

Abstract
Detailed mechanistic studies on the coupling of aryl halides with thiols catalyzed by palladium complexes of the alkylbisphosphine ligand CyPF-tBu (1-dicyclohexylphosphino-2-di-tert-butylphosphinoethylferrocene) are reported. The elementary steps that constitute the catalytic cycle, i.e. oxidative addition, transmetalation and reductive elimination, have been studied, and their relative rates are reported. Each of the steps of the catalytic process occurs at temperatures that are much lower than those required for the reactions catalyzed by a combination of palladium precursors and CyPF-tBu. To explain these differences in rates between the catalytic and stoichiometric reactions, studies were conducted to identify the resting state of the catalyst of the reactions catalyzed by a combination of Pd(OAc)2 and CyPF-tBu, a combination of Pd(dba)2 and CyPF-tBu, or the likely intermediate Pd(CyPF-tBu)(Ar)(Br). These show that the major palladium complex in each case lies off of the catalytic cycle. The resting state of the reactions catalyzed by Pd(OAc)2 and CyPF-tBu was the palladium bis-thiolate complex [Pd(CyPF-tBu)(SR)2] (R = alkyl or aryl). The resting state in reactions catalyzed by Pd2(dba)3 and CyPF-tBu was the binuclear complex [Pd(CyPF-tBu)]2(μ2, η2-dba) (9). The resting state of reactions of both aromatic and aliphatic thiols catalyzed by [Pd(CyPF-tBu)(p-tolyl)(Br)] (3a) was the hydridopalladium thiolate complex [Pd(CyPF-tBu)(H)(SR)] (R= alkyl and aryl). All these palladium species have been prepared independently, and the mechanisms by which they enter the catalytic cycle have been examined in detail. These features of the reaction catalyzed by palladium and CyPF-tBu have been compared with those of reactions catalyzed by the alkylbisphosphine DiPPF and Pd(OAc)2 or Pd(dba)2. Our data indicate that the resting states of these reactions are similar to each other and that our mechanistic conclusions about reactions catalyzed by palladium and CyPF-tBu can be extrapolated to reactions catalyzed by complexes of other electron-rich bisphosphines.
Introduction
The palladium-catalyzed coupling of aryl halides with thiols has developed from the early contributions of Migita and coworkers with tin thiolates1,2 to a method for the construction of aromatic C-S bonds with significant synthetic utility.3-9,10 Most experimental work on this process has focused on expanding the substrate scope and improving the turnover numbers of the catalytic process. Most recently, the authors' group reported the most active catalysts for this process.11-13 These catalysts, which contain the alkylbisphosphine CyPF-tBu14 (1-dicyclohexylphosphino-2-di-tert-butylphosphinoethylferrocene), couple aryl bromides and aryl chlorides with aromatic and aliphatic thiols with high functional group tolerance and high turnover numbers (eq 1). The process is now compatible with esters, nitriles, amides, amines and carboxylic acids; turnover numbers approaching 100,000 were obtained for reactions of aryl bromides, and turnover numbers approaching 10,000 were obtained for reactions of aryl chlorides.
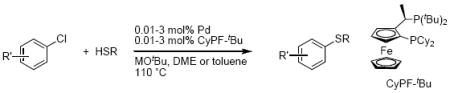 |
(1) |
Less effort has been spent to understand the mechanism of the coupling to form carbon-sulfur bonds, although individual steps of a likely mechanism have been reported (Figure 1). For example, studies from the authors' laboratory led to the observation of reductive eliminations from several palladium thiolate complexes to form the carbon-sulfur bond in thioethers.15,16 These studies established the influence of the electronic properties of the aryl and thiolate ligands on this reaction. The influence of several bidentate ligands, such as DPPE (1,2-bis(diphenylphosphino)ethane), DPPP (1,3-bis(diphenylphosphino)propane), DPPBz (1,2-bis(diphenylphosphino)benzene), DPPF (1,1′-bis(diphenylphosphino)ferrocene) and TRANSPHOS (2,11-bis(diphenylphosphinomethyl) benzo[c]phenanthrene) on the rate of reductive elimination were also examined.
Figure 1.

General mechanism for the palladium-catalyzed coupling of aryl halides with thiols.
In addition to these studies on elementary reactions, kinetic studies on a series of stoichiometric reactions proposed to constitute the coupling of phenyl iodide with a cysteine-derived thiol catalyzed by the combination of Pd(dba)2 and DPPF were reported by Campagne and Jutand.17 In these studies, no data that correlate the rates of the stoichiometric reactions and the rate of the catalytic process were reported. This connection can be complex, particularly for reactions of thiols that can become strong ligands for palladium and lead to catalyst deactivation.
One might anticipate several differences between the reactions catalyzed by complexes containing aromatic bisphosphines used previously and reactions catalyzed by complexes containing sterically hindered aliphatic bisphosphines developed more recently. For example, reductive elimination to form thioethers from arylpalladium thiolate complexes could be slower from the complexes containing the more electron-donating bisphosphines, and the steric hindrance of the ligand could lead to slower transmetalation. Moreover, the properties of the ligand could affect the formation of the active palladium(0) complex and could open new pathways to siphon the palladium from the catalytic cycle. The absence of data on the rates of oxidative addition of aryl halides, transmetalation to form palladium thiolates and reductive eliminations to form C-S bonds from CyPF-tBu-ligated palladium complexes prevents firm predictions of the relative rates of different steps of this catalytic process.
We report detailed mechanistic studies on the palladium-catalyzed coupling of aryl halides with thiols catalyzed by palladium complexes of CyPF-tBu and a related alkylbisphosphine DiPPF (1,1′-bis(diisopropylphosphino)ferrocene). Studies on the stoichiometric reactions involved in the catalytic cycle establish the relative rates of the individual steps. These data show that each step occurs at room temperature, whereas the reported catalytic reactions required hours at 110 °C to occur to completion. We also report the identification of the resting state of the reactions catalyzed by the combination of Pd(OAc)2 and CyPF-tBu, the combination of Pd(dba)2 and CyPF-tBu, or by Pd(CyPF-tBu)(Ar)(X). In all cases, the resting state lies off of the catalytic cycle, and we reveal the mechanism by which the resting states enter the cycle. Finally, we report qualitative mechanistic studies on the reactions catalyzed by the combination of DiPPF and Pd(OAc)2 or Pd(dba)2.3 These data indicate that the conclusions from studies of the CyPF-tBu-Pd catalyst can be extrapolated to reactions catalyzed by complexes of other electron-rich bisphosphines.
Results
1. Stoichiometric reactions of isolated complexes of the bidentate ligand CyPF-tBu
a. Oxidative addition of aryl chlorides
Oxidative addition of aryl chlorides to mono- or bisphosphine Pd(0) complexes is generally the turnover-limiting step of cross-coupling processes conducted with this class of aryl halide. No data have been published on the oxidative additions of chloroarenes to palladium(0) complexes of CyPF-tBu. However, Roy and Hartwig18,19 reported that the oxidative addition of phenyl tosylate to Pd(CyPF-tBu)[P(o-tolyl)3] occurs in less than 5 minutes at room temperature. Thus, one might expect that the oxidative addition of chloroarenes would be fast and would not be turnover limiting in the coupling of aryl halides with thiols at elevated temperatures.
To gather firm data on this issue, the reactions of 4-chlorotoluene with Pd(0) complexes of CyPF-tBu were studied. We first studied oxidative additions to Pd(CyPF-tBu)[P(o-tolyl)3] (1) generated in situ at room temperature from CyPF-tBu and Pd[P(o-tolyl)3]2.19 Subsequent treatment of this complex with 4-chlorotoluene afforded the corresponding arylpalladium chloride complex 2 in 91% yield in less than 15 min (eq 2).
| (2) |
Complex 2 was isolated and characterized by 31P{1H} and 1H NMR spectroscopy, as well as elemental analysis. Pd(CyPF-tBu)(p-tolyl)(Cl) (2) is a four-coordinate complex in which two isomers could be generated. However, only one isomer was detected by NMR spectroscopy. The arylpalladium bromide complex containing the same ligand exists as the isomer in which the palladium-bound aryl group is bound cis to the smaller phosphino group.18 We presume that the geometry of the arylpalladium chloride complex is the same as that of the arylpalladium bromide complex.
As expected,20,21 the oxidative addition of bromotoluene to the P(o-tolyl)3-stabilized Pd(0) complex 1 occurred faster than the oxidative addition of chlorotoluene. The reaction of the same Pd(0) complex 1 with 4-bromotoluene in C6D6 afforded the arylpalladium bromide complex 3a in 96% yield in less than 1 min at room temperature. Complex 3a was synthesized independently in 74% yield by allowing {Pd(μ-Br)(p-tolyl)[P(o-tolyl)3}2 to react with 2 equiv of CyPF-tBu in THF at room temperature.
b. Relative rates of transmetalation and C-S bond-forming reductive elimination of thioethers
To determine if these fast rates for oxidative addition caused a change in the typical turnover-limiting step for coupling of chloroarenes, we studied the reactions of the CyPF-tBu-ligated arylpalladium halide complexes with alkali metal thiolates and with the combination of thiols and tert-butoxide base. The reactions between arylpalladium halide complexes 2, 3a, and 3b with alkyl and aryl thiols in the presence of NaOtBu afforded the thioethers in high yields after less than 5 min at room temperature (eq 3).22 The thiol was added to a mixture of the arylpalladium halide complex, the alkali metal salt and P(o-tol)3. The Pd(0) product from these reactions was the P(o-tolyl)3-stabilized Pd(0) complex 1. The product was obtained in 80-99% yields,23 as determined by 31P{1H} NMR spectroscopy using an internal standard.
| (3) |
The reactions between sodium thiolates and arylpalladium halide complexes 2, 3a and 3b in the presence of P(o-tolyl)3 are summarized in eq 4. These reactions afforded the corresponding thioethers in 99% yield after 5 min at ambient temperature.24 Again, the palladium product from these reactions was the P(o-tolyl)3-stabilized Pd(0) complex 1, as determined by 31P{1H} NMR spectroscopy.
| (4) |
From these observations, it is clear that arylpalladium halide complexes 2, 3a and 3b are readily transformed into thiolate complexes by metathetical substitution reactions, despite the steric properties of CyPF-tBu. Moreover, these reactions imply that C-S bond-forming reductive elimination occurs rapidly at room temperature and in high yields from the arylpalladium thiolate complexes, as shown in eq 3 and 4. Even complex 3b, which possesses an electron rich aryl group bound to palladium25 underwent reductive elimination under mild conditions.
c. Independent synthesis and reactivity of the arylpalladium thiolate complexes
Because the arylpalladium thiolate complexes 4a and 4b were unstable at room temperature, they were generated and characterized in solution by NMR spectroscopy at low temperature. We envisioned forming complexes 4a and 4b independently by σ-bonded ligand exchange between the arylpalladium hydroxo complex [Pd(CyPF-tBu)(p-tolyl)(OH)] (5) and the corresponding thiol. The desired terminal palladium hydroxo complex26-30 5 was prepared in 60% yield by ligand substitution between 2 equiv of CyPF-tBu and the dimeric hydroxo complex {Pd(PPh3)(p-tolyl)(μ-OH)}231 in THF at room temperature.
| (5) |
The reaction of 1.1 equiv of thiol with the arylpalladium hydroxo complex 5 at -40 °C in THF-d8 formed the corresponding palladium tert-butyl- and anisyl-substituted thiolate complexes 4a and 4b, as shown in eq 5. These complexes were formed in less than 5 min in 94 and 99% yield, respectively, as determined by 1H NMR spectroscopy with 1,3,5-trioxane as internal standard. The equilibrium between the hydroxo complex and the thiolate complex lay far in the direction of the thiolate; no remaining hydroxo complex was detected.32
This process allowed for characterization of the thiolate complexes at low temperature because the proton transfer was fast, and water was the only byproduct. The arylpalladium thiolate complexes 4a and 4b were identified by 1H and 31P{1H} NMR spectroscopy at -40 °C. In the 31P{1H} NMR spectrum, the two sets of doublets at 66.0 and 19.5 ppm, and 64.4 and 25.0 ppm corresponding to the starting hydroxo compound 5 were replaced by two doublets at 68.5 and 12.5 ppm for 4a, and two doublets at 73.2 and 16.7 ppm for 4b. In the 1H NMR spectrum, the resonance corresponding to the hydroxo proton decayed along with the p-tolyl resonances corresponding to the palladium-bound aryl group. For the reaction yielding 4a, these resonances were replaced by a single tert-butyl resonance for the tert-butylthiolate group (δ 0.90) and a new set of p-tolyl signals corresponding to the palladium-bound aryl group in the thiolate complex. For the reaction yielding 4b, these resonances were replaced by a new singlet assigned to the methoxy group (δ 3.62) and new set of signals corresponding to the p-tolyl group.
Arylpalladium thiolate complexes 4a and 4b were unstable for even short periods of time at temperatures above -20 °C. The terminal hydroxo complex 5 reacted with 4-methoxybenzenethiol in less than 1 min at room temperature to form 95% yield of the thioether, as determined by GC using dodecane as an internal standard and 97% of a 10:1 isomeric mixture of the terminal hydridopalladium thiolate complex Pd(CyPF-tBu)(H)[S(C6H4-4-OMe)]33 (6), as determined by 31P{1H} NMR spectroscopy using an internal standard (eq 6). These data imply that reductive elimination from the presumed arylpalladium thiolate species formed from proton transfer between the thiol and the hydroxo complex occurs rapidly at room temperature and that the Pd(0) product reacts with the excess thiol to form complex 6.
| (6) |
Hydridopalladium thiolate complex 6 was independently synthesized in 60% yield as a 7.6 : 1 ratio of isomers by treating Pd(CyPF-tBu)[P(o-tolyl)3] (1) with 4-methoxybenzenethiol at room temperature. This material was characterized by 1H and 31P{1H} NMR spectroscopy, as well as elemental analysis. In the 31P{1H} NMR spectrum, a set of doublets at 101.2 and 11.8 ppm was observed for the major isomer and a set of doublets at 74.5 and 35.3 ppm was observed for the minor isomer. In the 1H NMR spectrum, the hydride resonances for the major and minor isomers appeared at -6.74 and -7.78 ppm, respectively.
2. Identification of the resting state of the palladium catalyst ligated by CyPF-iBu
Studies in section 1 on the individual reactions that constitute a likely catalytic cycle for the coupling of aryl halides with thiols showed that all of the reactions occur at room temperature. Yet, the reported conditions for the catalytic process involve elevated temperatures. To understand this discrepancy in rates, we studied the identity of the palladium complexes in solution. These complexes were identified by monitoring of the catalytic reactions by 31P{1H} NMR spectroscopy and by independently preparing the observed species. These catalytic reactions were conducted with 4-chlorotoluene and either 4-methoxythiophenol or 2-propanethiol in the presence of KOtBu and the combination of 10 mol % CyPF-tBu and 10 mol % of either Pd(OAc)2 or Pd(dba)2 (eq 7).
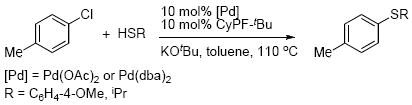 |
(7) |
a. The resting state in reactions catalyzed by a combination of Pd(OAc)2 and CyPF-tBu
The reaction of 4-chlorotoluene with 4-methoxybenzenethiol in the presence of KOtBu catalyzed by a combination of Pd(OAc)2 and CyPF-tBu at 110 °C contained a single soluble, phosphine-ligated palladium species, as determined by 31P{1H} NMR spectroscopy. The 31P{1H} NMR spectrum consisted of a doublet at 86.6 ppm (J = 29.1 Hz) and a broad resonance at 19.0 ppm. These signals correspond to the palladium bis-thiolate complex 7, which was independently prepared from a combination of (CyPF-tBu)PdCl2 and 4-methoxybenzenethiol in the presence of NEt3 in THF at room temperature (eq 8).
| (8) |
Likewise, the major species in solution in the reaction of 4-chlorotoluene with 2-propanethiol in the presence of KOtBu catalyzed by a combination of Pd(OAc)2 and CyPF-tBu at 100 °C was the palladium bis-thiolate complex 8. Complex 8 was prepared independently in 62% isolated yield from the reaction of (CyPF-tBu)PdCl2 and 2-propanethiol in the presence of NaOtBu in THF at room temperature (eq 8). The 31P{1H} NMR spectrum of complex 8 consisted of a doublet at 82.3 ppm (J = 26.1 Hz) and a broad signal at 14.3 ppm.
b. The resting state in reactions catalyzed by the combination of Pd(dba)2 and CyPF-tBu
The reaction of 4-chlorotoluene and 4-methoxybenzenethiol in the presence of KOtBu catalyzed by a combination of Pd(dba)2 and CyPF-tBu at 110 °C was monitored by 31P{1H} NMR spectroscopy. Instead of bis-thiolate complex 7, which was observed in the reactions catalyzed by a combination of Pd(OAc)2 and CyPF-tBu, a set of isomers of the binuclear Pd(0) complex possessing the general structure [(CyPF-tBu)Pd]2(dba) (9) were identified. Likewise, isomeric dba complexes 9 were the major species in solution in the reaction of 4-chlorotoluene and 2-propanethiol in the presence of KOtBu catalyzed by a combination of Pd(dba)2 and CyPF-tBu at 100 °C.
Dinuclear palladium(0) dba complex 9 was generated independently as a complex mixture of isomers in 39% yield by allowing (CyPF-tBu)Pd(dba)34 to react with NaOtBu and isopropylamine for 3 h at 100 °C. A related binuclear bisphosphine-palladium complex of dba, [(BINAP)Pd]2(dba), has been isolated and reported to be precatalyst for the palladium-catalyzed amination of aryl halides.35
c. Comparison of the rate of catalytic reactions conducted with Pd(OAc)2/CyPF-tBu, Pd(dba)2/CyPF-tBu and [Pd(CyPF-tBu)(p-tolyl)(Br)] as precatalysts
To gain more direct information on the rate of the actual catalytic process in the absence of the species formed from the precatalyst, we conducted reactions initiated with catalytic amounts of the proposed intermediate [Pd(CyPF-tBu)(p-tolyl)(Br)] (3a). Table 1 shows a comparison of the rates of the reactions of 4-chlorotoluene with 4-methoxybenzenethiol catalyzed by a combination of Pd(OAc)2 or Pd(dba)2 with CyPF-tBu vs 3a. When 1 mol % of the combination of Pd(OAc)2 and CyPF-tBu or Pd(dba)2 and CyPF-tBu were used as catalyst, no reaction occurred after 24 h at room temperature. On the other hand, when 1 mol % 3a was used as catalyst, full conversion of 4-chlorotoluene to the thioether occurred after 24 h at room temperature. Further studies on the scope of couplings of aryl chlorides with thiols to form thioethers will be reported in due course.
Table 1.
Comparison of the yield of the coupling of aryl halides with 4-methoxythiophenol after 24 h.
 | |||
|---|---|---|---|
| Entry[a] | X | [Pd] | Yield (%)[b] |
| 1 | Br | Pd(OAc)2, CyPF-tBu | 0 |
| 2 | Br | Pd(dba)2, CyPF-tBu | 0 |
| 3 | Br | Pd(CyPF-tBu)(p-tolyl)(Br) | 99 |
| 4 | Cl | Pd(OAc)2, CyPF-tBu | 0 |
| 5 | Cl | Pd(dba)2, CyPF-tBu | 0 |
| 6 | Cl | Pd(CyPF-tBu)(p-tolyl)(Br) | 99 |
Reaction conditions: ArX (1 mmol), thiol (1 mmol), KOtBu (1.4 mmol), toluene (1.5 mL), 24 h.
Determined by GC using dodecane as internal standard.
d. Reactions catalyzed by [Pd(CyPF-tBu)(p-tolyl)(Br)] (3a)
Having shown that the coupling of haloarenes with aromatic thiols is faster when initiated by a complex that lies directly on the catalytic cycle, we conducted experiments to identify the major species in this catalytic system. The reaction of 4-chlorotoluene with 4-methoxybenzenethiol in the presence of KOtBu (1.4 equiv) and 10 mol % of arylpalladium bromide complex 3a as catalyst was monitored by 31P{1H} NMR spectroscopy. The hydridopalladium thiolate complex Pd(CyPF-tBu)(H)[S(C6H4-4-OMe)] (6), which existed predominantly as one isomer, was the major species observed in the reactions conducted at room temperature. We presume this isomer contains the hydide cis to the larger di-tert-butylphosphino group.
A similar complex was observed during reactions of aliphatic thiols. The hydridopalladium thiolate complex [Pd(CyPF-tBu)(H)(SiPr)] (10) was the only palladium complex observed by 31P{1H} NMR spectroscopy in the reaction of 4-chlorotoluene with the aliphatic thiol 2-propanethiol catalyzed by 5 mol% 3a in toluene at room temperature. Complex 10 was isolated in 14% yield as a 15:1 mixture of isomers by reaction of Pd[P(o-tolyl)3]2 with CyPF-tBu in THF at room temperature, followed by subsequent treatment with 2-propanethiol. The 31P{1H} NMR spectrum of complex 10 consisted of a set of doublets at 99.8 and 13.1 ppm for the major isomer and two broad signals at 76.3 and 32.8 ppm, for the minor isomer. The 1H NMR spectrum contained resonances at -6.79 and -7.83 ppm for the hydrides of the major and minor isomers, respectively.
3. Reactions of hydridopalladium thiolate and palladium bis-thiolate complexes ligated by CyPF-tBu
Although Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) and Pd(CyPF-tBu)(H)[S(C6H4-4-OMe)] (6) are unlikely to lie directly on the catalytic cycle, and bis-thiolate complex 7 is not kinetically competent to be an intermediate in the catalytic cycle, these compounds could react with arylpalladium halides in the presence or absence of base to form thioether products. Yamamoto36 previously described the reaction of (bpy)Ni(SR)2 with aryl iodides to form thioethers. However the mechanism of this process was not evaluated, and analogous reactions with palladium thiolates have not been reported.
a. Reaction of Pd(CyPF-tBu)(H)[S(C6H4-4-OMe)] (6) with aryl halides
To test if hydridopalladium thiolate complex 6 reacts with haloarenes to form arylpalladium halide complexes 3 and thioethers, we conducted the reaction of 6 with 4-bromotoluene and NaOtPent in toluene. This reaction formed the arylpalladium bromide complex 3a in quantitative yield and the corresponding thioether in 88% yield after 1 h at 100 °C. In addition, complex 6 was converted to the P(o-tolyl)3-stabilized Pd(0) species 1 in quantitative yield in less than 1 h at 100 °C after treatment with a base in the presence of P(o-tolyl)3, as shown in Scheme 1.
Scheme 1.

b. Assessment of the catalytic competence of Pd(CyPF-tBu)(H)[S(C6H4-4-OMe)] (6)
The reactions of hydrido thiolate complex 6 with haloarenes to form thioethers and the reaction with base to form P(o-tolyl)3-stabilized Pd(0) species 1 implies that hydridopalladium thiolate complexes should be competent as precatalyst for the coupling of aryl halides with thiols. To test this hypothesis, the coupling of a chloroarene with an aromatic thiol was conducted with hydridopalladium arenethiolate complex 6 as catalyst. Indeed, the reaction of 4-chlorotoluene with 4-methoxybenzenethiol in the presence of KOtBu as base and 1 mol % 6 in toluene formed the coupled thioether product in 83% yield after 24 h at room temperature, as determined by GC analysis using an internal standard. Thus, these hydrido thiolate complexes and base appear to give rise to the Pd(CyPF-tBu) intermediate that lies on the cycle and adds haloarenes as the first step of the cycle.
c. Reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with aryl halides
The reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with 4-bromotoluene formed Pd(CyPF-tBu)(Br)2 (11) in 99% yield, together with the corresponding thioether in 99% yield after 12 h at 100 °C (eq 9). During this reaction, Pd(CyPF-tBu)[S(C6H4-4-OMe)](Br) (12) accumulated as an intermediate. Intermediate 12 was prepared independently by comproportionation of Pd(CyPF-tBu)(Br)2 and Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 at 100 °C. This comproportionation led to a 3.8 : 1 : 1 ratio of 12 : 7 : 11 after 4 h. This reaction of bis-thiolate complex 7 with chlorotoluene is much slower than that of hydridopalladium thiolate complex 6 with chlorotoluene and base.
| (9) |
The bis-thiolate complex 7 also reacted with aryl chlorides. The reaction of 7 with 4-chlorotoluene formed Pd(CyPF-tBu)(Cl)2 in 72% yield, together with the corresponding thioether in 85% yield after 48 h at 100 °C. The rate of this reaction is considerably slower than that of 7 with 4-bromotoluene. Under the same set of reaction conditions, ([Pd(CyPF-tBu)[S(C6H4-4-OMe)]2] = 2.6 × 10-2 M and [ArX] = 1.4 M), the starting bis-thiolate complex had completely decayed after 3 h of reaction with 4-bromotoluene, as determined by 31P{1H} NMR spectroscopy, whereas only 58% of this complex had decayed after 25 h of reaction with 4-chlorotoluene.
The addition of a base significantly increased the rate of the reaction of the palladium bis-thiolate complex 7 with aryl halides. The acceleration of the reaction of 7 with p-tolyl chloride was particularly evident. The reaction of [Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with p-tolyl chloride in the presence of 0.13 M of NaOtPent formed the thioether in 99% yield after 40 min at 100 °C, as determined by GC with an internal standard.37 In the absence of base, the yield of the thioether was only 32% after 25 h ([Pd(CyPF-tBu)[S(C6H4-4-OMe)]2] = 2.6 × 10-2 M and [4-chlorotoluene] = 1.4 M). The alkoxide complex Pd(CyPF-tBu)(OtPent)(p-tolyl) (13) was observed by 31P{1H} NMR spectroscopy as the palladium product of the reaction of the bis-thiolate complex 7 with p-tolyl chloride in the presence of NaOtPent (eq 10). Although we have not been able to isolate complex 13 in pure form, the closely related arylpalladium butoxide Pd(CyPF-tBu)(OtBu)(p-tolyl) (14) was isolated from the reaction of Pd(CyPF-tBu)(Cl)(p-tolyl) with NaOtBu in toluene solvent, and the 31P NMR spectra of 13 and 14 were nearly identical.38
| (10) |
d. Mechanistic studies on the reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with aryl halides
i. Kinetic study on the reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with 4-bromotoluene
In order to determine the mechanism by which the bis-thiolate complex 7 reacts with aryl halides and potentially enters the catalytic cycle, a series of kinetic studies were conducted on the reaction of 7 with 4-bromotoluene. These kinetic studies were conducted by monitoring the reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 with an excess of 4-bromotoluene at 100 °C in toluene-d8 solvent by 31P{1H} NMR spectroscopy over at least three half-lives. Excellent fits to a first order decay of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 were observed.
To determine the order of the reaction in aryl bromide, observed rate constants were measured for reactions conducted with concentrations of 4-bromotoluene varying from 0.27 to 1.4 M and a constant concentration of bis(4-methoxyphenyl) disulfide of 5.6 × 10-4 M. A plot of 1/kobs vs 1/[4-bromotoluene] is shown in Figure 2. These data reveal a positive dependence of the reaction on the concentration of this reagent, with a non-zero y-intercept that typically corresponds to the rate constant for a step that occurs prior to reaction with the incoming reagent.
Figure 2.
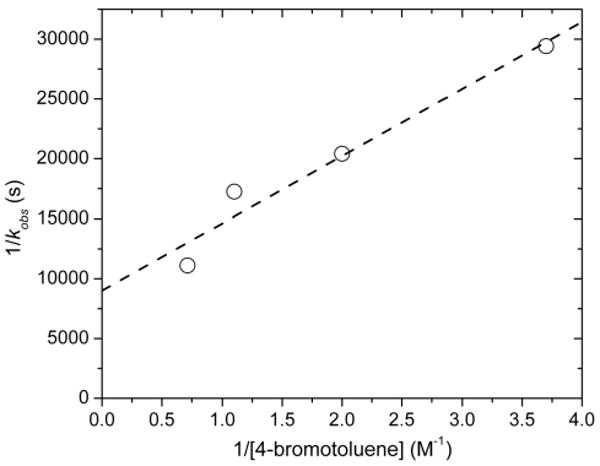
Plot of 1/kobs vs 1/[4-bromotoluene] for the reaction of the bis-thiolate complex 7 with 4-bromotoluene.
We considered that the initial step of the reaction could involve reversible reductive elimination of disulfide to form a Pd(0) species. To test this hypothesis, we conducted the reactions of 1.4 M 4-bromotoluene in the presence of 0–7.5 × 10-3 M added bis(4-methoxyphenyl) disulfide. A linear plot of 1/kobs vs [{S(C6H4-4-OMe)}2] shown in Figure 3 indicated that the reaction is inverse first order in bis(4-methoxyphenyl) disulfide.
Figure 3.
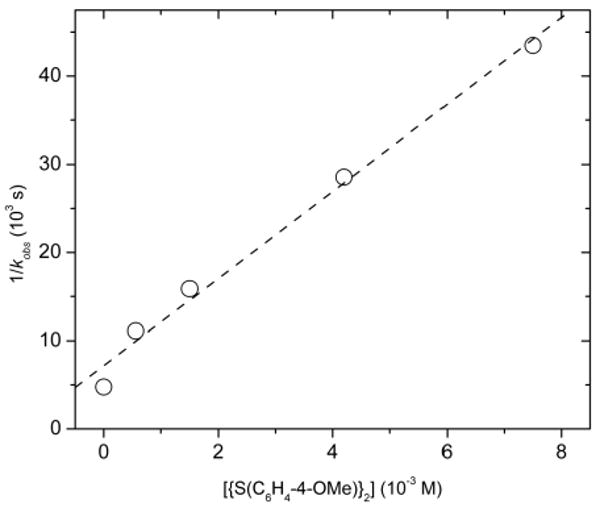
Plot of 1/kobs vs [{S(C6H4-4-OMe)}2] for the reaction of bis-thiolate complex 7 with 4-bromotoluene.
ii. Reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with 4-chlorotoluene
To determine if the kinetic data for the reactions of the bis-thiolate complex with chloroarenes parallel those for the reactions with bromoarenes, we conducted several qualitative rate measurements. The reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 with 4-chlorotoluene was conducted side by side in the presence and absence of added disulfide. Like the reactions with bromoarenes, the reactions with chloroarenes were inhibited by the added disulfide. As shown in Table 2, the reaction conducted without added disulfide occurred to higher conversions at various time points than did reactions conducted in the presence of 2.2 × 10-2 M of bis(4-methoxyphenyl) disulfide.
Table 2.
Influence of the addition of bis(4-methoxyphenyl) disulfide in the reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 with 4-chlorotoluene.
| t (min) | Conversion (%)[b] | |
|---|---|---|
| [(SR)2] = 0 | [(SR)2] = 0.022 M | |
| 560 | 17 | 5 |
| 1169 | 43 | 18 |
| 1500 | 57 | 25 |
Conditions: [Pd(CyPF-tBu)[S(C6H4-4-OMe)]2] = 2.6 × 10-2 M, [4-bromotoluene] = 1.4 M, 100 °C, toluene.
Determined by 31P{1H} NMR spectroscopy using PMes3 as internal standard.
iii. Probes for an equilibrium between Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) and Pd(CyPF-tBu)
To assess whether the initial step of the reaction of the bis-thiolate 7 with haloarenes was reductive elimination of disulfide to form a Pd(0) species, experiments were conducted to probe for a potential equilibrium between the bis-thiolate complex 7 and the combination of the disulfide and a Pd(0) complex containing the fragment Pd(CyPF-tBu). To do so, the reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with trans-stilbene to form the Pd(0) complex [Pd(CyPF-tBu)(trans-stilbene)] (15)34 was examined (eq 11). The oxidative addition of bis(4-methoxyphenyl) disulfide to stilbene complex 15 was complete at room temperature in less than 1 h. In contrast, treatment of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with trans-stilbene at 100 °C for prolonged times (>24 h) did not consume the bis-thiolate complex. These data show that the reductive elimination of disulfide from complex 7 to form the stilbene complex is disfavored thermodynamically.
 |
(11) |
However, the equilibrium between the bis-thiolate complex 7 and the Pd(0) stilbene complex 15 could be displaced if the reaction were conducted in the presence of a species that would consume the disulfide. It is known that the S-S bond in disulfides is cleaved by reaction with nucleophiles and bases, such as alkoxides.39,40 Therefore, we conducted the stoichiometric reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with NaOtPent and an excess of trans-stilbene in toluene at 100 °C (eq 12). Under these conditions, 29% yield of the Pd(0) stilbene complex 15 was observed after 6 h as part of a mixture consisting of 7 and 15 in a 2.8 : 1 ratio. Even in the presence of 9 equiv of alkoxide base, the reaction occurred to only 38% conversion after 20 h. The products of the reaction of bis(4-methoxyphenyl) disulfide with the base could not be identified by GC/MS analysis.
 |
(12) |
In the absence of base to cleave the S-S bond, we envisioned that the disulfide generated from reductive elimination in complex 7 could be trapped by reaction with an arylpalladium halide complex. To test this hypothesis, we conducted the reaction of the isolated arylpalladium bromide complex 3a with bis(4-methoxyphenyl) disulfide at 100 °C. As shown in eq 13, this transformation provided the diaryl thioether in 88% yield, together with 91% yield of a 3.7:1:1 mixture of complexes 12, 7 and 11. Little reaction was observed between complex 3a with bis(4-methoxyphenyl) disulfide at room temperature. These data indicate that the disulfide can react with an arylpalladium halide complex, such as 3a, to form the thioether product by some mechanism, but that this reaction requires elevated temperatures.
| (13) |
4. Reaction of [Pd(CyPF-tBu)]2(dba) (9) with aryl chlorides
When a combination of Pd(dba)2 and CyPF-tBu is used as precatalyst for the reaction of 4-chlorotoluene with 4-methoxybenzenethiol in the presence of KOtBu in toluene at 110 °C, the major palladium species in solution are the isomeric dinuclear, palladium dba adducts [Pd(CyPF-tBu)]2(dba) (9). To determine the relative rate of oxidative addition to this species and the catalytic coupling process, we examined qualitatively the rate of the stoichiometric reaction of 9 with an aryl chloride.
The reaction of dba complex 9 with 4-chlorotoluene in toluene was monitored at 60 °C. After 24 h, no reaction was observed by 31P{1H} NMR spectroscopy using an internal standard. Under the same conditions, but at 100 °C, the reaction proceeded slowly. After 1 h at 100 °C, 26% of the arylpalladium chloride complex 2 was observed, together with the starting complex 9 and Pd(CyPF-tBu)(dba). After 20 h at this temperature, complex 2 formed in 43% yield, and the solution contained a 2.8:1 ratio of 2 to the monomeric palladium(0) species Pd(CyPF-tBu)(dba).
5. Comparison of the stoichiometric reactions of DiPPF-ligated palladium complexes to those of CyPF-tBu-ligated palladium complexes
To determine if these processes are specific to the CyPF-tBu-Pd catalyst or if they occur with different palladium catalysts possessing properties related to those of CyPF-tBu, we conducted mechanistic experiments on the reactions catalyzed by the combination of palladium precursors and DiPPF (diisopropylphosphinoferrocene). Previous studies have shown that the combination of Pd(OAc)2 and DiPPF catalyzes the coupling of aromatic and aliphatic thiols with aryl bromides and electron poor aryl chlorides, albeit with rates that are slower than those of reactions catalyzed by the CyPF-tBu-Pd system.3
a. C-S bond-forming reductive elimination from DiPPF complexes of palladium
First, we compared the rates of reductive elimination from arylpalladium thiolate complexes ligated by DiPPF to those of reductive elimination from analogous complexes ligated by the CyPF-tBu. The arylpalladium bromide complex Pd(DiPPF)(C6H4-4-Me)(Br) (16) required for these studies was prepared by reaction of DiPPF with {Pd[P(o-tolyl)3](Ar)(μ-Br)}2 (Ar = C6H4-4-Me). Treatment of 16 with several thiols at room temperature for 2 h in the presence of NaOtBu and added DiPPF led to the formation of the corresponding free thioethers in 78-89% yields, as determined by GC analysis with an internal standard (eq 14). In these reactions, two DiPPF-Pd(0) complexes were formed in a 2:1 ratio, as determined by 31P{1H} NMR spectroscopy. A broad singlet at 21.0 ppm in the 31P{1H} NMR spectrum corresponded to the more abundant species, and a doublet at 21.0 and a triplet at 33.9 ppm corresponded to the less abundant species. These DiPPF-Pd(0) complexes were generated independently by treating Pd[P(o-tolyl)3]2 with 4.4 equiv of DiPPF in THF at room temperature. Based on the identity and corresponding 31P{1H} NMR spectra of known palladium(0) complexes of DPPF,41 we assign the structures of these Pd(0) complexes of DiPPF to be Pd(DiPPF)2 (17) and [(DiPPF)Pd]2(μ-DiPPF) (18).
| (14) |
The arylpalladium thiolate complexes of DiPPF are not stable at room temperature. Consequently, these complexes were prepared and characterized at -20 °C (eq 15). The reaction of the arylpalladium hydroxo complex 19 (prepared in 49% yield by reaction of {Pd(PPh3)(p-tolyl)(μ-OH)}231 with 2 equiv of the DiPPF ligand in THF at room temperature) with 1-1.1 equiv of the thiols at -20 °C in THF-d8 formed the corresponding arylpalladium thiolate complexes 20a and 20b in 96-99% yield, as determined by 1H NMR spectroscopy using 1,3,5-trioxane as internal standard.
| (15) |
Arylpalladium thiolate complexes 20a and 20b were identified by 1H and 31P{1H} NMR spectroscopy at -20 °C. The 31P{1H} NMR spectra for the arylpalladium thiolate complexes consisted of two doublets at 27.7 and 24.6 ppm for 20a or of two doublets at 32.2 and 26.2 ppm for 20b. The 1H NMR spectrum of 20a contained a single tert-butyl resonance for the tert-butylthiolate group (δ 0.95) and a new set of p-tolyl signals corresponding to the palladium-bound aryl group. In addition to these signals, the c NMR spectrum of 20b contained a singlet due to the methoxy group (δ 3.61) of the p-anisyl ligand.
Arylpalladium thiolate complexes ligated by DiPPF 20a and 20b underwent reductive elimination more slowly than the analogous CyPF-tBu complexes 4a and 4b. DiPPF-ligated arylpalladium thiolate complex 20b was generated from hydroxide complex 19 in THF-d8 at -20 °C. Reductive elimination of thioether from 20b was observed after warming the solution. Only 7% of the thioether was observed after 20 min at 5 °C. Reductive elimination from 20b occurred in 25% yield after 5 min and in 71% yield after 35 min at room temperature. As noted in section 1, the analogous reductive elimination from the CyPF-tBu complex was complete within 5 min at room temperature.
b. Determination of the resting state in reactions catalyzed by palladium complexes of DiPPF
i. Monitoring of the reactions of 4-chlorotoluene with thiols catalyzed by a combination of Pd(OAc)2 and DiPPF
In order to identify the resting state of the catalyst in reactions catalyzed by DiPPF-ligated palladium, reactions between 4-chlorotoluene and 4-methoxythiophenol in the presence of NaOtBu in dioxane at 110 °C were conducted with 10% of DiPPF and the palladium precatalysts Pd(OAc)2 or Pd(dba)2 (eq 16). A single species was observed by 31P{1H} NMR spectroscopy during the reaction of 4-chlorotoluene with 4-methoxybenzenethiol in the presence of NaOtBu and catalytic amounts of Pd(OAc)2 and DiPPF (eq 16). The major palladium complex in solution was the palladium bis-thiolate complex Pd(DiPPF)[S(C6H4-4-OMe)]2 (21). Complex 21 was independently prepared in 52% yield by treating Pd[P(o-tolyl)3]2 with DiPPF, followed by bis(4-methoxyphenyl) disulfide (see supporting information for characterization).
| (16) |
Like CyPF-tBu-ligated bis-thiolate 7, DiPPF-ligated bis-thiolate 21 reacted with 4-bromotoluene in toluene at 100 °C to form the palladium dibromide species Pd(DiPPF)(Br)2 (23) and the corresponding thioether after 12 h. The yield of thioether was 71%; the yield of dibromide 23 could not be quantified because most of this species precipitated from the reaction solution. However, dibromide 23 was prepared independently in 95% yield by the reaction of Pd(CH3CN)2Br2 with DiPPF in dichloromethane at room temperature. Like the reaction of CyPF-tBu-ligated bis-thiolate 7, this reaction of DiPPF-ligated bis-thiolate complex 21 with 4-bromotoluene was inhibited by added disulfide (Scheme 2). The reaction of 21 with 4-bromotoluene in toluene at 100 °C gave the thioether in 86% yield after 20 h, as determined by GC analysis with an internal standard. In contrast, the same reaction conducted with 3.7 × 10-2 M added disulfide did not lead to conversion of the starting complex, as determined by 31P{1H} NMR spectroscopy, and did not lead to quantities of thioether detectable by GC.
Scheme 2.

i. Monitoring of reactions catalyzed by a combination of Pd(dba)2 and DiPPF
The resting state of the palladium in reactions initiated by the combination of Pd(dba)2 and DiPPF as catalyst was also assessed. Like the reaction catalyzed by the combination of Pd(dba)2 and CyPF-tBu, the major species observed in the reaction catalyzed by Pd(dba)2 and DiPPF (eq 16) was the binuclear complex [(DiPPF)Pd]2(dba) (22), as determined by 31P{1H} NMR spectroscopy. This Pd(0) DiPPF complex was generated independently in 36% yield by allowing Pd(dba)2 to react with DiPPF in the presence of NaOtBu and isopropylamine (to consume the liberated dba) for 1 h at 100 °C.
DiPPF-ligated, dba-complex 22 reacted with 4-chlorotoluene to give a 1:2.3 mixture of Pd(DiPPF)(Cl)(p-tolyl) (24) and the starting complex 22 after 2.5 h at 100 °C. At longer reaction times (20 h), these complexes decomposed to give a complex mixture of unidentified products. Complex 24 was prepared independently in 32% yield by the reaction of Pd[P(o-tolyl)3]2, DiPPF, and 4-chlorotoluene for 3 h at 60 °C.
iii. Comparison of the rate of the catalytic reactions conducted with [Pd(CyPF-tBu)(p-tolyl)Br] and [Pd(DiPPF)(p-tolyl)Br] as precatalysts
In a previous section, we stated that the reaction of 4-chlorotoluene with 4-methoxybenzenethiol catalyzed by 1 mol % of the likely intermediate [Pd(CyPF-tBu)(p-tolyl)Br] (3a) occurred in 99% yield after 24 h at room temperature. To determine if the catalytic coupling could be conducted with DiPPF as ligand at room temperature, analogous data on reactions catalyzed by the likely intermediate [Pd(DiPPF)(p-tolyl)Br] (16) were obtained. The reaction of 4-chlorotoluene with 4-methoxybenzenethiol in the presence of KOtBu and 1 mol % of complex 16 was conducted in toluene at room temperature. However, under these reaction conditions, only 4% of the thioether was observed after 24 h. Reactions with 10% catalyst also occurred to low conversion. Thus, the catalytic cycle containing the palladium species ligated by CyPF-tBu occurs faster than that containing the palladium species ligated by DiPPF.
Discussion
The results section presented a body of qualitative and quantitative kinetic data on the coupling of aryl halides with thiols. Several pieces of data point to complexities that can arise when conducting coupling reactions of thiols. For example, the individual steps of the catalytic cycle have been shown, counterintuitively, to occur faster than the overall coupling reaction when typical precatalysts are used. As presented in this discussion section, this difference in rate results from the formation of several stable species that lie off of the catalytic cycle, such as stable thiolate complexes and stable Pd(0) complexes containing ligands from the catalyst precursor. A summary of the proposed mechanism is shown in Figure 4. This discussion section will analyze the relative rates of the stoichiometric reactions, the significance of the resting state of the catalyst, and the mechanism of the reaction of the palladium bis-thiolate complexes Pd(CyPF-tBu)(SR)2 with aryl halides. As part of this analysis, similarities and differences between the reactivity of palladium complexes of CyPF-tBu and the related alkylphosphine DiPPF are deduced.
Figure 4.

Proposed mechanism for the palladium-catalyzed thiation of aryl halides.
1. Rates of the stoichiometric reactions involved in the catalytic cycle
All the elementary steps that constitute the catalytic cycle for the thioetherification of aryl chlorides catalyzed by CyPF-tBu complexes of palladium are fast at room temperature. Oxidative addition of 4-chlorotoluene to the Pd(0) complex Pd(CyPF-tBu)[P(o-tolyl)3] (1) was complete in less than 15 min at 25 °C. The “transmetalation” step involving reaction of the arylpalladium halide complex 3a with thiol and NaOtBu occurs in less than 5 min at room temperature, and reductive elimination of aryl thioether from the resulting arylpalladium thiolate complex occurs in less than 1 min at room temperature.
The conditions required for reductive elimination of thioethers from the CyPF-tBu-ligated arylpalladium thiolates 4a and 4b can be compared with those for reductive elimination from other arylpalladium thiolate complexes containing different bisphosphines. For example, C-S bond formation from the arylpalladium thiolate complexes ligated by DiPPF 20a and 20b required longer reaction times (1.5 h) at room temperature than did 4a and 4b. Moreover, previous studies have shown that reductive eliminations of thioethers from Pd(DPPE)(SR)(Ar) complexes occur at 50 °C with half-lives ranging from 1 to 25 h.15,16 Only Pd(DPPE)(StBu)(C6H4-4-CF3) underwent reductive elimination at room temperature over the course of several hours. Hillhouse et al.42 have shown that reductive elimination from the dimeric complex [(PMe3)Ni(S-o-C6H4CMe2CH2)]2 occurs at ambient temperature after oxidation by oxygen or S8. However, reductive elimination from this complex in the absence of these oxidants required 24 h at 70 °C. Thus, reductive elimination of thioethers from the CyPF-tBu-ligated arylpalladium thiolate complexes is faster than reductive elimination from other complexes containing phosphines. This fast rate occurs despite the presence of dialkylphosphino groups on the phosphine ligand and is best attributed to the severe steric hindrance imparted by the combination of one di-tert-butylphosphino and one dicyclohexylphosphino substituent on the phosphine.
2. Identification of the resting state in reactions catalyzed by palladium complexes of CyPF-tBu
The apparent inconsistency between the fast rates of the stoichiometric reactions involved in the catalytic cycle for thioetherification of aryl chlorides catalyzed by CyPF-tBu-ligated palladium and the slower rate of the catalytic thioetherification of aryl chlorides catalyzed by the combination of CyPF-tBu and Pd(OAc)2 or Pd2(dba)3 can be explained by the identity of the resting state in the catalytic reactions. For reactions catalyzed by the combination of Pd(dba)2 and CyPF-tBu or Pd(OAc)2 and CyPF-tBu, the catalyst resting state lies off the catalytic cycle and enters it through a slow step. The complex [Pd(CyPF-tBu)]2(dba) (9) that is the resting state in reactions catalyzed by Pd(dba)2 and CyPF-tBu enters the catalytic cycle by slow dissociation of the dba ligand.43 A structurally related mononuclear complex, [Pd(CyPF-tBu)(dba)], generated in situ from Pd(dba)2 and the CyPF-tBu did not undergo oxidative addition of phenyl iodide, even after heating at 60 °C for 48 h.34 In our studies, oxidative addition of 4-chlorotoluene to 9 required prolonged reaction times at 100 °C. This slow rate appears to account for the slow rate of the thioetherifications catalyzed by Pd(dba)2 and CyPF-tBu.
Likewise the accumulation of the palladium as Pd(CyPF-tBu)(SR)2 in reactions catalyzed by the combination of Pd(OAc)2 and CyPF-tBu accounts for the slower rate of the reactions catalyzed by the combination of Pd(OAc)2 and CyPF-tBu than of the stoichiometric reactions. Palladium bis-thiolate complexes have been proposed as intermediates in several C-S bond forming reactions, such as the Pd-catalyzed azathiolation of carbon monoxide44 or the Pd-catalyzed addition of S-S bonds to alkynes.45,46 The reaction of bis-thiolate complexes with haloarenes, which had been reported by Yamamoto for Ni(SPh)2(bpy)2 with alkyl and aryl iodides,36 has not been considered as a potential step in palladium-catalyzed cross-coupling reactions prior to our work. Our data imply (vide infra) that reductive elimination of disulfide allows this species to enter the catalytic cycle and to react with haloarenes. This reductive elimination is slow and thermodynamically unfavorable, but it is driven by added base, and this effect of base helps to promote reactions initiated with this mixture of catalyst components.
3. Mechanism of the reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with aryl halides
Three potential classes of mechanisms for the reaction of Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) with haloarenes are shown in Scheme 3. Path A involves initial reversible dissociation of SR- from the bis-thiolate complex 7. Coordination of the aryl bromide to the resulting cationic species and nucleophilic attack of the thiolate could form Pd(CyPF-tBu)[S(C6H4-4-OMe)](Br) (12) and thioether. This mechanism predicts a positive dependence of the rate on the concentration of aryl bromide and zero-order dependence on the concentration of added disulfide. This predicted zero-order dependence on added disulfide is inconsistent with the observed inverse first-order dependence on added disulfide that was shown in Figure 3.
Scheme 3.

Path B involves initial, irreversible oxidative addition of aryl bromide to give a hexacoordinate Pd(IV) species. This species would undergo reductive elimination of thioether to form Pd(CyPF-tBu)[S(C6H4-4-OMe)](Br) (12). This pathway predicts a strict first-order dependence of the rate on the concentration of aryl bromide and a zero-order dependence on the concentration of added disulfide. The non-zero y-intercept in the plot of 1/kobs vs [ArBr] in Figure 2 and the inverse dependence of the rate on the concentration of disulfide are both inconsistent with the predicted rate behavior for Path B.
Path C consists of a reversible reductive elimination of disulfide, followed by an irreversible oxidative addition of aryl bromide to give an arylpalladium bromide complex. This complex would then react with the extruded disulfide by one of several mechanisms to give the bromopalladium thiolate 12, together with the thioether.47 This pathway predicts a positive order in the concentration aryl bromide and an inverse-first order in added disulfide. A plot of 1/kobs vs [ArBr] would be expected to be linear with a non-zero y-intercept. Our kinetic data are consistent with this pathway.
The rate equation corresponding to pathway C is shown in eq 17. It predicts that the reciprocals of the y-intercepts on the plots of 1/kobs vs 1/[ArBr] and 1/kobs vs [{S(C6H4-OMe)}2] correspond to the rate constant for reductive elimination of disulfide from complex 7 at 100 °C. These two values are in good agreement with each other [(1.1±0.2) ×10-4 s-1 and (1.4±0.2) ×10-4 s-1]. This mechanism also predicts that the ratio of rate constants (k6/k-5) for oxidative addition of 4-bromotoluene and bis(4-methoxyphenyl) disulfide can be determined from the slope of the plots. These values indicate that the oxidative addition of 4-bromotoluene is ca 1000 times slower than oxidative addition of the disulfide.
| (17) |
The first step in this Path C, the reductive elimination of disulfide, is less favorable thermodynamically than reductive elimination to form C-C or C-S bonds. Heating bis-thiolate complex 7 at 100 °C for prolonged reaction times with trans-stilbene did not lead to a palladium(0) alkene complex and free disulfide (eq 11) because the product of oxidative addition of the disulfide is favored thermodynamically. However, the equilibrium between Pd(CyPF-tBu)[S(C6H4-4-OMe)]2 (7) and (CyPF-tBu)Pd(stilbene) could be displaced by the addition of an alkoxide base to cleave the S-S bond in the disulfide product (eq 12).39,40
4. Significance of the hydridopalladium thiolate complex Pd(CyPF-tBu)(H)(SR) as catalyst resting state
The terminal hydrido complexes Pd(CyPF-tBu)(H)[S(C6H4-4-OMe)] (6) and Pd(CyPF-tBu)(H)(SiPr) (10) were the major species in solution during the thiation of 4-chlorotoluene with 4-methoxybenzenthiol and 2-propanethiol, respectively, catalyzed by Pd(CyPF-tBu)(p-tolyl)(Br) (3a). Complex 6, when prepared independently, catalyzed the reaction of 4-chlorotoluene and 4-methoxybenzenethiol in the presence of KOtBu at room temperature to form the thioether in 83% yield after 24 h. Although hydridopalladium thiolate complexes, such as Pd(H)(SAr)(PCy3)2,48 are known to form by reaction of thiols with palladium(0) species, the observation of the product of oxidative addition of the reactant X-H bond has not been observed previously as the resting state of cross-coupling reactions.
The hydrido palladium thiolate complexes Pd(CyPF-tBu)(H)(SR) likely enter the catalytic cycle by reaction with an aryl halide in the presence of base. This proposal is consistent with the reaction of hydridopalladium thiolate complex 6 with NaOtBu and P(o-tol)3 to form the palladium(0) species [Pd(CyPF-tBu)(P(o-tol)3)] (1), which we have shown to add chloroarenes at room temperature. It is also consistent with the reaction of complex 6 with an excess of 4-bromotoluene and NaOtPent to form the arylpalladium bromide complex 3a, together with the thioether in high yield (Scheme 1). This reaction likely occurs by elimination of the thiol induced by base to generate the combination of Pd(CyPF-tBu), thiolate, and chloroarene. Oxidative addition of the chloroarene, followed by transmetalation with the sodium thiolate and reductive elimination would then form the thioether and Pd(CyPF-tBu). Pd(CyPF-tBu) would add the second equiv of chloroarene to form the final arylpalladium chloride complex as product. Thus, there is a pathway for formation of the species within the catalytic cycle, but the accumulation of the palladium as the hydrido thiolate complex off the catalytic cycle does reduce the rate of the overall process.
5. Comparison of rates and catalyst resting states of reactions catalyzed by Pd(CyPF-tBu) complexes with those of reactions catalyzed by Pd(DiPPF) complexes
Our mechanistic data on the reactions catalyzed by palladium complexes of DiPPF show that our conclusions about the mechanism of the reaction of aryl halides and thiols catalyzed by the combination of Pd(OAc)2 and CyPF-tBu or the combination of Pd(dba)2 and CyPF-tBu can be extrapolated to the mechanism of reactions catalyzed by palladium complexes of other electron rich bisphosphines. First, C-S bond-forming reductive elimination takes place under mild conditions in good yields when the palladium is ligated by either phosphine. Moreover, the major palladium species in the reactions catalyzed by the combination of Pd(OAc)2 and DiPPF or the combination of Pd(dba)2 and DiPPF are analogous to those observed for reactions catalyzed by the same precursors with CyPF-tBu. The major palladium species in the reactions catalyzed by the combination of Pd(OAc)2 and DiPPF is the bis-thiolate complex Pd(DiPPF)(SR)2 (21), and the major palladium species in the reactions catalyzed by the combination of Pd(dba)2 and DiPPF is [Pd(DiPPF)]2(dba) (22).
Moreover, like the CyPF-tBu-ligated bis-thiolate complex 7, the DiPPF-ligated bis-thiolate complex 21 reacts with bromoarenes, and, like the reaction of CyPF-tBu-ligated 7 with bromoarenes, this transformation is inhibited by the addition of a disulfide (Scheme 2). These data imply that the reactions of the DiPPF-ligated bis-thiolate complex with haloarenes occur by the same mechanism involving reversible reductive elimination of disulfide, followed by irreversible oxidative addition of the haloarene, as was deduced for reactions of the CyPF-tBu-ligated bis-thiolate complex 7. Finally, the DiPPF-ligated dba complex 22, like the CyPF-tBu-ligated dba complex 9, reacts slowly with chloroarenes presumably because of slow dissociation of dba from the electron-rich DiPPF-Pd fragments.
More generally, these data show that the coupling of aryl bromides with thiols catalyzed by DiPPF-ligated palladium occurs by oxidative addition of the bromoarenes, transmetalation to form a thiolate complex and reductive elimination of thioether in a series of steps that occurs with rates that are faster than catalytic reactions reported previously. This difference in rates, like the difference in rates of reaction of CyPF-tBu complexes, results from the accumulation of the palladium as species that lie outside of the catalytic cycle.
The catalytic couplings of chloroarenes with thiols catalyzed by CyPF-tBu-ligated palladium are faster than the couplings catalyzed by DiPPF-ligated palladium. The reaction of 4-chlorotoluene with 4-methoxybenzenethiol catalyzed by the likely intermediate [(CyPF-tBu)Pd(Ar)(Br)] occurs to full conversion at room temperature but the same reaction catalyzed by [(DiPPF)Pd(Ar)(Br)] occurs to low conversion under these conditions. Because transmetalation and reductive elimination proceed at room temperature when palladium is ligated by either bisphosphine, and displacement of the bisphosphine ligand by a thiolate has not been observed in our experiments, the difference in rates of these catalytic reactions is best attributed to the slower oxidative addition of chloroarenes to palladium(0) complexes ligated by DiPPF.
Conclusions
We have reported a detailed mechanistic study on the coupling of aryl chlorides with thiols catalyzed by CyPF-tBu complexes of palladium. Our studies of the elementary steps involved in the catalytic cycle are summarized in Scheme 4 and led to the following conclusions.
Scheme 4.

The three steps of the catalytic cycle for the coupling of thiols with aryl chlorides, oxidative addition, transmetalation and reductive elimination proceed rapidly at or below room temperature with complexes ligated by CyPF-tBu.
The resting state for the reactions catalyzed by Pd(OAc)2/CyPF-tBu, Pd(dba)2/CyPF-tBu, and Pd(CyPF-tBu)(p-tolyl)(Br) are Pd(CyPF-tBu)(SR)2, [Pd(CyPF-tBu)]2(dba) and Pd(CyPF-tBu)(H)(SR), respectively, and each of these complexes lies off the catalytic cycle.
These species lying off of the catalytic cycle enter the cycle by reaction with aryl halides and, in the case of Pd(CyPF-tBu)(SR)2 and Pd(CyPF-tBu)(H)(SR), also with base.
These mechanistic conclusions pertain equally to reactions catalyzed by palladium complexes containing a different alkylbisphosphine, DiPPF.
Most generally, these studies show the importance of identifying the major complexes in the catalytic system to interpret rate data and the importance of determining the major complexes in the catalytic system to design improved catalysts.49 Efforts to increase the rate of oxidative addition, transmetalation, and reductive elimination would not improve the rates of the coupling of aryl chlorides with thiols catalyzed by the combination of Pd(OAc)2 and CyPF-tBu or Pd(dba)2 and CyPF-tBu. Instead, improvements in the efficiency of the process that generates the active catalyst are needed to improve the rates of these reactions. Studies on the coupling of chloroarenes with thiols in the presence of catalyst precursors that generate the active palladium species ligated by CyPF-tBu in a practical manner will be the subject of future studies.
Supplementary Material
Acknowledgments
We thank the NIH (GM-58108) for support of this work, Johnson-Matthey for PdCl2, and Solvias for CyPF-tBu. E. A. thanks the Fundación La Caixa and Ministerio de Educación for support. The authors thank Dr. Tokutaro Ogata for the independent synthesis of the DiPPF-Pd(0) complexes.
Footnotes
Supporting Information Available: Experimental procedures and characterization of products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kosugi M, Shimizu T, Migita T. Chem Lett. 1978;13 [Google Scholar]
- 2.Migita T, Shimizu T, Asami Y, Shiobara J, Kato Y, Kosugi M. Bull Chem Soc Jpn. 1980;53:1385. [Google Scholar]
- 3.Murata M, Buchwald SL. Tetrahedron. 2004;60:7397. [Google Scholar]
- 4.Mispelaere-Canivet C, Spindler JF, Perrio S, Beslin P. Tetrahedron. 2005;61:5253. [Google Scholar]
- 5.Zheng N, McWilliams JC, Fleitz FJ, Armstrong IJD, Volante RP. J Org Chem. 1998;63:9606. [Google Scholar]
- 6.Schopfer U, Schlapbach A. Tetrahedron. 2001;57:3069. [Google Scholar]
- 7.Moreau X, Campagne JM. J Organomet Chem. 2003;687:322. [Google Scholar]
- 8.Li GY. Angew Chem, Int Ed. 2001;40:1513. doi: 10.1002/1521-3773(20010417)40:8<1513::aid-anie1513>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 9.Itoh T, Mase T. Org Lett. 2004;6:4587. doi: 10.1021/ol047996t. [DOI] [PubMed] [Google Scholar]
- 10.Other transition-metal catalyzed couplings of aryl halides with thiols. Ni: Zhang Y, Ngeow KC, Ying JY. Org Lett. 2007;9:3495. doi: 10.1021/ol071248x.Cristau HJ, Chabaud B, Chene A, Christol H. Synthesis. 1981;892Gomez-Benitez V, Baldovino-Pantaleon O, Herrera-Alvarez C, Toscano RA, Morales-Morales D. Tetrahedron Lett. 2006;47:5059.. Cu: Sawada N, Itoh T, Yasuda N. Tetrahedron Lett. 2006;47:6595.Chen YJ, Chen HH. Org Lett. 2006;8:5609. doi: 10.1021/ol062339h.Palomo C, Oiarbide M, Lopez R, Gomez-Bengoa E. Tetrahedron Lett. 2000;41:1283.Wu YJ, He H. Synlett. 2003:1789.Savarin C, Srogl J, Liebeskind LS. Org Lett. 2002;4:4309. doi: 10.1021/ol026948a.Deng W, Zou Y, Wang YF, Liu L, Guo QX. Synlett. 2004:1254.Bates CG, Saejueng P, Doherty MQ, Venkataraman D. Org Lett. 2004;6:5005. doi: 10.1021/ol0477935.Bates CG, Gujadhur RK, Venkataraman D. Org Lett. 2002;4:2803. doi: 10.1021/ol0264105.Kwong FY, Buchwald SL. Org Lett. 2002;4:3517. doi: 10.1021/ol0266673.
- 11.Fernández-Rodríguez MA, Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:2180. doi: 10.1021/ja0580340. [DOI] [PubMed] [Google Scholar]
- 12.Fernández-Rodríguez MA, Shen QL, Hartwig JF. Chem Eur J. 2006;12:7782. doi: 10.1002/chem.200600949. [DOI] [PubMed] [Google Scholar]
- 13.Fernández-Rodríguez MA, Hartwig JF. J Org Chem. 2009;74:1663. doi: 10.1021/jo802594d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen Q, Shekhar S, Stambuli JP, Hartwig JF. Angew Chem, Int Ed. 2005;44:1371. doi: 10.1002/anie.200462629. [DOI] [PubMed] [Google Scholar]
- 15.Baranano D, Hartwig JF. J Am Chem Soc. 1995;117:2937. [Google Scholar]
- 16.Mann G, Baranano D, Hartwig JF, Rheingold AL, Guzei IA. J Am Chem Soc. 1998;120:9205. [Google Scholar]
- 17.Moreau X, Campagne JM, Meyer G, Jutand A. Eur J Org Chem. 2005:3749. [Google Scholar]
- 18.Roy AH, Hartwig JF. Organometallics. 2004;23:194. [Google Scholar]
- 19.Roy AH, Hartwig JF. J Am Chem Soc. 2003;125:8704. doi: 10.1021/ja035835z. [DOI] [PubMed] [Google Scholar]
- 20.Stille JK, Lau KSY. Acc Chem Res. 1977;10:434. [Google Scholar]
- 21.Barrios-Landeros F, Hartwig JF. J Am Chem Soc. 2005;127:6944. doi: 10.1021/ja042959i. [DOI] [PubMed] [Google Scholar]
- 22.See Supporting Information.
- 23.Variable amounts of terminal hydrido complex Pd(CyPF-tBu)(H)[S(C6H4-4-OMe)] (6) were observed in some cases by 31P{1H} NMR spectroscopy.
- 24.See Supporting Information.
- 25.Hartwig JF. Inorg Chem. 2007;46:1936. doi: 10.1021/ic061926w. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida T, Okano T, Otsuka S. J Chem Soc, Dalton Trans. 1976:993. [Google Scholar]
- 27.Grushin VV, Alper H. Organometallics. 1996;15:5242. [Google Scholar]
- 28.Akita M, Miyaji T, Muroga N, Mock-Knoblauch C, Adam W, Hikichi S, Morooka Y. Inorg Chem. 2000;39:2096. doi: 10.1021/ic991034e. [DOI] [PubMed] [Google Scholar]
- 29.Cámpora J, Palma P, del Río D, Alvarez E. Organometallics. 2004;23:1652. [Google Scholar]
- 30.Kraatz HB, Milstein D. J Organomet Chem. 1995;488:223. [Google Scholar]
- 31.Grushin VV, Alper H. Organometallics. 1993;12:1890. [Google Scholar]
- 32.Bryndza HE, Tam W. Chem Rev. 1988;88:1163. [Google Scholar]
- 33.Grushin VV. Chem Rev. 1996;96:2011. doi: 10.1021/cr950272y. [DOI] [PubMed] [Google Scholar]
- 34.Brunker TJ, Blank NF, Moncarz JR, Scriban C, Anderson BJ, Glueck DS, Zakharov LN, Golen JA, Sommer RD, Incarvito CD, Rheingold AL. Organometallics. 2005;24:2730. [Google Scholar]
- 35.Shekhar S, Ryberg P, Hartwig JF, Mathew JS, Blackmond DG, Strieter ER, Buchwald SL. J Am Chem Soc. 2006;128:3584. doi: 10.1021/ja045533c. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto T, Sekine Y. Inorg Chim Acta. 1984;83:47. [Google Scholar]
- 37.4-methylphenyl t-pentyl ether was observed in this reaction by GC/MS.
- 38.See Supporting Information for the characterization of Pd(CyPF-tBu)(OtBu)(p-tolyl).
- 39.Parker AJ, Kharasch N. Chem Rev. 1959;59:583. [Google Scholar]
- 40.Parker AJ, Kharasch N. J Am Chem Soc. 1960;82:3071. [Google Scholar]
- 41.Alcazar-Roman LM, Hartwig JF, Rheingold AL, Liable-Sands LM, Guzei IA. J Am Chem Soc. 2000;122:4618. [Google Scholar]
- 42.Han R, Hillhouse GL. J Am Chem Soc. 1998;120:7657. [Google Scholar]
- 43.Amatore C, Broeker G, Jutand A, Khalil F. J Am Chem Soc. 1997;119:5176. [Google Scholar]
- 44.Kuniyasu H, Kato T, Asano S, Ye JH, Ohmori T, Morita M, Hiralke H, Fujiwara S, Terao J, Kurosawa H, Kambe N. Tetrahedron Lett. 2006;47:1141. [Google Scholar]
- 45.Beletskaya IP, Ananikov VP. Eur J Org Chem. 2007:3431. [Google Scholar]
- 46.Ananikov VP, Kabeshov MA, Beletskaya IP, Khrustalev VN, Antipin MY. Organometallics. 2005;24:1275. [Google Scholar]
- 47.Because we have not been able to isolate the bromopalladium thiolato 12 in pure form, we have not been able to determine how this species is converted into Pd(CyPF-tBu)(Br)2. Presumably this transformation occurs by the reaction of complex 12 with another equivalent of 4-bromotoluene.
- 48.Osakada K, Hayashi H, Maeda M, Yamamoto T, Yamamoto A. Chem Lett. 1986;597 [Google Scholar]
- 49.For an example of this phenomenon during the formylation of aryl halides, see: Sergeev AG, Spannenberg A, Beller M. J Am Chem Soc. 2008;130:15549. doi: 10.1021/ja804997z.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.