

Abstract
The design, synthesis and biological evaluation of two potential (+)-spongistatin 1 analogs have been achieved. The analogs, incorporating tethers (red) in place of the ABCD and the CD components of the (+)-spongistatin 1 macrolide were designed such that the conformations of the retained skeleton (blue) would mimic the assigned major solution conformation of the natural product The nanomolar cytotoxicity observed for the ABEF analog, provides strong support for the assigned solution conformation.
Members of the spongistatin family of natural products were independently isolated by three research groups: Pettit1 (spongistatins), Kitigawa2 (altohyrtins), and Fusetani3 (cinachyrolide), from the marine sponges Spongia, Spirastrella, and Cinachyra respectively. Spongistatin 1[(+)-1], the most abundant congener, has been characterized as an antimitotic agent that inhibits tubulin polymerization by binding in the tubulin vinca alkaloid domain.4 Importantly subnanomolar cytotoxicity against several chemoresistant cancer cell lines has been demonstrated, making (+)-spongistatin 1 and congeners thereof important antitumor lead compounds.5 From the structural perspective the spongistatins also display significant architectural complexity; common features include the polyether backbone with 24 stereocenters, two spiroketals, a bis-pyran unit embedded within a 42-membered macrolide and a triene side chain bearing a vinyl chloride. The first total syntheses of (+)-spongistatin 2 by Evans6 and (+)-spongistatin 1 by Kishi,7 confirming each structure, have been followed by syntheses from the Smith,8 Paterson,9 Crimmins,10 Heathcock11 and the Ley12 Laboratories. A variety of synthetic approaches to various fragments of the spongistatins have also been reported,13 as well as the synthesis of 1 gram of (+)-spongistatin 1 (1) reported by this Laboratory.14
The design and synthesis of spongistatin analogs has been impeded by the molecular complexity, with only a few studies reported. Early on, Kishi and coworkers7 reported that epimerization of the CD spiroketal at C(23) affords an analog possessing cytotoxicity similar to (+)-spongistatin 1, while our laboratory reported that side chain analogs based on the α-D-glucose scaffold maintain modest micromolar activity, albeit most likely via a different mechanism.15 Subsequently Paterson et al.16 disclosed that unsaturation of the E-ring, achieved by elimination of water at C(35,36) and formation of a double bond affords an analog with increased cytotoxicity relative to (+)-spongistatin 1; a similar observation was made for spongistatin 2 by Heathcock and coworkers,11 while truncation of the triene side chain by Paterson resulted in a dramatic loss of activity.16
More recently, Heathcock reported spongistatin analogs, including acyclic congeners having only the E and F rings, as well as cyclic EF, ABEF and ABCD analogs, where at least one ring had been replaced with a polyethylene linker,17 do not maintain significant cytotoxicity.
Taken together, the available SAR data suggests that the E and F rings, as well as the triene side chain, are critical for biological activity. The question thus becomes: How important are the AB and CD spiroketals? Are these structural units required for potent cyctotoxicity, or simply to enforce the bioactive conformation, and in particular the nearly linear conformation of the western perimeter as observed in the assigned solution conformation.18
To achieve answers, we chose a minimalist design strategy, specifically to maintain a macrolide structure, first with excision of the AB and CD spiroketal units, and then an analog lacking only the CD unit. Two design criteria were foremost: (1) Analog construction would take advantage of advanced intermediates readily available either from our one-gram synthesis and/or commercially; and (2) We would select tethers that would enforce the western perimeter to maintain the same low energy linear conformation as defined by our solution conformation.18
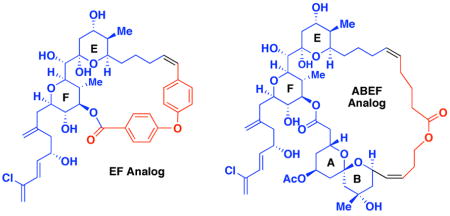
We first targeted macrolide 3 which maintained the EF bis-pyran system, but lacked the AB and CD spiroketals (Figure 2). Possible C(1)-C(28) tethers were selected based on length, orientation and flexibility, after in silico screening of known fragment libraries. We then performed Monte Carlo conformational searches (5000 steps) on analogs featuring these linkers. Analogs preserving the same low energy conformation were subjected to full conformational searches. These studies resulted in selection of the biaryl ether tether (Figure 2). Overlay of the resulting analog 3 with the twisted or “infinity” shaped major solution conformation of spongistatin in water revealed excellent overlap along the western perimeter specifically along the E, F and AB rings.
Figure 2.

(a) EF analog and (b) Overlay of EF analog and predicted solution conformation of (+)-spongistatin 1.
From the synthetic perspective, the requisite biaryl ether tether was anticipated to be available via a modified Ullmann ether synthesis,19 employing the TIPS ester of 6 and 4-formylphenyl boronic acid. With aldehyde 5 in hand, analog 3 would then be constructed using the same endgame strategy as in our gram synthesis of (+)-spongistatin 1, employing EF Wittig salt (+)-4.
Towards this end, deprotonation of (+)-4 was achieved with MeLi•LiBr, followed by union with 5 led to a 4:1 mixture of Z and E olefin isomers which could not be readily separated. The poor selectivity is in contrast to that observed in our (+)-spongistatin 1 synthesis which produced exclusively the Z-olefin isomer. The mixture of olefin isomers was next subjected to 3 equivalents of TBAF to remove the TIPS group, as well as the two F-ring TES groups; the seco-acid olefin isomers (8) still could not be separated. Pleasingly however macrocyclization occured only with the Z olefin isomer to furnish the desired product in 62% yield. Presumably the strain associated with incorporating an E-olefin prevented macrocyclization. Global deprotection led to an 11:1 mixture of products. However upon detailed 1- and 2-D NMR analysis, the major product, (+)-9, proved to be the ring-opened congener of the desired EF analog 3, as evidenced by the presence of a 13C NMR signal at 211.4 ppm. Presumably, the ring strain of the tether is sufficient to drive the equilibrium towards the open δ-hydroxyketone congener of 3.
We next turned our attention to an analog lacking only the CD spiroketal unit (e.g., ABEF analog 10, Figure 3). Conformational searches performed on the Heathcock ABEF spongistatin 2 analogs indicated that the linkers selected were not capable of preserving the nearly linear western perimeter of the spongistatins. We therefore undertook calculations that eventually led to a range of potential B to E tethers which were able to maintain the assigned solution conformation of (+)-spongistatin 1 (i.e., linear). Selection of the B to F tether illustrated in Figure 3 was based both on the ease of synthesis and the ability to engage in hydrogen bonding. That is, the length of the linker, as well as placement and orientation of the ester bond, were optimized to increase the probability of a hydrogen bond between the ester carbonyl and the C(42) hydroxyl group on the F ring, which would maintain the desired linear western perimeter conformation. The position of a Z-olefin adjacent to the B ring was also selected both to rigidify the linker by allylic strain and to simplify the synthesis.
Figure 3.

Retrosynthetic Analysis for ABEF analog 10 and overlay with (+)-spongistatin 1
Construction of the ABEF analog (10) was again envisioned to take advantage of EF Wittig salt (+)-4, now with AB aldehyde 11 (Figure 3). The tether would be incorporated into 11 in two segments; first a three carbon unit would be introduced by Wittig olefination of known AB aldehyde (−)-15,20 followed by deprotection and esterification of 12 with known acid 13,21 available from δ-valerolactone. Towards this end, Parikh-Doering oxidation22 of alcohol (−)-17 (Scheme 2), followed by olefination with Wittig salt 16 produced (−)-18 in 51% yield, along with deacylated (−)-19, which was re-acetylated to furnish an overall yield of 63%.
Scheme 2.

Synthesis of ABEF analog (−)-10.
At this juncture, the t-butyl ester was converted to a TIPS-ester, which was envisioned to be more compatible with our endgame strategy. A three step sequence was required: (1) treatment with TMSOTf, in the presence of 2,6-lutidine; (2) exposure of the reaction mixture to KF and (3) conversion to the TIPS-esterby treatment with TIPSCl and Et3N provided alcohol (−)-12 in 96% yield. For chain extension, alcohol (−)-12 was esterified with carboxylic acid 13 (88%), the latter available in one step via hydrolysis of δ-valerolactone in the presence of PMBCl.21b Oxidative removal of the PMB ether from (−)-12, followed by Parikh-Doering oxidation22 then provided (−)-11.
With aldehyde (−)-11 in hand, union with EF Wittig salt (+)-4 furnished the full seco ABEF carbon skeleton, possessing exclusively the Z-olefin adjacent to the AB ring, which upon treatment with 3 equivalents of TBAF, Yamaguchi macrocyclization of the derived seco-acid (−)-22, and global deprotection at low temperature afforded ABEF analog (−)-10. The structure, and in particular the integrity of the E ring hemiketal, was confirmed by NMR spectroscopy. Importantly, assignment of the solution conformation of analog (−)-10, defined exploiting NMR methods and deconvolution tactics employed in our solution assignment of (+)-spongistatin 1, are in accordance with the initially calculated conformations.
Biological evaulation reveal that analog (+)-9 (Table 1), not surprisingly given the missing E ring hemiketal structural component, was inactive against all four cell lines tested. However analog (−)-10, wherein the AB spiroketal is retained, but lacks the CD spiroketal displayed nanomolar activities against the tested cancer cell lines. Studies demonstrating that mode of action of (−)-10 is similar to (+)-spongistatin 1 (1) will be published in due course.
Table 1.
IC50 (nM) values of spongistatin 1 and analogs in the cell growth inhibition assay with different cancer cell lines.
| MDA-MB-435 | HT-29 | H522-T1 | U937 | |
|---|---|---|---|---|
| (+)-Spongistatin 1 | 0.0225 | 0.058 | 0.16 | 0.059 |
| (+)-9 | >1000 | >1000 | >1000 | >1000 |
| (−)-10 | 82.8 | 161.2 | 297.2 | 60.5 |
In summary, the design and synthesis of a (+)-spongistatin 1 ABEF analog (−)-10, based on the assigned solution conformation of (+)-spongistatin 1 (1), possessing nanomolar cell growth inhibitory activity against several human tumor cell lines, has been achieved, and as such provides circumstantial evidence for the assigned solution conformation of spongistatin 1 (1). Or as often stated by Ralph Hirschmann: “Let the receptor be the judge.”
Supplementary Material
Figure 1.

(a) Spongistatin 1 and 2 (b) Calculated Hydrogen Bonding Network of (+)-Spongistatin 1.
Scheme 1.

Synthesis of EF-analog 3
Acknowledgments
Financial support was provided by the NIH (NCI) through Grant No. CA-70329. CAR was supported by the American Cancer Society-Michael Schmidt Postdoctoral Fellowship.
Footnotes
Supporting Information Available: Spectroscopic and analytical data and selected experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Pettit GA, Cichacz ZA, Gao F, Herald CL, Boyd MR. J Chem Soc, Chem Commun. 1993:1166. [Google Scholar]; (b) Pettit GR, Cichacz ZA, Gao F, Herald CL, Boyd MR. J Chem Soc, Chem Commun. 1993:1805. [Google Scholar]
- 2.(a) Kobayashi M, Aoki S, Sakai H, Kawazoe K, Kihara N, Sasaki T, Kitagawa I. Tetrahedron Lett. 1993;34:2795. [Google Scholar]; (b) Kobayashi M, Aoki S, Sakai H, Kihara N, Sasaki T, Kitigawa I. Chem Pharm Bull. 1993;41:989. doi: 10.1248/cpb.41.989. [DOI] [PubMed] [Google Scholar]
- 3.Fusetani N, Shinoda K, Matsunaga S. J Am Chem Soc. 1993;115:3977. [Google Scholar]
- 4.(a) Bai R, Cichacz ZA, Herald CL, Pettit GR, Hamel E. Mol Pharmacol. 1993;44:757. [PubMed] [Google Scholar]; (b) Bai R, Taylor GF, Cichacz ZA, Herald CL, Kepler JA, Pettit GR, Hamel E. Biochemistry. 1995;34:9714. doi: 10.1021/bi00030a009. [DOI] [PubMed] [Google Scholar]
- 5.(a) Pettit GA, Cichacz ZA, Gao F, Herald CL, Boyd MR, Schmidt JM, Hooper JN. J Org Chem. 1993;58:1302. [Google Scholar]; (b) Pettit GR. Pure Appl Chem. 1994;66:2271. [Google Scholar]
- 6.(a) Evans DA, Coleman PJ, Dias LC. Angew Chem, Int Ed. 1997;36:2738. [Google Scholar]; (b) Evans DA, Trotter BW, Côté B, Coleman PJ. Angew Chem, Int Ed. 1997;36:2741. [Google Scholar]; (c) Evans DA, Trotter BW, Côté B, Coleman PJ, Dias LC, Tyler AN. Angew Chem, Int Ed. 1997;36:2744. [Google Scholar]; (d) Evans DA, Trotter BW, Coleman PJ, Côté B, Dias LC, Rajapakse HA, Tyler AN. Tetrahedron. 1999;55:8671. [Google Scholar]
- 7.(a) Guo J, Duffy KJ, Stevens KL, Dalko PI, Roth RM, Hayward MM, Kishi Y. Angew Chem, Int Ed. 1998;37:187. [Google Scholar]; (b) Hayward MM, Roth RM, Duffy KJ, Dalko PI, Stevens KL, Guo J, Kishi Y. Angew Chem, Int Ed. 1998;37:192. [Google Scholar]
- 8.(a) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew Chem, Int Ed. 2001;40:196. [PubMed] [Google Scholar]; (b) Smith AB, III, Zhu W, Shirakami S, Sfouggatakis C, Doughty VA, Bennett CS, Sakamoto Y. Org Lett. 2003;5:761. doi: 10.1021/ol034037a. [DOI] [PubMed] [Google Scholar]
- 9.Paterson I, Chen DYK, Coster MJ, Acena JL, Bach J, Gibson KR, Keown LE, Oballa RM, Trieselmann T, Wallace DJ, Hodgson AP, Norcross RD. Angew Chem, Int Ed. 2001;40:4055. [PubMed] [Google Scholar]
- 10.Crimmins MT, Katz JD, Washburn DG, Allwein SP, McAtee LF. J Am Chem Soc. 2002;124:5661. doi: 10.1021/ja0262683. [DOI] [PubMed] [Google Scholar]
- 11.Heathcock CH, McLaughlin M, Medina J, Hubbs JL, Wallace GA, Scott R, Claffey MM, Hayes CJ, Ott GR. J Am Chem Soc. 2003;125:12844. doi: 10.1021/ja030317+. [DOI] [PubMed] [Google Scholar]
- 12.Ball M, Gaunt MJ, Hook DF, Jessiman AS, Kawahara S, Orsini P, Scolaro A, Talbot AC, Tanner HR, Yamanoi S, Ley SV. Angew Chem, Int Ed. 2005;44:5433. doi: 10.1002/anie.200502008. [DOI] [PubMed] [Google Scholar]
- 13.Recent Reviews with references: Yeung KS, Paterson I. Chem Rev. 2005;105:4237. doi: 10.1021/cr040614c.Pietruszka J. Angew Chem Int Ed. 1998;37:2629. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2629::AID-ANIE2629>3.0.CO;2-A.
- 14.Smith AB, III, Tomioka T, Risatti CA, Sperry JB, Sfouggatakis C. Org Lett. 2008;10(19):4359. doi: 10.1021/ol801792k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith AB, III, Lin Q. Bioorg Med Chem Lett. 1998:567. doi: 10.1016/s0960-894x(98)00050-x. [DOI] [PubMed] [Google Scholar]
- 16.(a) Paterson I, Chen DYK, Coster MJ, Acena JL, Bach J, Wallace DJ. Org Biomol Chem. 2005;3:2431. doi: 10.1039/b504151a. [DOI] [PubMed] [Google Scholar]; (b) Paterson I, Acena JL, Bach J, Chen DYK, Coster MJ. Chem Commun. 2003:462. [PubMed] [Google Scholar]
- 17.Wagner CE, Wang Q, Melamed A, Fairchild CR, Wild R, Heathcock CH. Tetrahedron. 2008;64:124. [Google Scholar]
- 18.See preceding article.
- 19.Evans DA, Katz JL, West TR. Tetrahedron Lett. 1998;39:2937. [Google Scholar]
- 20.Hubbs JL, Heathcock CH. J Am Chem Soc. 2003;125:12836. doi: 10.1021/ja030316h. [DOI] [PubMed] [Google Scholar]
- 21.(a) Hoye T, Kurth MJ, Lo V. Tetrahedron Lett. 1981;22(9):815. [Google Scholar]; (b) Jacobi PA, Li Y. Org Lett. 2003;5:701. doi: 10.1021/ol0275116. [DOI] [PubMed] [Google Scholar]
- 22.Parikh JP, Doering WE. J Am Chem Soc. 1967;89:5505. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.