proliferation, demonstrating the interaction of the CDK pathway and telomerase. Telomeres are the DNA TTAGGG-repeat sequences that cap and stabilize chromosomes. Traditionally thought to fend off senescence it appears increasingly likely they also regulate cell proliferation. Telomerase reverse transcriptase (TERT) is the catalytic factor that leads to telomerase activation. Relevant to its role in vascular disease, TERT is activated by mitogens, which are upregulated in diseases characterized by VSMC proliferation. There is emerging evidence that inhibiting telomerase in vivo may reduce vascular disease 5, 6.
In this context, Gizard et al define the mechanism by which PPARα impairs cell proliferation in human coronary VSMC. They demonstrate that PPARα activation inhibits mitogen-induced telomerase activity by transcriptionally repressing TERT. Their methodical approach revealed that PPARα’s effect was indirect and related to inhibition of E2F binding sites in the TERT promoter. These sites had been described previously7, but not in vascular cells. Gizard found that PPARα activation inhibits TERT transcription by blocking the binding of E2F-1 to its binding sites in the proximal TERT promoter. In addition, p16-mediates some of the repression of TERT by recruiting p107 and p130 to the proximal E2F-1 site further blocking TERT transcription. PPARα activation by fibrates inhibits telomerase activity by inducing p16 resulting in inhibition of E2F-dependent transcriptional activation of the TERT promoter. The authors present convincing loss- and gain-of-function studies to support a key role of E2F in the regulation of telomerase activity and prove that E2F is required for the repression of TERT promoter activity by PPARα. Moreover, their demonstration that fenofibric acid and gemfibrozil, inhibit telomerase activation in a femoral artery endothelial-denudation model, suggests this as a potential therapy in humans. This is feasible, since these drugs are widely used to increase high-density lipoprotein (HDL) in humans-also through a PPARα-dependent mechanism. Thus, it appears that the antiproliferative effects of PPARα are caused by impingement on the p16/Rb/E2F transcriptional cascade and are ultimately mediated by suppression of telomerase (in vitro and in vivo). What are the intersections of this work with several newly-identified pathways of VSMC proliferation (Figure)?
Interactions of other pathways regulating VSMC proliferation with the telomerase pathway.
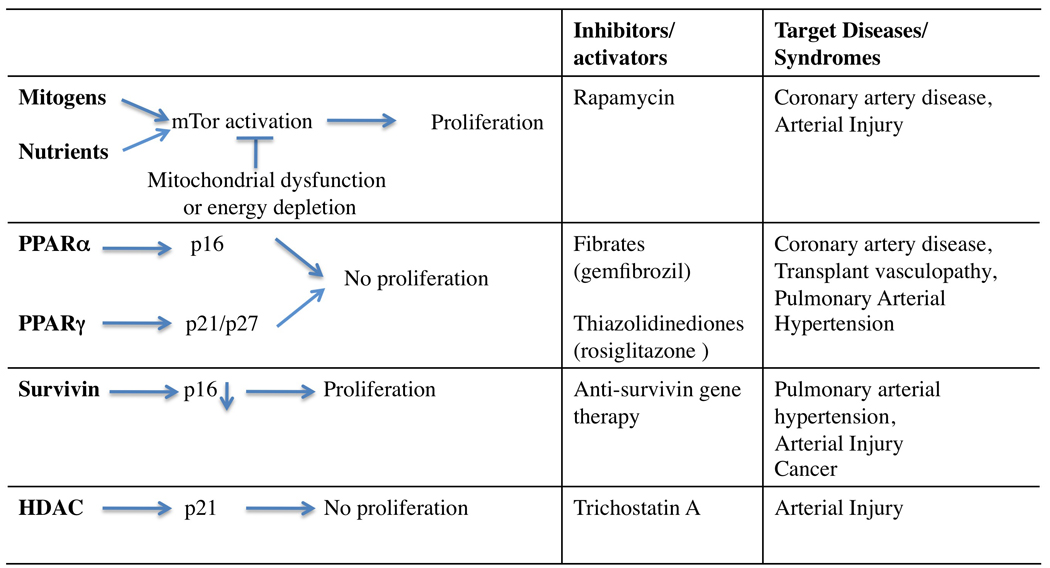
Several pathways interact to control VSMC proliferation. External mitogenic signals activate mTor, leading to proliferation. PPARα and PPARγ as well as inhibitors of survivin and HDAC increase CDK inhibitor levels.
Regulation of VSMC Proliferation
While it is not possible to discuss all molecules involved in controlling VSMC proliferation, some are noteworthy because they are recently recognized or are targeted by drugs that are either in clinical use or preclinical development. These are the low hanging fruit that may be harvested in the attempts to selectively decrease VSMC proliferation in vascular disease.
mTor (mammalian target of rapamcyin)
mTor is an ubiquitously expressed kinase that integrates cellular energy and nutrient status with external mitotic signals.8 When sufficient nutrients and appropriate mitogens are present, mTor will lead to proliferation by the expression of several proteins including cyclin D1. Mitochondrial dysfunction, energy depletion and amino acid deprivation all lead to mTor inactivation, effectively blocking proliferation.9 The interaction of mTor with mitochondria raises the possibility that changes in mitochondrial function would alter VSMC proliferation. Impaired mitochondrial function and fusion has been proposed to be responsible for the development of certain forms of pulmonary arterial hypertension10. In a potential link between this pathway and the current study, rapamycin upregulates CDK inhibitors p21WAF1 and p27KIP1 in VSMCs.11
PPARγ Agonists
Thiazolidinediones, such as rosiglitazone, are PPARγ agonists developed for the therapy of type II diabetes mellitus. They decrease VSMC proliferation by inhibiting mitogen-induced degradation of the CDK inhibitors p21WAF1 and p27KIP1.12 PPARγ agonists might have a role in preventing transplant vasculopathy and neointima formation in PAH. For example, rosiglitazone reduces both pulmonary hypertension and vascular remodeling in pulmonary hypertensive ApoE knockout mice fed a high-fat diet.13
Survivin
Survivin, a known apoptosis inhibitor, was traditionally thought to be exclusively expressed in cancers but more recently has been found in vascular injury14 and PAH15. However, survivin not only impairs apoptosis, it also increases proliferation by initiating cell cycle entry. When survivin is translocated to the nucleus it competes with CDK4 for interaction with p16INK4a16, a potential link to the pathway described by Gizard et al. Inhibition of survivin can be therapeutically exploited to prevent neointima formation14 and reverses pulmonary hypertension and vascular obstruction in experimental models.15
Pre–B-cell Colony-enhancing Factor (PBEF) and Histone Deacetylase (HDAC)
The PBEF is a regulator of SMC proliferation that increases nicotinamide phosphoribosyltransferase activity, upregulating supplies of nicotinamide adenine dinucleotide (NAD+) for the SIRT transcription regulators. SIRT family members regulate transcription and apoptosis through their ability to deacetylate histones and nonhistone proteins (e.g. p53 and TERT). PBEF is able to shift VSMC from a proliferative to a contractile phenotype. PBEF over-expression enhances cell survival whereas PBEF knockdown increases SMC apoptosis17. Through regulation of HDAC activity, PBEF modulates VSMC proliferation and survival.17 There is a potential interaction between the cyclin-CDK-telomerase pathway and HDAC. The HDAC inhibitor, trichostatin A, results in histone hypermethylation of the p21WAF1 promoter,18 leading to increased p21WAF1 expression and cell cycle arrest in a variety of cells, including VSMCs.19
Bone Morphogenetic Protein Receptor Type 2 (BMPR2)
Loss of function mutations in BMPR2 are common in familial PAH, a disease characterized by excessive VSMC proliferation, particularly in response to transforming growth factor-beta (TGFβ)20. Signalling from the BMPR2 receptor involves the SMAD signalling cascade. The connection of BMPR2 signalling to the cyclin-CDK-telomerase pathway is unknown; however BMPR2 inactivation, which decreases VSMC differentiation, does lowers p27KIP1 expression.21
Future studies
There are several areas which were not addressed by Gizard et al that merit further study.
First, TERT activity can also be regulated at the post-translational level by phosphorylation. For example, hypoxia (a well-established cause of pulmonary hypertension and excessive PASMC proliferation) increases TERT phosphorylation, which increases SMC proliferation.22 Thus, both activity and expression of TERT modulate telomerase activity and the role of “activity” deserves further study.
Second, there are many parallels between proliferation of VSMC and cancer cells23. A recent publication demonstrated reduced TERT expression in cancer cells exposed to TGFβ, which was dependent on the −252 to +3 region of the TERT promoter.24 Comparative studies of this pathway in cancer and vascular disease may be profitable.
Third, as with most pathways there is tissue heterogeneity in this pathway. Depending on cell type, E2F transcription factors can promote or inhibit TERT expression7 and different E2F transcription family members can have opposing effects on TERT promoter activity. The complexity of TERT expression needs further study to allow this mechanism to be therapeutically exploited.
Conclusion
Several signalling pathways exist in VSMCs to control proliferation (Figure 1). Although there are many upstream initiators of proliferation, there are fewer crucial downstream decision steps for determining whether VSMCs will proliferate. First, mTor activation only occurs when the cellular environment is conducive for proliferation (sufficient nutrients/energy and absence of prohibitive signals). Second, many signalling pathways, including PPARα, converge in having effects of the CDK inhibitors, including p16INK4a, p21WAF1 and p27KIP1. Third, we now have another potential common point at which to attack proliferation-TERT.
Acknowledgments
Funding: This work was supported by NIH-RO1-HL071115.
Footnotes
Disclosures: The authors have no conflicts of interest.
References
- 1.Morice MC, Serruys PW, Sousa JE, Fajadet J, Ban Hayashi E, Perin M, Colombo A, Schuler G, Barragan P, Guagliumi G, Molnar F, Falotico R. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. N Engl J Med. 2002;346(23):1773–1780. doi: 10.1056/NEJMoa012843. [DOI] [PubMed] [Google Scholar]
- 2.Gizard F, Nomiyama T, Zhao Y, Findeisen HM, Heywood EB, Jones KL, Staels B, Bruemmer D. The PPAR{alpha}/p16INK4a Pathway Inhibits Vascular Smooth Muscle Cell Proliferation by Repressing Cell Cycle-Dependent Telomerase Activation. Circ Res. 2008;103 doi: 10.1161/CIRCRESAHA.108.186205. XXX–XXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gizard F, Amant C, Barbier O, Bellosta S, Robillard R, Percevault F, Sevestre H, Krimpenfort P, Corsini A, Rochette J, Glineur C, Fruchart JC, Torpier G, Staels B. PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J Clin Invest. 2005;115(11):3228–3238. doi: 10.1172/JCI22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005;23(36):9408–9421. doi: 10.1200/JCO.2005.01.5594. [DOI] [PubMed] [Google Scholar]
- 5.Fuster JJ, Andres V. Telomere biology and cardiovascular disease. Circ Res. 2006;99(11):1167–1180. doi: 10.1161/01.RES.0000251281.00845.18. [DOI] [PubMed] [Google Scholar]
- 6.Minamino T, Komuro I. Role of telomeres in vascular senescence. Front Biosci. 2008;13:2971–2979. doi: 10.2741/2902. [DOI] [PubMed] [Google Scholar]
- 7.Won J, Yim J, Kim TK. Opposing regulatory roles of E2F in human telomerase reverse transcriptase (hTERT) gene expression in human tumor and normal somatic cells. FASEB J. 2002;16(14):1943–1945. doi: 10.1096/fj.02-0311fje. [DOI] [PubMed] [Google Scholar]
- 8.Martin KA, Blenis J. Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv Cancer Res. 2002;86:1–39. doi: 10.1016/s0065-230x(02)86001-8. [DOI] [PubMed] [Google Scholar]
- 9.Desai BN, Myers BR, Schreiber SL. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc Natl Acad Sci U S A. 2002;99(7):4319–4324. doi: 10.1073/pnas.261702698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROSHIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294(2):H570–H578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 11.Martin KA, Rzucidlo EM, Merenick BL, Fingar DC, Brown DJ, Wagner RJ, Powell RJ. The mTOR/p70 S6K1 pathway regulates vascular smooth muscle cell differentiation. Am J Physiol Cell Physiol. 2004;286(3):C507–C517. doi: 10.1152/ajpcell.00201.2003. [DOI] [PubMed] [Google Scholar]
- 12.Wakino S, Kintscher U, Kim S, Yin F, Hsueh WA, Law RE. Peroxisome proliferator-activated receptor gamma ligands inhibit retinoblastoma phosphorylation and G1--> S transition in vascular smooth muscle cells. J Biol Chem. 2000;275(29):22435–22441. doi: 10.1074/jbc.M910452199. [DOI] [PubMed] [Google Scholar]
- 13.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115(10):1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- 14.Simosa HF, Wang G, Sui X, Peterson T, Narra V, Altieri DC, Conte MS. Survivin expression is up-regulated in vascular injury and identifies a distinct cellular phenotype. J Vasc Surg. 2005;41(4):682–690. doi: 10.1016/j.jvs.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 15.McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Puttagunta L, Michelakis ED. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. 2005;115(6):1479–1491. doi: 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki A, Hayashida M, Ito T, Kawano H, Nakano T, Miura M, Akahane K, Shiraki K. Survivin initiates cell cycle entry by the competitive interaction with Cdk4/p16(INK4a) and Cdk2/cyclin E complex activation. Oncogene. 2000;19(29):3225–3234. doi: 10.1038/sj.onc.1203665. [DOI] [PubMed] [Google Scholar]
- 17.van der Veer E, Nong Z, O'Neil C, Urquhart B, Freeman D, Pickering JG. Pre-B-cell colony-enhancing factor regulates NAD+-dependent protein deacetylase activity and promotes vascular smooth muscle cell maturation. Circ Res. 2005;97(1):25–34. doi: 10.1161/01.RES.0000173298.38808.27. [DOI] [PubMed] [Google Scholar]
- 18.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97(18):10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okamoto H, Fujioka Y, Takahashi A, Takahashi T, Taniguchi T, Ishikawa Y, Yokoyama M. Trichostatin, A an inhibitor of histone deacetylase, inhibits smooth muscle cell proliferation via induction of p21(WAF1) J Atheroscler Thromb. 2006;13(4):183–191. doi: 10.5551/jat.13.183. [DOI] [PubMed] [Google Scholar]
- 20.Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, Trembath RC. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104(7):790–795. doi: 10.1161/hc3201.094152. [DOI] [PubMed] [Google Scholar]
- 21.Tada Y, Majka S, Carr M, Harral J, Crona D, Kuriyama T, West J. Molecular effects of loss of BMPR2 signaling in smooth muscle in a transgenic mouse model of PAH. Am J Physiol Lung Cell Mol Physiol. 2007;292(6):L1556–L1563. doi: 10.1152/ajplung.00305.2006. [DOI] [PubMed] [Google Scholar]
- 22.Minamino T, Mitsialis SA, Kourembanas S. Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol Cell Biol. 2001;21(10):3336–3342. doi: 10.1128/MCB.21.10.3336-3342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 24.Lacerte A, Korah J, Roy M, Yang XJ, Lemay S, Lebrun JJ. Transforming growth factor-beta inhibits telomerase through SMAD3 and E2F transcription factors. Cell Signal. 2008;20(1):50–59. doi: 10.1016/j.cellsig.2007.08.012. [DOI] [PubMed] [Google Scholar]