

Abstract
The Notch pathway has recently been implicated in mesenchymal progenitor cell (MPC) differentiation from bone marrow-derived progenitors. However, whether Notch regulates MPC differentiation in an RBPjκ-dependent manner, specifies a particular MPC cell fate, regulates MPC proliferation and differentiation during early skeletal development or controls specific Notch target genes to regulate these processes remains unclear. To determine the exact role and mode of action for the Notch pathway in MPCs during skeletal development, we analyzed tissue-specific loss-of-function (Prx1Cre; Rbpjkf/f), gain-of-function (Prx1Cre; Rosa-NICDf/+) and RBPjκ-independent Notch gain-of-function (Prx1Cre; Rosa-NICDf/+; Rbpjkf/f) mice for defects in MPC proliferation and differentiation. These data demonstrate for the first time that the RBPjκ-dependent Notch signaling pathway is a crucial regulator of MPC proliferation and differentiation during skeletal development. Our study also implicates the Notch pathway as a general suppressor of MPC differentiation that does not bias lineage allocation. Finally, Hes1 was identified as an RBPjκ-dependent Notch target gene important for MPC maintenance and the suppression of in vitro chondrogenesis.
Keywords: RBPjκ, Notch, Hes1, Mesenchymal progenitor cell, Limb development, Mouse
INTRODUCTION
The Notch pathway is an evolutionarily conserved signaling system that regulates cell proliferation, differentiation, cell fate determination and stem/progenitor cell self-renewal in both embryonic and adult organs (Artavanis-Tsakonas et al., 1999; Chiba, 2006; Lai, 2004). In mammals, Notch signaling is initiated when a ligand (jagged 1, jagged 2, delta-like 1 or delta-like 4) binds to a single-pass transmembrane cell surface Notch receptor (Notch1-4) on the neighboring cell. Ligand-receptor interactions ultimately induce cleavage of the Notch receptors via the gamma-secretase complex, releasing the Notch intracellular domain (NICD) to activate both canonical and non-canonical Notch signaling mechanisms. During canonical Notch signaling, the NICD translocates to the nucleus and binds the transcriptional repressor, RBPjκ, converting it into an activator and inducing the expression of downstream target genes (Kopan and Ilagan, 2009; Lai, 2002). Some of the most well-defined canonical, or RBPjκ-dependent, Notch target genes include specific members of the Hes/Hey family of basic helix-loop-helix transcription factors: Hes1, Hes5, Hes7, Hey1, Hey2 and HeyL, which are thought to mediate much of Notch function (Iso et al., 2003; Iso et al., 2001).
The NICD also has the ability to function independently of RBPjκ via non-canonical interactions with proteins and complexes of other signaling pathways (Martinez Arias et al., 2002). RBPjκ-independent Notch signaling was originally identified in Drosophila when specific Notch alleles were shown to induce Deltex-dependent Notch signaling events and the repression of neural cell fate (Matsuno et al., 1997; Ramain et al., 2001). Mammalian NICDs also bind to many other signaling molecules and transcriptional regulators such as SMADs 1, 5 and 8 (Dahlqvist et al., 2003; Itoh et al., 2004), SMADs 2 and 3 (Blokzijl et al., 2003), Lef1 (Ross and Kadesch, 2001), β-catenin (Hayward et al., 2005), dishevelled (Dvl) (Axelrod et al., 1996), IκB kinase α subunit (IKKα) (Vacca et al., 2006; Vilimas et al., 2007), the p65/p50 subunits of the NF-κB complex (Wang et al., 2001) and deltex (Axelrod et al., 1996; Diederich et al., 1994; Hayward et al., 2005; Ramain et al., 2001; Vacca et al., 2006; Vilimas et al., 2007), potentially regulating these pathways in an RBPjκ-independent manner. Recently, compelling data for RBPjκ-independent Notch signaling in the skin was demonstrated by comparing conditional genetic mouse models removing core components of the gamma-secretase complex (presinilin 1/2; PS1/PS2), the Notch receptors (Notch1/2; N1/N2) or the RBPjκ transcriptional repressor (Demehri et al., 2009). These reports highlight the complexity of RBPjκ-dependent and -independent Notch signaling in various cell systems and the reasons why detailed genetic studies are required to identify the mechanisms by which Notch regulates context-dependent cell proliferation and differentiation.
The Notch pathway is important in regulating stem/progenitor cell self-renewal, proliferation and differentiation from various tissues including: hematopoietic (Hadland et al., 2004; Kunisato et al., 2003; Robert-Moreno et al., 2005; Stier et al., 2002; Varnum-Finney et al., 2000), neural (de la Pompa et al., 1997; Hatakeyama et al., 2004; Hitoshi et al., 2002; Ohtsuka et al., 1999), pancreatic (Apelqvist et al., 1999; Jensen et al., 2000) and intestinal (Fre et al., 2005). However, genetic investigations into Notch regulation of mesenchymal stem/progenitor cells (MPCs) have gone largely unexplored. This has probably been owing to the fact that many mutant mice for crucial components of the Notch pathway are embryonic-lethal prior to MPC differentiation and overt skeletogenesis (Donoviel et al., 1999; Hamada et al., 1999; Hrabe de Angelis et al., 1997; Oka et al., 1995; Swiatek et al., 1994; Xue et al., 1999). Recently, work by Hilton et al. (Hilton et al., 2008) used conditional genetic approaches to demonstrate that “upstream” components of the Notch pathway (PS1/PS2 and N1/N2) were crucial in regulating osteoblastic differentiation of bone marrow-derived MPCs in mice. In vitro studies using human bone marrow-derived MPCs (hMPCs) have also implicated Notch signaling and jagged 1 in the induction of hMPC proliferation and a context-dependent regulation of differentiation (Oldershaw et al., 2008; Vujovic et al., 2007). Collectively, these data suggest that various Notch signaling components might play crucial and differential roles in MPC self-renewal, proliferation and differentiation.
MPCs are not only multi-potent cells found in various adult tissues including bone, cartilage, muscle and fat, but are also prominent cells populating the embryonic limb-bud, which are required for chondrogenic and osteogenic differentiation during skeletal development. During limb development, various cell types migrate into the limb field, a great many of which include MPCs of the lateral plate mesoderm, which begin rapid proliferation driving growth of the limb. During early phases of limb development, the apical ectodermal ridge (AER) maintains an apical zone of cells in an undifferentiated state primarily via fibroblast growth factor (Fgf) signals (Niswander et al., 1994). As MPCs withdraw from control of the AER, many of the cells are recruited into mesenchymal condensations that undergo chondrogenic and osteogenic differentiation, giving rise to the skeletogenic elements. The MPC cell fate decision to undergo chondrogenesis versus osteogenesis is dictated by a balance between both the expression and activity of Sox9 and Runx2 (Akiyama et al., 2005). Sox9 is expressed in all MPCs, maintaining their ability to undergo both chondrogenesis and osteogenesis (Akiyama et al., 2002; Bi et al., 1999). MPCs localized near the center of condensations strongly upregulate Sox9 expression and activity, inducing important downstream targets such as type II collagen (Col2a1) and aggrecan (Agc1; Acan – Mouse Genome Informatics), and therefore undergo the process of chondrogenesis. Meanwhile, Sox9 “low”-expressing cells around the borders of the condensations upregulate Runx2 expression and activity, thereby inducing osteogenic molecules such as type I collagen (Col1a1), osterix (Osx) and osteocalcin (Oc), and suppressing the chondrogenic fate (Akiyama et al., 2005; Drissi et al., 2000). Although Sox9 and Runx2 are crucial regulators of MPC differentiation, very few additional molecules have been identified to control MPC differentiation at or upstream of Sox9 and/or Runx2.
To determine the exact role and mode of action for the Notch pathway in MPCs we analyzed tissue-specific loss-of-function (Prx1Cre; Rbpjkf/f), gain-of-function (Prx1Cre; Rosa-NICDf/+) and RBPjκ-independent Notch gain-of-function (Prx1Cre; Rosa-NICDf/+; Rbpjkf/f) mice for defects in MPC proliferation and differentiation during early limb development. Additionally, we used limb-bud tissue sections, in vitro limb-bud-derived MPC cultures and C3H10T1/2 mesenchymal cell cultures to identify the expression of specific Notch pathway components, to determine if the Notch pathway was a generic regulator of MPC differentiation and to identify potential Notch target gene(s) regulating MPC differentiation.
MATERIALS AND METHODS
Mouse strains
All mouse strains including Rosa-NICD, Rbpjk and Prx1Cre are as previously described (Han et al., 2002; Logan et al., 2002; Murtaugh et al., 2003). Prx1Cre mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA), whereas Rosa-NICD and Rbpjk floxed mice were generous gifts from Dr Douglas Melton (Harvard University, MA, USA) and Dr Tasuku Honjo (Kyoto Graduate School of Medicine, Japan), respectively.
Analyses of mouse embryos
Embryonic tissues were harvested at E11.0-E12.5 in PBS, fixed in 10% neutral buffered formalin, then processed and embedded in paraffin prior to sectioning at 4 μm. Standard Alcian Blue/Orange G (AB/OG) staining was performed in order to analyze tissue architecture and cartilage composition of the limb-buds. In situ hybridization was performed as described previously (Hilton et al., 2005; Hilton et al., 2007; Hilton et al., 2008), using 35S-labeled riboprobes. Unpublished riboprobes were generated from the following cDNA clones: Sox9 (4165469), Agc1 (5345931), Hes1 (10469606), Hey1 (9792713), jagged 1 (Jag1) (10699187), delta-like 1(Dll1; 10698888), and delta-like 4 (Dll4; 7492828), available from Open Biosystems or ATCC. The Gfp probe was generated by cloning the enhanced Gfp coding sequence into the pGEM-T Easy vector. Notch1, Notch2, Notch3, Fgf8 and Fgf10 cDNAs and riboprobes are as described (Bellusci et al., 1997; Crossley and Martin, 1995; Mitsiadis et al., 1995). For BrdU immunostaining analyses, pregnant females were injected with BrdU at 0.1 mg/g body weight 2 hours prior to harvest. BrdU detection was performed on paraffin sections using a kit from Invitrogen as per manufacturer's instructions. Analyses of apoptotic MPCs were performed using both anti-cleaved caspase 3 immunostaining (Cell Signaling) and TUNEL staining (Roche Cell Death In Situ Kit) on limb-bud sections according to the manufacturers' instructions. Wholemount skeletal staining of embryos was performed as previously described (Hilton et al., 2005; McLeod, 1980).
Limb-bud MPC and C3H10T1/2 cell culture
Limb-bud derived MPCs were isolated from E11.5 CD1 mouse embryos as previously described (Zhang et al., 2004). For chondrogenic differentiation, MPCs were seeded in micromass (1 × 105 cells in 10 μl) for 1.5 hours before adding standard media, media containing the gamma secretase and Notch inhibitor N-(3,5-difluorophenylacetyl-L-alanyl)]-S-phenylglycine t-butyl ester (DAPT; 1 μM; Calbiochem), or media containing Hes1/control shRNA lentivirus. Cells were cultured for a time-course of 6 hours, at 3, 5 and 7 days prior to harvest for cartilage staining (ABH/OG) or total RNA isolations. C3H10T1/2 micromass chondrogenic cultures were treated and harvested in a similar manner (Denker et al., 1999; Haas and Tuan, 1999). Limb-bud-derived MPCs were also cultured in monolayer for 21 days and treated with either osteogenic (10 nM dexamethasone; 50 μM ascorbic acid; 10 mM β-glycerolphosphate) or adipogenic medium (Millipore) in the presence and absence of DAPT. Fixed MPCs were stained for osteoblastic differentiation using an Alkaline Phosphatase stain (nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyhosphate P-toluidine salt) or adipogenic differentiation using an Oil Red-O staining solution (Millipore). Total RNA was isolated from monolayer cultures at day 21 for use in real-time RT-PCR analyses.
Real-time RT-PCR
Real-time RT-PCR was performed on RNA extracted from both embryonic limb-bud tissues or micromass cultures as previously described (Dong et al., 2005). Primer sequences are available upon request. Mouse-specific PCR primers were developed for: Sox9, Runx2, Col2a1, Agc1, Col1a1, alkaline phosphatase (Ap), Oc, Pparg, jagged 1, jagged 2, delta-like 1, delta-like 3, delta-like 4, Notch1, Notch2, Notch3, Notch4, Hes1, Hes3, Hes5, Hes7, Hey1, Hey2, HeyL and cyclin D1. Gene expression was normalized to β-actin expression levels and then normalized to control samples.
Western blot analyses
Total protein was isolated from either whole mouse limb-bud tissue or cultured limb-bud-derived MPCs in the presence and absence of DAPT (1 μm). Protein samples (~100 μg) from each isolation were subsequently separated on 10% SDS-polyacrylamide and subjected to standard western blotting procedures. NICD1 and NICD2 cleaved proteins were detected using the bTAN 20 (Notch1) and C651.6DdHN (Notch2) primary antibodies (0.4 μg/ml) and then further probed with appropriate secondary antibody (1:3000). Anti-β-actin antibody (Sigma) was used as a control for equal protein loading.
RESULTS
Expression of Notch pathway components during MPC differentiation in vitro and in vivo
We performed real-time RT-PCR to identify the exact temporal expression of the five murine Notch ligands (Jag1,2 and Dll1,3,4), the four Notch receptors (Notch1-4), and the six canonical Notch target genes of the Hes/Hey family (Hes1, Hes5, Hes7, Hey1, Hey2 and HeyL) during limb-bud MPC differentiation and in vitro chondrogenesis. Limb-bud MPCs were isolated from E11.5 mouse embryos and cultured for 6 hours, 3 days and 7 days in micromass. Of the five possible Notch ligands, only Jag1, Dll1 and Dll4 were detected at significant levels, with Jag1 showing the highest level of expression at all time-points (Fig. 1A). Only 3 of the 4 Notch receptors (N1, N2 and N3) were detected during limb-bud MPC differentiation, with Notch2 displaying dramatically higher levels of expression at each time-point as compared with the other Notch receptors (Fig. 1B). To begin understanding which “downstream” components of the Notch signaling pathway are important during limb-bud MPC differentiation and chondrogenesis, we examined the expression of classical RBPjκ-dependent Notch target genes. Of the six possible targets within the Hes/Hey family, only Hes1, Hey1 and HeyL were identified. Hey1 and HeyL were the most abundant Notch target genes, showing similar levels of expression at each time-point that increased during MPC differentiation in vitro (Fig. 1C). Whereas Hes1 displayed a lower level of expression as compared with Hey1 and/or HeyL, Hes1 expression was most pronounced in early limb-bud MPCs, with declining expression levels during MPC differentiation, indicating a potential role in regulating the earliest stages of MPC commitment to the chondrocyte lineage (Fig. 1C).
Fig. 1.

Notch pathway component expression during MPC differentiation and chondrogenesis both in vitro and in vivo. (A-C) Real-time RT-PCR gene expression analyses of the Notch ligands (A), Notch receptors (B) and the Hes/Hey family of RBPjκ-dependent Notch target genes (C). (Da-j) In situ hybridization for the indicated Notch pathway components at E11.5 (Da-Dh) and for N2 (Di) and Hes1 (Dj) at E12.0. Insets show high magnification of N1- and Dll4-associated endothelial cells surrounding vascular canals on alternative sections. (E) Western blot for cleaved Notch2 protein (NICD2) isolated from limb-bud-derived MPCs (LB-MPCs) cultured in the presence and absence of DAPT or from whole limb-bud (WLB). The y-axis of graphs is the relative gene expression normalized to β-actin represented in arbitrary units. d, days; hr, hours.
We also performed in situ hybridization analyses on E11.5 and E12.0 limb-bud sections to identify the exact in vivo spatial expression pattern for the Notch signaling molecules identified in our real-time RT-PCR analyses. These data demonstrated that Notch ligands Jag1, Dll1 and Dll4 all had very different expression profiles. At E11.5, Jag1 was expressed moderately throughout much of the limb-bud mesenchyme but was highly expressed in a concentrated region of the distal medial mesenchyme adjacent to the apical zone (Fig. 1Da). Of the other two Notch ligands, Dll1 was sporadically expressed throughout the limb-bud mesenchyme (Fig. 1Db), while Dll4 demonstrated a more concentrated expression pattern around vascular structures (Fig. 1Dc, high magnification insert) at E11.5. Dll4 is a well-known regulator of angiogenesis, which, along with Notch1, have been documented previously as crucial factors expressed in the vascular endothelium (Hellstrom et al., 2007; Shutter et al., 2000). The Notch receptor, Notch1, was also primarily expressed in regions of vascular tissues (Fig. 1Dd, high magnification insert) and the early ectoderm in E11.5 limb-buds, with lower levels of expression observed throughout some of the mesenchyme. Notch2 was expressed ubiquitously throughout most of the limb-bud MPCs at the same stage (Fig. 1De). Notch3 was expressed sporadically in the limb-bud mesenchyme, with higher concentrations in the proximal and peripheral MPCs. The Notch target genes, Hes1 and Hey1, each had expression patterns similar to that of Notch2 at E11.5 (Fig. 1De,g,h), although a slight elevation of Hes1 expression could be observed in the distal medial MPCs overlapping regions where Jag1 expression is concentrated (Fig. 1Da,g). By E12.0-E12.5, most of the Notch pathway components are difficult to detect via in situ hybridization. Interestingly, only Notch2 and Hes1 expression were maintained in limb-bud MPCs surrounding chondrogenic condensations, but showed significant downregulation within the condensations themselves (Fig. 1Di,j, black and white contours), whereas components like Hey1 maintained a more ubiquitous expression pattern (data not shown).
To determine which Notch receptor is active in the limb-bud mesenchyme, we isolated total protein from cultured MPCs in the presence and absence of the gamma-secretase and Notch inhibitor, DAPT. We also directly isolated protein from wild-type E11.5 whole limb-bud tissue and performed western blot analyses for each sample using Notch1 and Notch2 antibodies to detect the active (NICD) form of the receptor. Western blot analyses revealed that Notch2 was the prominent receptor activated in E11.5 limb-bud MPCs, and that DAPT treatment of cultured MPCs can reduce the abundance of cleaved Notch2 (NICD2; Fig. 1E). Notch1 (NICD1) was nearly undetectable at total protein concentrations up to 100 μg (data not shown) and, unfortunately, a Notch3 antibody that can specifically detect active Notch3 signaling does not exist. Therefore, taken together, these data suggest that Notch2 is the primary Notch receptor activated in MPCs, whereas the other components of the Notch pathway (Jag1, Dll1, N3, Hes1, Hey1 and HeyL) might also be important mediators of MPC proliferation and differentiation during limb development.
Notch signaling is a general regulator of MPC differentiation
To determine the role of Notch signaling in MPCs, we performed Notch loss-of-function assays on E11.5 limb-bud derived MPC cultures using the Notch inhibitor, DAPT. We first examined chondrogenesis in limb-bud micromass cultures by measuring cartilage nodule formation in the presence and absence of 1 μM DAPT. DAPT treatment significantly enhanced cartilage nodule formation (Fig. 2A), suggesting that Notch inhibition accelerates commitment of MPCs to the chondrocyte lineage, a finding that is consistent with a prior study (Fujimaki et al., 2006). We also assessed the effect of DAPT on the expression of the chondrogenic markers Sox9, Col2a1 and Agc1 via real-time RT-PCR. Compared with untreated cultures, DAPT enhanced Sox9, Col2a1 and Agc1 expression (Fig. 2A) within the first 3-5 days of culture, although Agc1 expression was significantly reduced by day 7, indicating that Notch might play a later role in chondrocyte maturation or maintenance of the committed chondrocyte phenotype.
Fig. 2.

DAPT-mediated Notch inhibition enhances limb-bud MPC differentiation without biasing lineage determination. (A) Alcian Blue/Orange G (AB/OG) staining of limb-bud MPC micromass cartilage nodules and real-time RT-PCR for Sox9, Col2a1 and Agc1 at 3, 5 and 7 days. (B) Alkaline Phosphatase staining of limb-bud MPC osteogenic cultures and real-time RT-PCR for Col1a1, AP and Oc at 21 days. (C) Oil Red-O staining of limb-bud MPC adipogenic cultures and real-time RT-PCR for Pparg at 21 days. The y-axis of graphs is the relative gene expression normalized to β-actin and to the control. *, P<0.05 versus control. d, days; hr, hours.
To determine whether Notch specifically regulates chondrogenesis or generally controls MPC differentiation, we performed limb-bud MPC differentiation assays in both osteogenic and adipogenic conditions. We plated limb-bud MPCs in monolayer and cultured the cells for 21 days in osteogenic media in the absence and presence of DAPT (1 μM; Fig. 2B). DAPT treatment enhanced normal osteoblastic differentiation of MPCs. Cultures displayed elevated Alkaline Phosphatase staining and real-time RT-PCR analyses demonstrated a significant increase in the expression of osteoblast marker genes: Col1a1, AP and Oc (Fig. 2B). Finally, we plated limb-bud MPCs in monolayer and cultured the cells for 21 days in adipogenic media in the absence and presence of DAPT (1 μM; Fig. 2C). DAPT treatment similarly enhanced normal adipogenic differentiation of MPCs, which displayed elevated Oil Red-O staining and increased expression of the adipocyte marker gene, Pparg (Fig. 2C). These data demonstrate that inhibition of Notch signaling in vitro enhances limb-bud MPC differentiation toward the chondrocyte, osteoblast and apipocyte lineages, suggesting a general role for Notch signaling in the maintenance of MPCs.
RBPjκ-dependent Notch signaling suppresses MPC differentiation during chondrogenesis
As a first step in assessing the requirement for Notch signaling during limb-bud MPC differentiation and chondrogenesis in vivo, we analyzed embryonic mouse limb-buds in which the canonical Notch effector, Rbpjk, was selectively deleted in the early limb mesenchyme using the Prx1Cre transgene (Prx1Cre; Rbpjkf/f, where “f” represents the floxed allele; Fig. 3). The Prx1Cre mouse line was used in this study because it specifically targets MPCs of the lateral plate mesoderm that give rise to chondrocytes, osteoblasts and connective tissue cells, but not myoblasts, blood lineage cells or vascular endothelial cells within the developing limb (see Fig. S1 in the supplementary material; data not shown) (Durland et al., 2008; Logan et al., 2002; Martin and Olson, 2000). To assay for changes in the commitment of limb-bud MPCs to cells of the chondrocyte lineage, we performed Alcian Blue staining, in situ hybridization and real-time RT-PCR for Sox9, Col2a1 and Agc1. Prx1Cre; Rbpjkf/f mutant (RBPjκ) limb-buds at E12.5 exhibited an increase in Alcian Blue staining of chondrogenic rudiments as compared with controls that demonstrated nearly undetectable levels of Alcian Blue staining (Fig. 3Aa,b). In situ hybridization analyses revealed an increase in both Col2a1 and Agc1 expression in RBPjκ mutant sections. Interestingly, all of the mutant Col2a1-positive cells also expressed Agc1, indicating that these cells are now fully committed chondrocytes (Fig. 3Af,h). Wild-type sections at this stage demonstrated that only a central core of Col2a1-positive cells expressed Agc1, highlighting the normal progression of chondrocyte differentiation (Fig. 3Ae,g). Additionally, RBPjκ mutant sections displayed reduced levels of Sox9 expression, suggesting that the mutant cells have progressed beyond the earliest stages of chondrogenesis. Real-time RT-PCR analyses performed on mRNA isolated from E12.5 whole limb-buds are consistent with the in situ hybridization results for each of the chondrogenic marker genes – Sox9, Col2a1 and Agc1 (Fig. 3B). Real-time RT-PCR performed on earlier limb-buds (E11.5) demonstrated elevated expression of all chondrogenic markers from RBPjκ mutant samples (data not shown). These data suggest that RBPjκ-dependent Notch signaling normally maintains limb-bud MPCs and that loss of RBPjκ results in accelerated chondrogenic differentiation for those cells determined to undergo the process of chondrogenesis.
Fig. 3.

Loss of RBPjκ-dependent Notch signaling in vivo accelerates chondrogenesis during limb development. (Aa,b) AB/OG staining of wild-type (WT) and Prx1Cre; Rbpjkf/f (RBPjκ) E12.5 limb-bud sections. (Ac-h) In situ hybridization for Sox9 (Ac,d), Col2a1 (Ae,f) and Agc1 (Ag,h). (B) Real-time RT-PCR analyses from limb-buds of WT and RBPjκ mutant E12.5 hindlimbs. The y-axis of graphs is the relative gene expression normalized to β-actin and to the WT control. *, P<0.05 versus control.
Sustained Notch activation suppresses MPC differentiation and enhances proliferation in an RBPjκ-dependent manner
We next performed Notch gain-of-function experiments to determine whether Notch activation in vivo could suppress or delay MPC differentiation and chondrogenesis in the developing limb. Gain-of-function experiments were performed using a mouse model system in which the intracellular domain of mouse Notch1 and GFP (NICD-IRES-GFP) were targeted to the Rosa26 reporter locus containing upstream transcriptional stop sequences flanked by loxP sites (Rosa-NICD-IRES-GFP). It has been established that following Cre activation, NICD and GFP expression are sustained specifically within Cre-expressing cell populations (Murtaugh et al., 2003). We again used the Prx1Cre transgene to induce NICD expression and sustained Notch activity within the early limb-bud MPCs prior to chondrogenesis (Prx1Cre; Rosa-NICDf/+), hereafter referred to as NICD mutants. Analyses of NICD mutant E18.5 skeletal preparations demonstrated a clear suppression of normal limb, skull and sternum formation, all specific areas of Prx1Cre expression (Fig. 4Aa,b). Closer examination of the limbs revealed that only a few of the most proximal and distal cartilaginous rudiments developed in NICD mutants, although even these elements were hypoplastic with evidence of delayed cartilage development (Fig. 4Ac-f). To determine if the limb phenotypes arose from the inhibition of MPC differentiation during chondrogenesis, we analyzed E12.5 limb-buds from NICD and wild-type control littermates. Sections from the NICD mutant limb-buds exhibited fewer condensations and thereby showed reduced Alcian Blue staining as compared with controls (Fig. 4Ba,b). Mutants always displayed three digit condensations (apparent loss of first and fifth digits) and often did not develop more proximal condensations. To assess for disruptions in chondrogenesis and MPC differentiation, we performed in situ hybridization for Sox9, Col2a1 and Agc1. NICD mutant sections showed a near complete suppression of these marker genes, although the rudimentary digit condensations that did form seemed to express significant levels of each gene (Fig. 4Bc-h). To investigate why these rudimentary condensations formed at all in NICD mutants, we performed in situ hybridization for Gfp, which marks MPCs that actively express the NICD-IRES-GFP transcript and therefore have Notch activation. Interestingly, each of the rudimentary condensations did not display evident Gfp expression, whereas most other MPCs within the limb-bud showed robust Gfp expression, suggesting that the Prx1Cre transgene did not target this population of cells efficiently (Fig. 4Bi,j). We also performed real-time RT-PCR analyses on mRNA isolated from E12.5 whole limb-buds. These data are consistent with the in situ hybridization results, showing significant decreases in Sox9, Col2a1 and Agc1 expression (Fig. 4C). We also analyzed the expression of the early osteoblast differentiation regulator, Runx2, which like Sox9, showed significantly reduced levels of expression in the NICD mutants (Fig. 4C). Analyses of the RBPjκ-dependent Notch target genes Hes1, Hey1 and HeyL demonstrated increased levels of expression in NICD mutants (Fig. 4C). These data suggest that Notch signaling suppresses MPC differentiation in a localized and possibly cell-autonomous manner acting upstream of Sox9 and Runx2, potentially via RBPJκ-dependent signaling mechanisms.
Fig. 4.

Sustained activation of Notch signaling suppresses MPC differentiation during skeletal development. (Aa-f) Alcian Blue/Alizarin Red staining of WT and Prx1Cre; Rosa-NICDf/+ (NICD) mutant E18.5 skeletons (Aa,b), forelimbs (Ac,d) and hindlimbs (Ae,f). Black arrows indicate NICD mutant limbs, green arrow indicates the open sternum of the NICD mutant. (Ba-h) AB/OG staining of WT and NICD hindlimb sections at E12.5 (Ba,b). In situ hybridization for Sox9 (Bc,d), Col2a1 (Be,f) and Agc1 (Bg,h). (Bi,j) Gfp expression indicated NICD expression/activity in WT (Bi) and NICD (Bj) sections. (C) Real-time RT-PCR for Sox9, Col2a1, Agc1, Runx2, Hes1, Hey1 and HeyL from limb-buds. The y-axis of graphs is the relative gene expression normalized to β-actin and to the WT control. *, P<0.05 versus control. d, digits; fe, femur; fi, fibula; h, humerus; il, illium; pu, pubic; r, radius; s, scapula; t, tibia; u, ulna.
To exclude the possibility that sustained Notch activation impaired skeletal patterning and growth or massively induced MPC apoptosis, we analyzed the expression of limb patterning regulators and assessed alterations in proliferation and apoptosis. We first performed in situ hybridization studies on E11.0 hindlimb sections for the FGF and Shh signaling molecules Fgf8, Fgf10 and Patched1 (Ptc1) to determine whether crucial regulators of limb development and patterning were significantly affected by NICD overexpression (Niswander et al., 1994). Although we observed a slight thickening of the AER and an apparent increase in Fgf8 and Fgf10 expression (Fig. 5Aa-d), we do not believe that this can account for the localized suppression of MPC differentiation previously observed in these animals. Additionally, Ptc1 expression was unchanged between NICD mutant and wild-type sections (Fig. 5Ae,f), indicating uninterrupted Shh activity, which is crucial for normal digit patterning and identity. We next performed TUNEL labeling and cleaved caspase 3 immunohistochemistry experiments to detect apoptotic MPCs on E11.0 hindlimb sections. NICD mutant sections showed no significant change in MPC apoptosis as compared with wild-type littermate controls (Fig. 5B; see also Fig. S2 in the supplementary material). We also detected no significant change in apoptosis at later time-points of MPC differentiation (data not shown). Finally, we performed BrdU labeling experiments on E11.5 sections to determine whether sustained Notch activation has an adverse effect on MPC proliferation and limb growth. Interestingly, our data showed that NICD mutant sections displayed a significant increase in the percentage of BrdU labeled nuclei throughout the limb-bud, but was very evident in regions (red dashed boxes) proximal to the highly proliferative apical zone (AZ) or progress zone (Fig. 5Ca-c). To verify the BrdU data, we performed real-time RT-PCR for the proliferation and cell cycle regulator, cyclin D1. NICD mutants exhibited a greater than 30% increase in cyclin D1 expression as compared with controls (Fig. 5Cd). These data indicated that the limb phenotype in NICD mutants is probably caused by the suppression of MPC differentiation and not due to perturbations in limb patterning, MPC apoptosis or MPC proliferation.
Fig. 5.

Sustained activation of Notch signaling in the limb mesenchyme does not significantly affect limb patterning or apoptosis, but increases MPC proliferation during limb development. (Aa-f) In situ hybridization for Fgf8 (Aa,b), Fgf10 (Ac,d), and Ptc1 (Ae,f) on WT (Aa,c,e) and Prx1Cre; Rosa-NICDf/+ mutant (NICD; Ab,d,f) sections at E11.0. (B) TUNEL staining and statistical analyses of MPC apoptosis for WT and NICD sections at E11.0. (Ca-c) BrdU immunohistochemistry (Ca,b) and statistical analyses of MPC proliferation (Cc) for WT (Ca) and NICD (Cb) sections at E11.5. (Cd) Real-time RT-PCR for the proliferation marker, CyclinD1. *, P<0.05 versus control. AZ, apical zone. Red dashed boxes denote regions analyzed for MPC proliferation.
To determine whether Notch suppression of MPC differentiation and chondrogenesis was mediated solely via RBPjκ-dependent signaling mechanisms, we performed Notch gain-of-function experiments in the absence of the RBPjκ transcriptional effector (Prx1Cre; Rosa-NICDf/+; Rbpjkf/f) (NICD; RBPjκ). Analyses of Alizarin Red and Alcian Blue-stained skeletons at E18.5 demonstrated that in contrast to the NICD mutants, which lacked normal limbs, specific skull bones and sternum, the NICD; RBPjκ mutant animals failed to show a similar arrest in the development of these elements (Fig. 6Aa,b,d). Upon closer examination, the NICD; RBPjκ mutant animals closely resembled the RBPjκ mutant skeletons, such that they had shorter skeletal elements (red arrows highlight tibiae lengths) as compared with wild-type littermates (Fig. 6Aa,c,d). Detailed histological and molecular analyses of E12.5 hindlimb sections from wild-type, NICD; RBPjκf/+, and NICD; RBPjκf/f mutant littermates further demonstrated that suppression of MPC differentiation via Notch activation requires RBPjκ. NICD; RBPjκf/+ mutants, which for this experiment had the genotype Prx1Cre; Rosa-NICDf/+; Rbpjkf/+, displayed an identical phenotype to the previously described Prx1Cre; Rosa-NICDf/+ mutant mice (Fig. 6 NICD; RBPjκf/+ mutant compared with Fig. 4 NICD mutant). NICD mutants lacking a single Rbpjk allele (NICD; RBPjκf/+) again demonstrated a near complete suppression of MPC differentiation, resulting in limbs with only three distal digit condensations. E12.5 NICD; RBPjκf/+ limb-bud sections exhibited reduced Alcian Blue staining and complete loss of chondrogenic marker gene expression (Sox9, Col2a1 and Agc1), except for within cells confined to the three distal digits (Fig. 6Bb,e,h,k). When Gfp expression was assessed, once again the three digit condensations showed the near absence of Gfp expression and therefore lacked sustained NICD activation (Fig. 6Bn). Interestingly, NICD mutants lacking both Rbpjk alleles (NICD; RBPjκf/f) demonstrated a complete rescue of MPC differentiation and chondrogenesis. E12.5 NICD; RBPjκf/f mutant limb-bud sections showed the reappearance of all chondrogenic elements with slightly expanded and more robust Alcian Blue staining when compared with wild-type littermate controls (Fig. 6Ba,c). Additionally, in situ hybridization analyses of NICD; RBPjκf/f mutant sections demonstrated that the double mutants displayed accelerated and expanded Sox9, Col2 and Agc1 expression as compared with wild-type littermate controls, phenotypes strikingly similar to RBPjκ mutant littermates (Fig. 6Bd,f,g,i,j,l; data not shown). To determine that the genetic rescue of MPC differentiation in NICD; RBPjκf/f mutants was not due to inefficient recombination and loss of NICD expression, we performed in situ hybridization analyses for Gfp expression on adjacent sections. NICD; RBPjκf/f mutant sections displayed robust levels of Gfp expression, and therefore NICD activation, throughout much of the limb-bud mesenchyme, except for those regions previously identified in NICD; RBPjκf/+ mutant sections (Fig. 6Bn,o). Therefore, these data demonstrate for the first time that Notch suppression of MPC differentiation and chondrogenesis is solely mediated via RBPjκ-dependent signaling mechanisms.
Fig. 6.

Notch signaling suppresses MPC differentiation in an RBPJκ-dependent manner. (Aa-d) Alcian Blue/Alizarin Red staining of WT and Prx1Cre; Rosa-NICDf/+ (NICD), Prx1Cre; Rbpjkf/f (RBPjκ) and Prx1Cref; Rosa-NICDf/+; Rbpjkf/f (NICD; RBPjκ) mutant E18.5 skeletons. Black arrows indicate NICD mutant limbs. Red arrows mark the length of tibiae. Asterisks identify parietal bones. (Ba-c) AB/OG staining of WT, NICD; RBPjκf/+, and NICD; RBPjκf/f littermate hindlimb sections at E12.5. (Bd-l) In situ hybridization for Sox9 (Bd-f), Col2a1 (Bg-i), and Agc1 (Bj-l). (Bm-o) Gfp expression assesses NICD activity in WT (Bm), NICD; RBPjκf/+ (Bn), and NICD; RBPjκf/f (Bo) sections.
The RBPjκ-dependent Notch target gene, Hes1, is an important suppressor of MPC differentiation during chondrogenesis
Our data indicate that Notch regulation of MPC differentiation is mediated via RBPjκ-dependent Notch signaling mechanisms. Several RBPjκ-dependent Notch target genes of the Hes/Hey family mediate Notch control of stem/progenitor cell differentiation in several organ systems. Hes1, Hey1 and HeyL are the only Hes/Hey family members significantly expressed in limb-bud MPCs and C3H10T1/2 mesenchymal cells cultured in high-density micromass (Fig. 1B; data not shown). Therefore, we first performed loss-of-function experiments by infecting the easily transducible C3H10T1/2 mesenchymal cells with Hes1, Hey1 and HeyL shRNA viruses and then cultured cells in high-density micromass. Similar to limb-bud MPCs, the multi-potent mesenchymal cell line, C3H10T1/2, undergoes chondrogenesis when cultured in high-density micromass over a 2-week culture period (Denker et al., 1999; Haas and Tuan, 1999). C3H10T1/2 cells transduced with Hes1 shRNA virus, and not Hey1 or HeyL shRNA virus, resulted in an acceleration of chondrogenesis as assayed by Alcian Blue staining and real-time RT-PCR for Sox9, Col2a1 and Agc1 (see Fig. S3Aa-f,C in the supplementary material; data not shown) similar to our other Notch loss-of-function studies. Hey1 and/or HeyL shRNA-transduced cultures exhibited no significant change in Alcian Blue staining, with inconsistent and relatively unchanged chondrogenic gene expression (data not shown). Additionally, we performed transient Hes1 overexpression gain-of-function experiments in C3H10T1/2 micromass cultures, which demonstrated a significant suppression of chondrogenesis as assessed by Alcian Blue staining (see Fig. S3Ba-f in the supplementary material) and real-time RT-PCR analyses for Sox9, Col2a1 and Agc1 (see Fig. S3D in the supplementary material), similar to our other Notch gain-of-function studies. As Hes1 appeared to be an important regulator of mesenchymal cell differentiation using the C3H10T1/2 cell model, we performed analogous Hes1 shRNA loss-of-function studies using limb-bud-derived MPCs cultured in high-density micromass for 3, 5 and 7 days. Significant reductions in Hes1 expression resulted in accelerated chondrogenesis as observed by enhanced Alcian Blue staining (Fig. 7Aa-f) and elevated gene expression of Sox9, Col2a1 and Agc1 at nearly all time-points in Hes1 shRNA cultures (Fig. 7B). At the later time-points, days 5 and 7, Agc1 expression is unchanged or mildly suppressed, suggesting a potential role for Hes1 in promoting chondrocyte maturation or maintaining the committed chondrocyte phenotype. This is consistent with our experiments in which limb-bud-derived MPCs cultured in high-density micromass were treated with the Notch inhibitor, DAPT (Fig. 2A). Collectively, these data suggest that Hes1 is an RBPjκ-dependent Notch target gene of the Hes/Hey family expressed in MPCs and important for Notch-mediated suppression of MPC differentiation during chondrogenesis.
Fig. 7.

Hes1 is an important RBPjκ-dependent Notch target gene that suppresses MPC differentiation and chondrogenesis. (Aa-f) Alcian Blue staining of control infected (Aa,c,e) and Hes1 shRNA-infected (shHes1; Ab,d,f) limb-bud MPC micromass cultures. (B) Real-time RT-PCR for Sox9, Col2a1, Agc1 and Hes1. The y-axis of graphs is the relative gene expression normalized to β-actin and to the control at day 3. *, P<0.05 versus control. d, days.
DISCUSSION
In this study, we provided the first genetic evidence that RBPjκ-dependent Notch signaling regulates MPC proliferation and differentiation during skeletal development. Use of Notch gain- and loss-of-function genetic approaches demonstrated that the RBPjκ-dependent Notch pathway is the sole Notch signaling mechanism regulating MPCs during early skeletal development. Additional in vitro data also indicated that manipulation of the Notch pathway does not bias lineage commitment, but rather suppresses the differentiation of MPCs prior to their commitment to become osteoblasts, chondrocytes or adipocytes. Finally, our in vivo and in vitro results argue that Notch regulation of MPC differentiation acts upstream of both Sox9 and Runx2 during skeletogenic differentiation, and is potentially mediated by the RBPjκ-dependent Notch target gene, Hes1.
Several reports have indicated that Notch signaling components are expressed during early vertebrate limb development. These data have analyzed the expression of a few select Notch molecules using varying methodologies. Wholemount in situ hybridization analyses for Notch1 and Jag2 demonstrated in vivo expression of these genes within the AER and overlying ectoderm of the developing limb-bud (Francis et al., 2005; Williams et al., 1995), whereas semi-quantitative RT-PCR studies using limb-bud MPC micromass cultures indicated that Notch1, Notch2, Notch3, delta-like 1, jagged 1 and jagged 2 were detectable to varying degrees within the first 5 days of culture (Fujimaki et al., 2006). Here, we have expanded upon these studies by characterizing the expression of all Notch ligands, receptors and classical target genes of the Hes/Hey family by assessing their quantitative expression in limb-bud MPC micromass cultures using real-time RT-PCR, as well as in situ hybridization on tissue sections to determine more precisely their cellular/tissue distribution. Although much of our data is consistent with the previous studies, our combined approach using quantitative real-time RT-PCR and in situ hybridization localization studies have identified Jag1 as the primary Notch ligand robustly expressed in the medial and distal limb-bud mesenchyme, whereas Dll1 shows lower levels of expression throughout the limb-bud mesenchyme. Our gene expression and western blot data further demonstrated that Notch2 is the prominent, active receptor expressed in the limb-bud mesenchyme both in vitro and in vivo. Finally, we identified Hes1 as a candidate Notch target gene that is expressed primarily in undifferentiated MPCs as compared with Hey1 and HeyL, which are upregulated in differentiating MPCs and ubiquitously expressed during early limb development. Based on the overlapping expression domains of Jag1, Notch2, Hes1 and Hey1 proximal to the apical zone, it is interesting to speculate that the jagged 1 ligand might signal through the Notch2 receptor to activate Hes/Hey target genes in an RBPjκ-dependent manner during limb development. This jagged 1-Notch2/RBPjκ-Hes/Hey signal might function to maintain MPCs in an undifferentiated state as they exit cellular regions under the influence of the AER and become exposed to external signals provoking their differentiation. Consistent with this hypothesis, transient viral overexpression of a Hes/Hey transcription factor homologue, Hairy1a, in chick limb-bud mesenchyme delays the progression of MPC differentiation and chondrogenesis (Vasiliauskas et al., 2003). It is also of note that Notch2 acts as a genetic modifier of Jag1 mutations in a mouse model for Alagille syndrome, which is an autosomal dominant developmental disorder characterized by craniofacial, rib, vertebral and limb skeletal abnormalities, as well as other non-skeletal-related anomalies (McCright et al., 2002; McDaniell et al., 2006).
To determine whether the Notch signaling molecules expressed in limb-bud-derived MPCs were important regulators of MPC differentiation, we performed in vitro MPC differentiation assays using the gamma-secretase and Notch inhibitor, DAPT. Our data indicate that by inhibiting gamma-secretase and/or Notch signals in MPCs, we effectively removed a “break” or “pacemaker” on differentiation and thereby allowed MPCs to more rapidly respond to differentiating cues provided in the chondrogenic, osteogenic or adipogenic cultures. Consistent with these data, several groups have reported on the ability of Notch signaling to regulate in vitro chondrogenic, osteogenic and adipogenic differentiation from various progenitor and immature cell types (Fujimaki et al., 2006; Hilton et al., 2008; Oldershaw et al., 2008; Ross et al., 2006). Similar effects were also observed in vivo from our conditional RBPjκ loss-of-function mice, such that mutant embryos formed mesenchymal condensations earlier than control littermates and MPCs within condensations underwent a more rapid progression of chondrogenic differentiation. Conversely, our conditional NICD gain-of-function mice demonstrated that targeted MPCs with sustained Notch activation are maintained in an undifferentiated state, even in the presence of relatively normal patterning and differentiation signals. It is also worth noting that these targeted in vivo gain-of-function phenotypes might have resulted from additional secondary effects to surrounding cell populations within the developing limb. Collectively, these data, along with our expression studies, further support the notion that jagged 1/Notch2/RBPjκ signaling during early limb development might aid in controlling the onset and pace of differentiation for MPCs as they exit control from the AER. Normal transient downregulation of the Notch signal in some MPCs would allow cells to respond to both patterning and differentiating cues during limb-bud development, whereas Notch active cells would be more resistant to these signals and thereby be maintained in a more immature or undifferentiated state.
During endochondral bone development, skeletal cells (chondrocytes and osteoblasts) are derived from the earliest common mesenchymal progenitor population originating in the lateral plate mesoderm. As these MPCs differentiate toward the osteoblast or chondrocyte lineages, they pass through at least one bi-potential intermediate known as the chondro-osteo progenitor (COP) cell. Whereas MPCs can be targeted by the Prx1Cre transgene (Durland et al., 2008; Logan et al., 2002; Martin and Olson, 2000) (see Fig. S1 in the supplementary material), COP cells are targeted by the Col2a1Cre transgene (Hilton et al., 2007; Szabova et al., 2009) (Fig. 8). Here, we have used Prx1Cre mice to perform both Notch gain- and loss-of-function studies in vivo, demonstrating the importance of RBPjκ-dependent Notch signaling in controlling MPCs prior to chondrocyte or osteoblast differentiation (Fig. 8A). These data demonstrated that Notch activation in MPCs inhibits development of COP cells, and thereby inhibits formation of any cartilage or bone. Moreover, we determined that this regulation occurs at or upstream of Sox9 expression, which is subsequently required for Runx2 expression and COP differentiation. Recently, the Notch pathway has also been implicated in regulating later stages of chondrocyte development (Mead and Yutzey, 2009). Col2a1Cre; Rosa-NICD mice were used to activate Notch in COPs, resulting in delays to both chondrocyte and osteoblast maturation (Fig. 8B), although cartilage and bone formation do eventually occur, unlike in our Prx1Cre model. Interestingly, activation of Notch in COPs led to reductions in chondrocyte proliferation (Mead and Yutzey, 2009) (Fig. 8B), whereas Notch activation in MPCs enhanced proliferation of limb-bud mesenchyme (Fig. 5Ca-d; Fig. 8A). RBPjκ-dependent Notch loss-of-function in COPs, using the same Col2a1Cre, resulted in increased chondrocyte proliferation and delayed maturation (Mead and Yutzey, 2009), whereas removal of upstream Notch components (N1/N2 or PS1/PS2), using the Prx1Cre, resulted in decreased chondrocyte proliferation and delayed maturation (Hilton et al., 2008). The varied chondrocyte proliferation effects observed in these Notch loss-of-function animal models might be due to alterations in the different Cre-targeted cell populations or, alternatively, might suggest additional RBPjκ-independent Notch signaling effects within chondrocytes. It is also of note that the severe phenotypic effects observed in our Prx1Cre; Rbpjk mutant mice could be due to the combined effects on both the MPC population and the more differentiated COP cells. The Notch effects described here during differentiation of the chondrocyte and osteoblast lineages are summarized in Fig. 8. Investigations into Notch regulation directly within the osteoblast lineage have also uncovered various and sometimes opposing Notch effects on osteoblast proliferation, differentiation and function, which is probably due to the timing at which Notch signaling was removed or activated during maturation of the osteoblast lineage (Engin et al., 2008; Hilton et al., 2008; Zanotti et al., 2008). All of these data, as well as our studies described above, have recently been summarized in several reviews (Engin and Lee, 2009; Zanotti and Canalis, 2009). Notch signaling within the skeletal system is complex, and only more detailed genetic studies will be able to determine whether the Notch-specific effects on MPCs, chondrocytes and osteoblasts are mediated via RBPjκ-dependent or -independent mechanisms, at which specific stage of differentiation and within which cell type Notch regulates context-specific proliferation and maturation, and which Notch target genes are the crucial mediators of Notch function in skeletal cells.
Fig. 8.
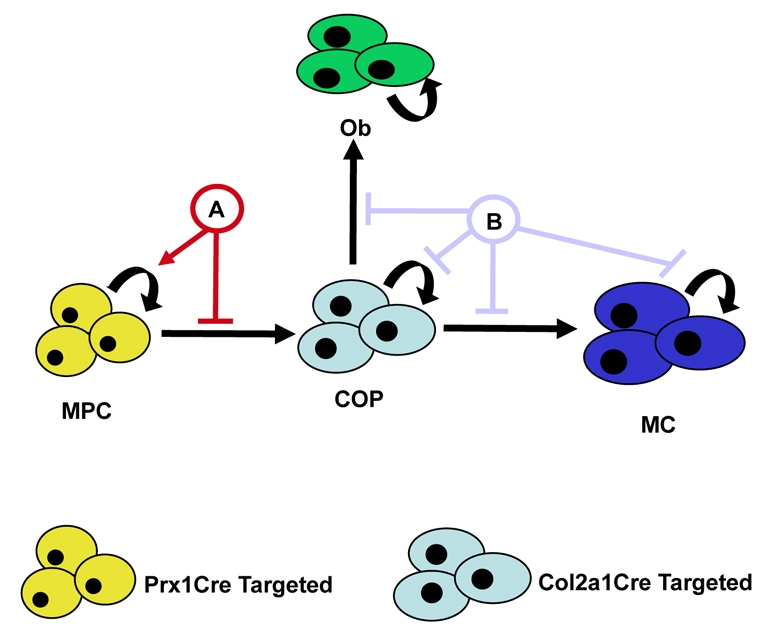
Model for Notch regulation of MPC and COP differentiation during cartilage development. Cartilage and bone development begins from the earliest common precursor, the MPC (yellow), which differentiates to become a more lineage-restricted COP (light blue) cell. Further differentiation allows COPs to adopt either an osteoblastic (Ob, green) fate or that of a maturing chondrocyte (MC, dark blue). Arrows indicate induction of proliferation or differentiation; perpendicular lines indicate suppression. Curved arrows indicate cell proliferation/self-renewal. A, RBPjκ-dependent/Hes1 regulation of MPC proliferation and differentiation (Figs 1, 2, 3, 4, 5, 6 and 7); B, Notch regulation of COP differentiation and chondrocyte proliferation (Mead and Yutzey, 2009).
Supplementary Material
Acknowledgements
We thank Dr Doug Melton for providing Rosa-NICD floxed mice. We also thank Drs Urban Lendahl, David Ornitz and Fanxin Long for plasmid constructs. We gratefully acknowledge the technical expertise of Ryan Tierney and Nehal Porecha within the Center for Musculoskeletal Research Histology Core for processing all tissue samples. The bTAN 20 (Notch1) and C651.6DdHN (Notch2) antibodies developed by Spyros Artavanis-Tsakonas were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained at the University of Iowa. This work was supported by start-up funds from the Department of Orthopaedics at the University of Rochester Medical Center to M.J.H. A.K. is supported as a doctoral student by the NIH T32 training grant AR053459 (R.J.O.). Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.042911/-/DC1
References
- Akiyama H., Chaboissier M. C., Martin J. F., Schedl A., de Crombrugghe B. (2002). The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813-2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H., Kim J. E., Nakashima K., Balmes G., Iwai N., Deng J. M., Zhang Z., Martin J. F., Behringer R. R., Nakamura T., et al. (2005). Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA 102, 14665-14670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apelqvist A., Li H., Sommer L., Beatus P., Anderson D. J., Honjo T., Hrabe de Angelis M., Lendahl U., Edlund H. (1999). Notch signalling controls pancreatic cell differentiation. Nature 400, 877-881 [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S., Rand M. D., Lake R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science 284, 770-776 [DOI] [PubMed] [Google Scholar]
- Axelrod J. D., Matsuno K., Artavanis-Tsakonas S., Perrimon N. (1996). Interaction between Wingless and Notch signaling pathways mediated by dishevelled. Science 271, 1826-1832 [DOI] [PubMed] [Google Scholar]
- Bellusci S., Grindley J., Emoto H., Itoh N., Hogan B. L. (1997). Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 124, 4867-4878 [DOI] [PubMed] [Google Scholar]
- Bi W., Deng J. M., Zhang Z., Behringer R. R., de Crombrugghe B. (1999). Sox9 is required for cartilage formation. Nat. Genet. 22, 85-89 [DOI] [PubMed] [Google Scholar]
- Blokzijl A., Dahlqvist C., Reissmann E., Falk A., Moliner A., Lendahl U., Ibanez C. F. (2003). Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 163, 723-728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S. (2006). Notch signaling in stem cell systems. Stem Cells 24, 2437-2447 [DOI] [PubMed] [Google Scholar]
- Crossley P. H., Martin G. R. (1995). The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development 121, 439-451 [DOI] [PubMed] [Google Scholar]
- Dahlqvist C., Blokzijl A., Chapman G., Falk A., Dannaeus K., Ibanez C. F., Lendahl U. (2003). Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development 130, 6089-6099 [DOI] [PubMed] [Google Scholar]
- de la Pompa J. L., Wakeham A., Correia K. M., Samper E., Brown S., Aguilera R. J., Nakano T., Honjo T., Mak T. W., Rossant J., et al. (1997). Conservation of the Notch signalling pathway in mammalian neurogenesis. Development 124, 1139-1148 [DOI] [PubMed] [Google Scholar]
- Demehri S., Morimoto M., Holtzman M. J., Kopan R. (2009). Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol. 7, e1000067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker A. E., Haas A. R., Nicoll S. B., Tuan R. S. (1999). Chondrogenic differentiation of murine C3H10T1/2 multipotential mesenchymal cells: I. Stimulation by bone morphogenetic protein-2 in high-density micromass cultures. Differentiation 64, 67-76 [DOI] [PubMed] [Google Scholar]
- Diederich R. J., Matsuno K., Hing H., Artavanis-Tsakonas S. (1994). Cytosolic interaction between deltex and Notch ankyrin repeats implicates deltex in the Notch signaling pathway. Development 120, 473-481 [DOI] [PubMed] [Google Scholar]
- Dong Y., Drissi H., Chen M., Chen D., Zuscik M. J., Schwarz E. M., O'Keefe R. J. (2005). Wnt-mediated regulation of chondrocyte maturation: modulation by TGF-beta. J. Cell Biochem. 95, 1057-1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoviel D. B., Hadjantonakis A. K., Ikeda M., Zheng H., Hyslop P. S., Bernstein A. (1999). Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 13, 2801-2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drissi H., Luc Q., Shakoori R., Chuva De Sousa Lopes S., Choi J. Y., Terry A., Hu M., Jones S., Neil J. C., Lian J. B., et al. (2000). Transcriptional autoregulation of the bone related CBFA1/RUNX2 gene. J. Cell. Physiol. 184, 341-350 [DOI] [PubMed] [Google Scholar]
- Durland J. L., Sferlazzo M., Logan M., Burke A. C. (2008). Visualizing the lateral somitic frontier in the Prx1Cre transgenic mouse. J. Anat. 212, 590-602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F., Lee B. (2009). NOTCHing the bone: Insights into multi-functionality. Bone 46, 274-280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F., Yao Z., Yang T., Zhou G., Bertin T., Jiang M. M., Chen Y., Wang L., Zheng H., Sutton R. E., et al. (2008). Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med. 14, 299-305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis J. C., Radtke F., Logan M. P. (2005). Notch1 signals through Jagged2 to regulate apoptosis in the apical ectodermal ridge of the developing limb bud. Dev. Dyn. 234, 1006-1015 [DOI] [PubMed] [Google Scholar]
- Fre S., Huyghe M., Mourikis P., Robine S., Louvard D., Artavanis-Tsakonas S. (2005). Notch signals control the fate of immature progenitor cells in the intestine. Nature 435, 964-968 [DOI] [PubMed] [Google Scholar]
- Fujimaki R., Toyama Y., Hozumi N., Tezuka K. (2006). Involvement of Notch signaling in initiation of prechondrogenic condensation and nodule formation in limb bud micromass cultures. J. Bone Miner. Metab. 24, 191-198 [DOI] [PubMed] [Google Scholar]
- Haas A. R., Tuan R. S. (1999). Chondrogenic differentiation of murine C3H10T1/2 multipotential mesenchymal cells: II. Stimulation by bone morphogenetic protein-2 requires modulation of N-cadherin expression and function. Differentiation 64, 77-89 [DOI] [PubMed] [Google Scholar]
- Hadland B. K., Huppert S. S., Kanungo J., Xue Y., Jiang R., Gridley T., Conlon R. A., Cheng A. M., Kopan R., Longmore G. D. (2004). A requirement for Notch1 distinguishes 2 phases of definitive hematopoiesis during development. Blood 104, 3097-3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada Y., Kadokawa Y., Okabe M., Ikawa M., Coleman J. R., Tsujimoto Y. (1999). Mutation in ankyrin repeats of the mouse Notch2 gene induces early embryonic lethality. Development 126, 3415-3424 [DOI] [PubMed] [Google Scholar]
- Han H., Tanigaki K., Yamamoto N., Kuroda K., Yoshimoto M., Nakahata T., Ikuta K., Honjo T. (2002). Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int. Immunol. 14, 637-645 [DOI] [PubMed] [Google Scholar]
- Hatakeyama J., Bessho Y., Katoh K., Ookawara S., Fujioka M., Guillemot F., Kageyama R. (2004). Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development 131, 5539-5550 [DOI] [PubMed] [Google Scholar]
- Hayward P., Brennan K., Sanders P., Balayo T., DasGupta R., Perrimon N., Martinez Arias A. (2005). Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development 132, 1819-1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom M., Phng L. K., Hofmann J. J., Wallgard E., Coultas L., Lindblom P., Alva J., Nilsson A. K., Karlsson L., Gaiano N., et al. (2007). Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776-780 [DOI] [PubMed] [Google Scholar]
- Hilton M. J., Tu X., Cook J., Hu H., Long F. (2005). Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development 132, 4339-4351 [DOI] [PubMed] [Google Scholar]
- Hilton M. J., Tu X., Long F. (2007). Tamoxifen-inducible gene deletion reveals a distinct cell type associated with trabecular bone, and direct regulation of PTHrP expression and chondrocyte morphology by Ihh in growth region cartilage. Dev. Biol. 308, 93-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton M. J., Tu X., Wu X., Bai S., Zhao H., Kobayashi T., Kronenberg H. M., Teitelbaum S. L., Ross F. P., Kopan R., et al. (2008). Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med. 14, 306-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitoshi S., Alexson T., Tropepe V., Donoviel D., Elia A. J., Nye J. S., Conlon R. A., Mak T. W., Bernstein A., van der Kooy D. (2002). Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 16, 846-858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabe de Angelis M., McIntyre J., 2nd, Gossler A. (1997). Maintenance of somite borders in mice requires the Delta homologue DII1. Nature 386, 717-721 [DOI] [PubMed] [Google Scholar]
- Iso T., Sartorelli V., Poizat C., Iezzi S., Wu H. Y., Chung G., Kedes L., Hamamori Y. (2001). HERP, a novel heterodimer partner of HES/E(spl) in Notch signaling. Mol. Cell. Biol. 21, 6080-6089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso T., Kedes L., Hamamori Y. (2003). HES and HERP families: multiple effectors of the Notch signaling pathway. J. Cell. Physiol. 194, 237-255 [DOI] [PubMed] [Google Scholar]
- Itoh F., Itoh S., Goumans M. J., Valdimarsdottir G., Iso T., Dotto G. P., Hamamori Y., Kedes L., Kato M., ten Dijke P. (2004). Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J. 23, 541-551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J., Pedersen E. E., Galante P., Hald J., Heller R. S., Ishibashi M., Kageyama R., Guillemot F., Serup P., Madsen O. D. (2000). Control of endodermal endocrine development by Hes-1. Nat. Genet. 24, 36-44 [DOI] [PubMed] [Google Scholar]
- Kopan R., Ilagan M. X. (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisato A., Chiba S., Nakagami-Yamaguchi E., Kumano K., Saito T., Masuda S., Yamaguchi T., Osawa M., Kageyama R., Nakauchi H., et al. (2003). HES-1 preserves purified hematopoietic stem cells ex vivo and accumulates side population cells in vivo. Blood 101, 1777-1783 [DOI] [PubMed] [Google Scholar]
- Lai E. C. (2002). Keeping a good pathway down: transcriptional repression of Notch pathway target genes by CSL proteins. EMBO Rep. 3, 840-845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai E. C. (2004). Notch signaling: control of cell communication and cell fate. Development 131, 965-973 [DOI] [PubMed] [Google Scholar]
- Logan M., Martin J. F., Nagy A., Lobe C., Olson E. N., Tabin C. J. (2002). Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77-80 [DOI] [PubMed] [Google Scholar]
- Martin J. F., Olson E. N. (2000). Identification of a prx1 limb enhancer. Genesis 26, 225-229 [PubMed] [Google Scholar]
- Martinez Arias A., Zecchini V., Brennan K. (2002). CSL-independent Notch signalling: a checkpoint in cell fate decisions during development? Curr. Opin. Genet. Dev. 12, 524-533 [DOI] [PubMed] [Google Scholar]
- Matsuno K., Go M. J., Sun X., Eastman D. S., Artavanis-Tsakonas S. (1997). Suppressor of Hairless-independent events in Notch signaling imply novel pathway elements. Development 124, 4265-4273 [DOI] [PubMed] [Google Scholar]
- McCright B., Lozier J., Gridley T. (2002). A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 129, 1075-1082 [DOI] [PubMed] [Google Scholar]
- McDaniell R., Warthen D. M., Sanchez-Lara P. A., Pai A., Krantz I. D., Piccoli D. A., Spinner N. B. (2006). NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 79, 169-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod M. J. (1980). Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology 22, 299-301 [DOI] [PubMed] [Google Scholar]
- Mead T. J., Yutzey K. E. (2009). Notch pathway regulation of chondrocyte differentiation and proliferation during appendicular and axial skeleton development. Proc. Natl. Acad. Sci. USA 106, 14420-14425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiadis T. A., Lardelli M., Lendahl U., Thesleff I. (1995). Expression of Notch 1, 2 and 3 is regulated by epithelial-mesenchymal interactions and retinoic acid in the developing mouse tooth and associated with determination of ameloblast cell fate. J. Cell Biol. 130, 407-418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh L. C., Stanger B. Z., Kwan K. M., Melton D. A. (2003). Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. USA 100, 14920-14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswander L., Jeffrey S., Martin G. R., Tickle C. (1994). A positive feedback loop coordinates growth and patterning in the vertebrate limb. Nature 371, 609-912 [DOI] [PubMed] [Google Scholar]
- Ohtsuka T., Ishibashi M., Gradwohl G., Nakanishi S., Guillemot F., Kageyama R. (1999). Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. EMBO J. 18, 2196-2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka C., Nakano T., Wakeham A., de la Pompa J. L., Mori C., Sakai T., Okazaki S., Kawaichi M., Shiota K., Mak T. W., et al. (1995). Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development 121, 3291-3301 [DOI] [PubMed] [Google Scholar]
- Oldershaw R. A., Tew S. R., Russell A. M., Meade K., Hawkins R., McKay T. R., Brennan K. R., Hardingham T. E. (2008). Notch signaling through Jagged-1 is necessary to initiate chondrogenesis in human bone marrow stromal cells but must be switched off to complete chondrogenesis. Stem Cells 26, 666-674 [DOI] [PubMed] [Google Scholar]
- Ramain P., Khechumian K., Seugnet L., Arbogast N., Ackermann C., Heitzler P. (2001). Novel Notch alleles reveal a Deltex-dependent pathway repressing neural fate. Curr. Biol. 11, 1729-1738 [DOI] [PubMed] [Google Scholar]
- Robert-Moreno A., Espinosa L., de la Pompa J. L., Bigas A. (2005). RBPjkappa-dependent Notch function regulates Gata2 and is essential for the formation of intra-embryonic hematopoietic cells. Development 132, 1117-1126 [DOI] [PubMed] [Google Scholar]
- Ross D. A., Kadesch T. (2001). The notch intracellular domain can function as a coactivator for LEF-1. Mol. Cell. Biol. 21, 7537-7544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross D. A., Hannenhalli S., Tobias J. W., Cooch N., Shiekhattar R., Kadesch T. (2006). Functional analysis of Hes-1 in preadipocytes. Mol. Endocrinol. 20, 698-705 [DOI] [PubMed] [Google Scholar]
- Shutter J. R., Scully S., Fan W., Richards W. G., Kitajewski J., Deblandre G. A., Kintner C. R., Stark K. L. (2000). Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 14, 1313-1318 [PMC free article] [PubMed] [Google Scholar]
- Stier S., Cheng T., Dombkowski D., Carlesso N., Scadden D. T. (2002). Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood 99, 2369-2378 [DOI] [PubMed] [Google Scholar]
- Swiatek P. J., Lindsell C. E., del Amo F. F., Weinmaster G., Gridley T. (1994). Notch1 is essential for postimplantation development in mice. Genes Dev. 8, 707-719 [DOI] [PubMed] [Google Scholar]
- Szabova L., Yamada S. S., Wimer H., Chrysovergis K., Ingvarsen S., Behrendt N., Engelholm L. H., Holmbeck K. (2009). MT1-MMP and type II collagen specify skeletal stem cells and their bone and cartilage progeny. J. Bone Miner. Res. 24, 1905-1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacca A., Felli M. P., Palermo R., Di Mario G., Calce A., Di Giovine M., Frati L., Gulino A., Screpanti I. (2006). Notch3 and pre-TCR interaction unveils distinct NF-kappaB pathways in T-cell development and leukemia. EMBO J. 25, 1000-1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum-Finney B., Xu L., Brashem-Stein C., Nourigat C., Flowers D., Bakkour S., Pear W. S., Bernstein I. D. (2000). Pluripotent, cytokine-dependent, hematopoietic stem cells are immortalized by constitutive Notch1 signaling. Nat. Med. 6, 1278-1281 [DOI] [PubMed] [Google Scholar]
- Vasiliauskas D., Laufer E., Stern C. D. (2003). A role for hairy1 in regulating chick limb bud growth. Dev. Biol. 262, 94-106 [DOI] [PubMed] [Google Scholar]
- Vilimas T., Mascarenhas J., Palomero T., Mandal M., Buonamici S., Meng F., Thompson B., Spaulding C., Macaroun S., Alegre M. L., et al. (2007). Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat. Med. 13, 70-77 [DOI] [PubMed] [Google Scholar]
- Vujovic S., Henderson S. R., Flanagan A. M., Clements M. O. (2007). Inhibition of gamma-secretases alters both proliferation and differentiation of mesenchymal stem cells. Cell Prolif. 40, 185-195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Shelly L., Miele L., Boykins R., Norcross M. A., Guan E. (2001). Human Notch-1 inhibits NF-kappa B activity in the nucleus through a direct interaction involving a novel domain. J. Immunol. 167, 289-295 [DOI] [PubMed] [Google Scholar]
- Williams R., Lendahl U., Lardelli M. (1995). Complementary and combinatorial patterns of Notch gene family expression during early mouse development. Mech. Dev. 53, 357-368 [DOI] [PubMed] [Google Scholar]
- Xue Y., Gao X., Lindsell C. E., Norton C. R., Chang B., Hicks C., Gendron-Maguire M., Rand E. B., Weinmaster G., Gridley T. (1999). Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 8, 723-730 [DOI] [PubMed] [Google Scholar]
- Zanotti S., Canalis E. (2009). Notch and the skeleton. Mol. Cell. Biol. 30, 886-896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S., Smerdel-Ramoya A., Stadmeyer L., Durant D., Radtke F., Canalis E. (2008). Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology 149, 3890-3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Ziran N., Goater J. J., Schwarz E. M., Puzas J. E., Rosier R. N., Zuscik M., Drissi H., O'Keefe R. J. (2004). Primary murine limb bud mesenchymal cells in long-term culture complete chondrocyte differentiation: TGF-beta delays hypertrophy and PGE2 inhibits terminal differentiation. Bone 34, 809-817 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.