

Abstract
JAK3 is a non-receptor tyrosine kinase, predominantly expressed in hematopoietic cells and that has been implicated in the signal transduction of the common gamma chain subfamily of cytokine receptors. As a result, JAK3 plays an essential role in hematopoieisis during T cell development. JAK3 inactivating mutations result in immunodeficiency syndromes (SCID) in both humans and mice. Recent data indicate that abnormal activation of JAK3 due to activating mutations is also found in human hematological malignancies, including acute megakaryoblastic leukemia (AMKL) and cutaneous T celI lymphoma (CTCL). After a brief summary of the JAK3 structure and function, we will review the evidence on the emerging role of JAK3 activation in hematological malignancies that warrant further studies to test the relevance of specific inhibition of JAK3 as a therapeutic approach to these challenging clinical entities.
Keywords: Hematopoiesis, Lymphocytes, Tyrosine kinase, Cytokine signaling
1. Introduction
Janus Kinase 3 (JAK3) also known as Leucocyte JAK (L-JAK) was cloned in 1994 from natural killer cells by the group of John O'Shea (Kawamura et al., 1994). JAK3 belongs to a family of four membrane-associated intracellular non-receptor tyrosine kinase proteins (JAK1, JAK2, JAK3, and TYK2) that mediate signals initiated by cytokine and growth factor receptors. This review highlights the recent observation that abnormal activation of JAK3 is also found in human hematologic malignancies, indicating that a tight balance in its activity is essential for normal hematopoietic development.
2. Structure
The JAK3 gene is located on human chromosome 19p13.1 and comprises 24 exons (Fig. 1). The commonly described isoform (JAK3S) encodes an 1124-amino acid (aa) protein with a predicted molecular mass of 125 kD. Alternative splicing in exon 24 also leads to the expression of JAK3B (a 1094-aa protein lacking part of the kinase domain) and JAK3 M (a 1131-aa protein).
Fig. 1.
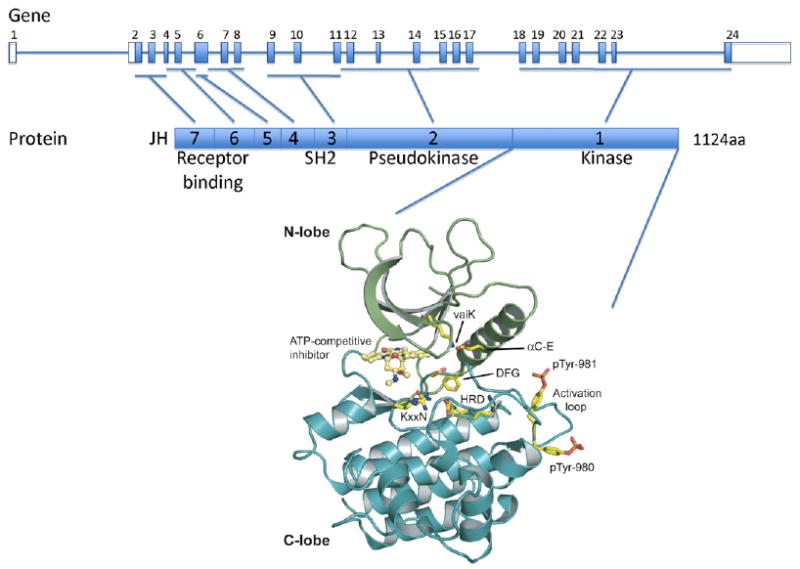
Structure of the JAK3 gene and protein. The human JAK3 genomic structure is shown in the upper panel and the JAK3 protein structure is represented in the middle panel. JAK3 is comprised of 7 JAK homology (JH) domains. JH1 contains the C terminus kinase domain; JH2 contains a pseudokinase domain and the alanine 572 residue which mutation to valine leads to constitutive activation of the kinase in acute megakaryoblastic leukemia and cutaneous T cell lymphoma. The N terminus region (JH6 and JH7) is critical for γc receptor binding and signal transduction. Y100C mutation found in SCID patients abolishes the interaction between JAK3 and the common γc chain. A cartoon representation of the JH1 domain of Jak3 (PDB code: 1YVJ) is shown (N-lobe is in green and C-lobe in blue) in the lower panel. A small molecule staurosporine analogue is seen bound in the kinase catalytic cleft in yellow. Conserved kinase domain residues are shown in stick representation and indicated HRD, KxxN, DFG, αC-E, vaiK and are important for orientation of the substrate residue hydroxyl group (HRD), γ-phosphate neutralization (KxxN), α- and β- phosphate positioning (αC-E, vaiK), metal binding (DFG), and conformational transition between active and inactive states (DFG). Human Jak3 varies from canonical kinase domains with substitutions of the KxxN and vaiK motifs to xxRN and vavK, both commonly seen substitutions. Phosphotyrosines pTyr-980 and pTyr-981 critical for JAK3 activity are shown in stick representation. Figure was made using Pymol (www.pymol.org).
The JAK3 protein presents seven JAK homology (JH) domains common with the other JAK proteins (Baker et al., 2007). Although the crystal structure of the entire JAK3 protein, or any other JAK, has not yet been published, the structure of the JH1 kinase domain has been solved (Fig. 1) (Boggon et al., 2005). The JH1 domain is a kinase domain whose activity is regulated in part by phosphorylation of key tyrosine residues (Y980 and Y981) in its kinase activation loop. This domain bears a bilobar structure, characteristic of kinase domains. Its role is to catalyze the transfer of the γ-phosphate from the ATP phosphate donor onto a protein substrate. In this process, ATP complexed with a divalent cation is recruited to the ATP binding pocket, a common feature of kinases that can be targeted by small molecule inhibitors to block catalytic kinase activity. The JH2 or pseudokinase domain, which has no kinase activity, is thought to interact with signal transducers and activators of transcription (STAT) and negatively regulate kinase activity of the JH1 domain. The JH3-JH7 domains are predicted to fold as SH2 (JH3-JH4) and FERM (JH4-JH7) domains. JH6 and JH7 play an important role in binding to the common gamma chain, or γc, receptor and a mutation at the position Y100 abolishes this interaction and inhibits JAK3 activation.
3. Expression and activation
In sharp contrast with the ubiquitous expression of the other JAKs, JAK3 is predominantly expressed in hematopoietic tissues (Ghoreschi et al., 2009; Thomis and Berg, 1997), but has also been detected in brain, spinal cord, heart, skeletal muscle, liver, pancreas, prostate, kidney, and lung. JAK3 expression is also found in epithelial cancer cells, including primary breast cancer and cell lines (SW480: colorectal adenocarcinoma, A549: lung carcinoma, and G361: melanoma).
In addition to its restricted expression, the discrete function of JAK3 in hematopoiesis is also due to its unique ability to bind only one cytokine receptor, the common gamma chain or γc, whose expression is also restricted to hematopoietic tissues. The γc subunit is shared by several heteromeric cytokine receptors important for the development of lymphoid cells, including IL2-IL4-, IL7-, IL9-, IL-13, IL15-, and IL21-receptors (Rochman et al., 2009). JAK1 binds to the β-subunit of these cytokine receptors. Once the receptors are engaged by their ligands, conformational changes lead to the activation and auto-transphosphorylation of JAK3 (Fig. 2). Then, phosphorylation of the intracellular part of the receptor by JAK3 creates docking sites for several signaling molecules, including Signal Transducers and Activators of Transcription (STAT) factors, Phosphatidyl-Inositol 3-Kinase (PI3K) and Insulin Receptor Substrate (IRS). Phosphorylation of STAT factors allows their translocation to the nucleus to regulate the transcription of a wide variety of target genes that are highly dependent on the STAT factor and celI-context (Ihle, 2001).
Fig. 2.
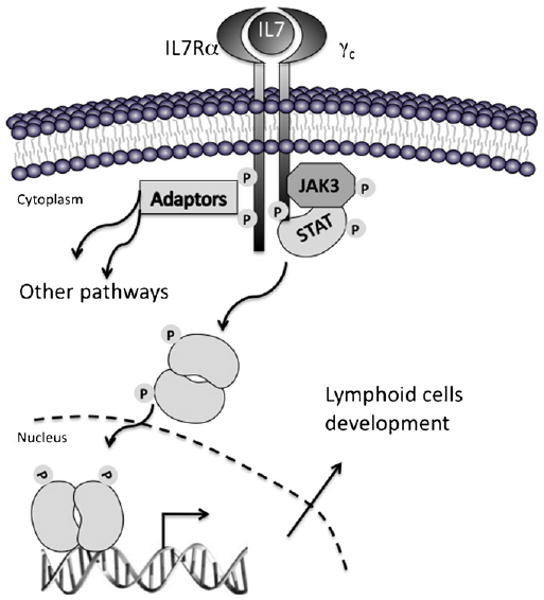
Schematic representation of the signal transduction initiated by cytokine receptors through JAK3. The heterodimeric IL7 receptor is used as an example. Conformational changes resulting from the binding of the cytokine to its receptor induce autophosphorylation of JAK3 and phosphorylation of the cytokine receptor by JAK3, which lead to recruitment of STAT and adaptor proteins that are then phosphorylated by JAK3 and other JAKs. Phosphorylated STAT factors migrate to the nucleus where they act as transcription factors of genes important for survival, proliferation and differentiation of lymphoid cells.
Several factors are responsible for attenuating the signals initiated by cytokine receptor–ligand interactions in order to ensure a transient activation of the pathway and controlled cellular responses. These factors include tyrosine phosphatases (e.g. Srchomology 2 domain-containing phosphatase-1: SHP-1) that allow dephosphorylation of JAK3 (Han et al., 2006) or E3 ubiquitin ligases (i.e. suppressors of cytokine signaling or SOCS and cytokine signaling inducible SH2 domain-containing proteins or CIS), which are responsible for ubiquitination and proteasome-dependent degradation of the kinase (Piessevaux et al., 2008; Yasukawa et al., 2000).
4. Biological function and mutations in hematologic disorders
The function of JAK3 is linked to the function of the cytokine receptors that use the γc receptor chain. As a result, the best-characterized role of JAK3 in hematopoiesis is during lymphocyte development in which these receptors were shown to have an important role (Ghoreschi et al., 2009; Thomis and Berg, 1997). The function of JAK3 in non-hematopoietic cells remains to be determined, as is whether γc is required for its activation in these cells.
The role of JAK3 in hematopoiesis is highlighted by the presence of germline inactivating mutations (missense/nonsense, splicesite, deletion or insertion mutations) on both copies of JAK3 in about 10% of patients with the autosomal recessive T- and NK-cell negative/B-cell positive type of severe combined immunodeficiency (SCID); a condition characterized by a profound defect in mature T and NK cells, and to a lesser extent B lymphocytes (Russell et al., 1995). Patients present life-threatening infections in the first months of life, which can often be cured by hematopoietic stem celI transplantation, suggesting that JAK3 does not have an essential role outside of hematopoiesis. A similar phenotype is observed in JAK3-deficient mice that have a striking deficiency in thymic progenitor celI development, an absence of lymph nodes and a severely reduced number of circulating CD8+ T and NK cells (Nosaka et al., 1995).
Patients with inactivating mutations of γc have a similar SCID phenotype to that of JAK3-SCID patients. Furthermore, γc-deficient mice have an identical phenotype to JAK3-deficient animals indicating that JAK3 requires the scaffold structure of γc to become activated and that JAK3 is likely to be the only JAK to transduce γc signals (Ghoreschi et al., 2009; Rochman et al., 2009; Thomis and Berg, 1997). It is thought that inhibition of the IL-7 receptor, which is also mutated in about 10% of autosomal recessive SCID patients, is the basis for most of the abnormalities associated with a JAK3 or γc deficiency.
Using an unbiased mass spectrometry approach to identify novel tyrosine kinase mutations in myeloid leukemia, a novel JAK3A572V mutation was identified in the CMK cell line derived from a patient with acute megakaryoblastic leukemia (AMKL) (Walters et al., 2006). Although this alanine to valine substitution in the JH2 pseudokinase domain of JAK3 appears quite conservative, it affects a conserved amino acid predicted to be on the cleft side of the C helix at the same position as the catalytic glutamic acid residue in active kinase domains. This catalytic cleft region of the JH2 domain is thought to interact with the JH1 domain and play a role in regulation of the kinase activity. JAK3A572V mediates proliferation of the CMK cells, induces cytokine independent growth of BaF3 cells in vitro and leads to constitutive autophosphorylation of the JAK3 kinase and phosphorylation of several downstream effectors, including STAT5, AKT or ERK (Cornejo et al., 2009; Walters et al., 2006). Together, JAK3A572V is a bona fide activating mutation of JAK3, which is predicted to disrupt an important autoregulatory interaction between the JH2 and JH1 domains. Using resequencing strategies, other groups have reported additional activating mutations in AMKL celI lines and patients (De Vita et al., 2007; Sato et al., 2008), including a JAK3A573V mutation (Malinge et al., 2008), targeting the neighboring conserved amino acid, while other groups did not find JAK3 mutations in their cohort of patients (Norton et al., 2007). Although other genetic lesions (e.g. chromosomal translocations) that would lead to JAK3 aberrant activation are not detected with classical sequencing approaches, these observations indicate that JAK3 activating mutations constitute rare events in AMKL.
The finding of JAK3 mutations in megakaryoblastic malignancies was unexpected as JAK3 is generally associated with lymphoid development and was not previously shown to participate in myeloid cell development. Interestingly, expression of the JAK3A572V mutant allele in a murine bone marrow transplant model not only showed a subtle megakaryocyte hyperplasia (Walters et al., 2006), but also a more striking lymphoproliferative disease characterized by the expansion of CD8+TCRαβ+CD44+CD122+Ly-6C+ T cells that closely resemble an effector/memory T cell subtype (Cornejo et al., 2009). In addition, prominent skin infiltration reminiscent of Pautrier's microabcesses, a morphologic feature characteristic of several forms of human cutaneous T cell lymphoma (CTCL), was visible in JAK3A572V animals. Subsequently, a JAK3A572V mutation was found in 1 of 30 cutaneous T cell lymphoma patients, and who was diagnosed with a severe CD4+ mycosis fungoides (Cornejo et al., 2009). This incongruence between the mouse model and the human phenotype suggests that the cell-context in which the mutation arises is important for the cellular phenotype of the disease. In support of this hypothesis, when JAK3A572V expressing bone marrow cells were introduced into Kb−/− Db−/− (i.e. MHC class l-deficient) syngeneic animals that cannot develop CD8+ T cells, recipients developed a CD4+ lymphoproliferative disease (Cornejo et al., 2009). Although functional analysis are required to confirm the role of JAK3 activation in the initiation or progression of human CTCL, these results show that constitutive JAK3 activity could also drive CD4+ T cell lymphoproliferation in mice.
These observations indicate that JAK3A572V mutation is found in 1/30 cases of CTCL and that its expression in murine hematopoietic progenitors is sufficient to efficiently induce a lymphoproliferative disorder. However, although rare JAK3 activating mutations are associated with AMKL, expression of JAK3A572V in a retroviral transduction/bone marrow transplant model does not result in megakaryoblastic leukemia (Cornejo et al., 2009; Walters et al., 2006). Therefore, JAK3 mutations likely arise as a secondary/late oncogenic hit during megakaryoblastic transformation after acquisition of other critical mutations that confer altered self-renewal properties and a megakaryocyte phenotype to the malignant clone. In this context, the receptor scaffold required for JAK3 activity remains to be identified.
5. Therapeutic use of JAK3 inhibitors
As a kinase that can be targeted at the level of the ATP binding pocket, JAK3 has historically been a very attractive target for the development of small molecule inhibitors. Indeed, its restricted expression and function within the hematopoietic compartment should in principle result in very limited side effects on other organs. Therefore, based on the essential role of JAK3 in T and NK-cell development, the original idea was to obtain inhibitors that could serve as immunosuppressors (Pesu et al., 2008). Several small molecule compounds have been reported to inhibit JAK3, although selectivity of some of these small molecules against JAK3 as compared to the other JAK family members and tyrosine kinases in general remains to be established (Changelian et al., 2008). Nevertheless, some JAK3 inhibitors have already shown efficacy in preventing graft rejection and autoimmune disease in animal models of organ transplants (Changelian et al., 2003) and are now being evaluated in Phase 1 and Phase 2 clinical trials for kidney transplants. In addition, JAK3 inhibitors (i.e. CP-690, 550, R348 and VX509) are also being tested in the context of other inflammatory diseases, including psoriasis and rheumatoid arthritis.
Interestingly, JAK3 inhibitors could also benefit anti-cancer therapies. In addition to JAK3 activating mutations found in rare cases of acute megakaryoblastic leukemia and cutaneaous T cell lymphoma, JAK3 activation has been reported in several lymphoproliferative disorders, including mantle-cell lymphoma (MCL), Burkitt's lymphoma, HTLV-1 induced adult T cell lymphoma/leukemia (ATLL), and anaplastic large cell lymphoma (ALCL). Except in the context of ALCL, in which the NPM-ALK fusion protein was reported to physically interact with and activate JAK3 (Crockett et al., 2004), the mechanistic basis for constitutive JAK3 activation, as well as its precise contribution to other lymphoid disorders, is unclear. Of note, inhibition of JAK3 appears important for the growth of NPM-ALK+ lymphoma celIs in vitro (Amin et al., 2003; Dien Bard et al., 2009). In addition, a pan-JAK inhibitor efficiently inhibited the proliferative phenotype of isolated murine CD8+ T cells expressing JAK3A572 V in vitro (Cornejo et al., 2009). Based on these observations, we suggest that selective inhibition of JAK3 or downstream signaling pathways could be a novel therapeutic strategy for the treatment of a subset of these malignancies generally associated with poor prognosis. Together, a precise understanding of the incidence of aberrant activation of JAK3 in patients as well as the analysis of physiological models of constitutive JAK3 activation in mice will help validate the therapeutic potential of JAK3 inhibitors in the treatment of human hematologic malignancies.
Acknowledgments
We apologize to those colleagues whose work couId not be cited due to space constraints. We thank Pr. Gary Gilliland for critical discussions and support and Pr. Jon Aster, Pr. Harvey Cantor, Pr. Jean-Philippe Merlio, Dr. Scott Rodig and Dr. Olivier Bernard for helpful discussions. TJB is funded by NIAID, R01 AI075133. TM is an INSERM investigator and is funded by a European Society of Hematology (EHA) José Carreras fellowship and an Agence Nationale Recherche grant.
References
- Amin HM, Medeiros LJ, Ma Y, Feretzaki M, Das P, Leventaki V, Rassidakis GZ, O'Connor SL, McDonnell TJ, Lai R. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene. 2003;22:5399–407. doi: 10.1038/sj.onc.1206849. [DOI] [PubMed] [Google Scholar]
- Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26:6724–37. doi: 10.1038/sj.onc.1210757. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, Li Y, Manley PW, Eck MJ. Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood. 2005;106:996–1002. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, Rizzuti BJ, Sawyer PS, Perry BD, Brissette WH, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302:875–8. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- Changelian PS, Moshinsky D, Kuhn CF, Flanagan ME, Munchhof MJ, Harris TM, Doty JL, Sun J, Kent CR, Magnuson KS, et al. The specificity of JAK3 kinase inhibitors. Blood. 2008;111:2155–7. doi: 10.1182/blood-2007-09-115030. [DOI] [PubMed] [Google Scholar]
- Cornejo MG, Kharas MG, Werneck MB, Le Bras S, Moore SA, Ball B, Beylot-Barry M, Rodig SJ, Aster JC, Lee BH, et al. Constitutive JAK3 activation induces lymphoproliferative syndromes in murine bone marrow transplantation models. Blood. 2009;113:2746–54. doi: 10.1182/blood-2008-06-164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crockett DK, Lin Z, Elenitoba-Johnson KS, Lim MS. Identification of NPM-ALK interacting proteins by tandem mass spectrometry. Oncogene. 2004;23:2617–29. doi: 10.1038/sj.onc.1207398. [DOI] [PubMed] [Google Scholar]
- De Vita S, Mulligan C, McElwaine S, Dagna-Bricarelli F, Spinelli M, Basso G, Nizetic D, Groet J. Loss-of-function JAK3 mutations in TMD and AMKL of Down syndrome. Br J Haematol. 2007;137:337–41. doi: 10.1111/j.1365-2141.2007.06574.x. [DOI] [PubMed] [Google Scholar]
- Dien Bard J, Gelebart P, Anand M, Zak Z, Hegazy SA, Amin HM, Lai R. IL-21 contributes to JAK3/STAT3 activation and promotes cell growth in ALK-positive anaplastic large cell lymphoma. Am J Pathol. 2009;175:825–34. doi: 10.2353/ajpath.2009.080982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–87. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Amin HM, Franko B, Frantz C, Shi X, Lai R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood. 2006;108:2796–803. doi: 10.1182/blood-2006-04-017434. [DOI] [PubMed] [Google Scholar]
- Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–7. doi: 10.1016/s0955-0674(00)00199-x. [DOI] [PubMed] [Google Scholar]
- Kawamura M, McVicar DW, Johnston JA, Blake TB, Chen YQ, Lal BK, Lloyd AR, Kelvin DJ, Staples JE, Ortaldo JR, et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc Natl Acad Sci USA. 1994;91:6374–8. doi: 10.1073/pnas.91.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinge S, Ragu C, Della-Valle V, Pisani D, Constantinescu SN, Perez C, Villeval JL, Reinhardt D, Landman-Parker J, Michaux L, et al. Activating mutations in human acute megakaryoblastic leukemia. Blood. 2008;112:4220–6. doi: 10.1182/blood-2008-01-136366. [DOI] [PubMed] [Google Scholar]
- Norton A, Fisher C, Liu H, Wen Q, Mundschau G, Fuster JL, Hasle H, Zeller B, Webb DK, O'Marcaigh A, et al. Analysis of JAK3, JAK2, and C-MPL mutations in transient myeloproliferative disorder and myeloid leukemia of Down syndrome blasts in children with Down syndrome. Blood. 2007;110:1077–9. doi: 10.1182/blood-2007-03-080374. [DOI] [PubMed] [Google Scholar]
- Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Doherty PC, Grosveld GC, Ihle JN. Defective lymphoid development in mice lacking Jak3. Science. 1995;270:800–2. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O'Shea JJ. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223:132–42. doi: 10.1111/j.1600-065X.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piessevaux J, Lavens D, Peelman F, Tavernier J. The many faces of the SOCS box. Cytokine Growth Factor Rev. 2008;19:371–81. doi: 10.1016/j.cytogfr.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9:480–90. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, Migone TS, Noguchi M, Markert ML, Buckley RH, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995;270:797–800. doi: 10.1126/science.270.5237.797. [DOI] [PubMed] [Google Scholar]
- Sato T, Toki T, Kanezaki R, Xu G, Terui K, Kanegane H, Miura M, Adachi S, Migita M, Morinaga S, et al. Functional analysis of JAK3 mutations in transient myeloproliferative disorder and acute megakaryoblastic leukaemia accompanying Down syndrome. BrJ Haematol. 2008;141:681–8. doi: 10.1111/j.1365-2141.2008.07081.x. [DOI] [PubMed] [Google Scholar]
- Thomis DC, Berg LJ. The role of Jak3 in lymphoid development, activation, and signaling. Curr Opin Immunol. 1997;9:541–7. doi: 10.1016/s0952-7915(97)80108-2. [DOI] [PubMed] [Google Scholar]
- Walters DK, Mercher T, Gu TL, O'Hare T, Tyner JW, Loriaux M, Goss VL, Lee KA, Eide CA, Wong MJ, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006;10:65–75. doi: 10.1016/j.ccr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Yasukawa H, Sasaki A, Yoshimura A. Negative regulation of cytokine signaling pathways. Annu Rev Immunol. 2000;18:143–64. doi: 10.1146/annurev.immunol.18.1.143. [DOI] [PubMed] [Google Scholar]