

Abstract
Insulinomas are rare neuroendocrine tumors of pancreatic islet cells that retain the ability to produce and secrete insulin. In contrast to normally differentiated β-cells, insulinoma cells continue to secrete insulin and proinsulin at low blood glucose. This deregulated insulin secretion manifests clinically as fasting hypoglycemia. The molecular pathways that characterize normal insulin secretion and β-cell growth are reviewed and contrasted to the biology of insulinomas. The second half of this review summarizes the clinical approach to the disorder. The diagnosis of insulinoma is established by demonstrating inappropriately high insulin levels with coincident hypoglycemia at the time of a supervised fast. Localization of insulinomas is challenging owing to their small size but should be attempted to maximize the chance for successful surgical resection and avoid risks associated with reoperation. In the majority of cases, successful surgical resection leads to lifelong cure.
Keywords: β-cell, hypoglycemia, insulinoma, insulin secretion, nesidioblastosis
The concentration of insulin in plasma reflects the balance between rates of appearance (i.e., secretion) and disappearance (i.e., distribution and degradation) of insulin from the vascular space. Although changes in the volume of distribution and rate of degradation of the hormone can influence insulin levels in some conditions, these factors are generally not responsible for exerting rapid changes in plasma insulin levels in the physiologic state. A change in the rate of insulin secretion, triggered by fuel, entero-incretin and/or neural signals, is the factor principally responsible for acutely modifying plasma insulin concentration. The β-cell, a cell uniquely specialized to synthesize, process, store and secrete insulin, is central to this function. Pancreatic β-cells sense ambient glucose levels and secrete insulin in an amount precisely adjusted to match the magnitude of the glucose stimulus. This function imparts β-cells with a prominent role in whole-body glucose homeostasis. The insulin–glucose dose–response relationship, established from rodent pancreas perfusion studies [1], is sigmoidal (Figure 1). Although the magnitude of the insulin secretory response differs at low or high glucose levels, little to no change in the rate of insulin secretion is observed outside the physiologic range. This contrasts to the large increases in the rate of insulin secretion seen as glucose rises within the physiologic range (~70–400 mg/dl [~5–25 mM]). The half-maximal insulin secretory response in this ex vivo setting, an index of β-cell sensitivity to glucose, is observed at a glucose concentration between approximately 140 and 180 mg/dl (~8–10 mM). Physiologic (i.e., nutrient, neural, entero-incretin signals), therapeutic or pathologic processes that change the characteristics of this dose–response relationship impact whole-body glucose homeostasis. A clinical and diagnostic feature of subjects harboring insulinomas is the continued secretion of insulin at glucose concentration below the physiologic range. Although the insulin–glucose dose–response for human insulinomas has not been defined and probably differs among individual tumors, several rodent insulinoma cell lines (HIT-T15, INS-1, β-TC6 and MIN-6) have an abnormal insulin–glucose dose–response, characterized by either an upward (increased responsiveness) and/or leftward (increased sensitivity) departure from the normal β-cell insulin–glucose dose–response at low glucose levels.
Figure 1. Insulin–glucose dose–response.
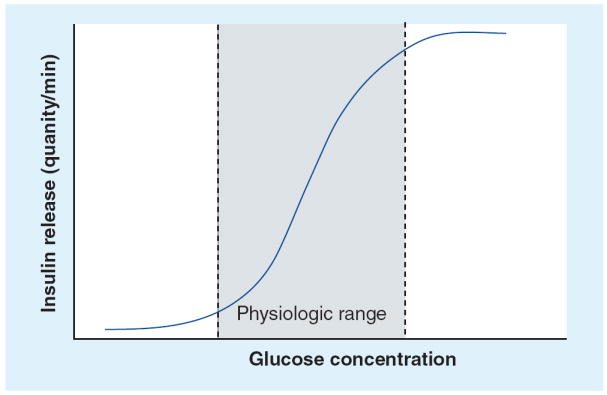
The relationship between glucose and the rate of insulin secretion is sigmoidal. The maximal change in the rate of insulin secretion occurs when glucose is within the physiologic range (shaded area). The half maximal and maximal response are indicated by marks on the y-axis and indicate the responsiveness of β-cells to glucose. The glucose concentration that elicits the half maximal response is an index of β-cell sensitivity to glucose. Physiologic and pathologic factors can influence the sensitivity and/or responsiveness of β-cells to glucose.
Molecular events regulating glucose-stimulated insulin secretion
Our current understanding of the molecular events associated with insulin secretion (Figure 2) is derived from in vitro rodent models. Several discoveries were crucial in defining the steps involved in glucose-stimulated insulin secretion. Of particular importance were the realizations that β-cells were electrically excitable [2], glucose controlled β-cell electrical activity [3], β-cells expressed potassium channels (Kir6.2) responsible for generating a hyperpolarizing current that could be inhibited by both intracellular ATP [4] as well as glucose [5] and Kir6.2 channels formed a larger hetero-octomeric complex (KATP channels) with the receptor for sulfonylurea drugs, SUR1 [6,7]. More recently, advances in the field of biological imaging has permitted direct visualization of insulin-granule secretion from β-cells [8-11], and has refined our understanding of insulin exocytosis. Insulin secretion from mouse β-cells exposed to a rapid increase in glucose (from 50 to 300 mg/dl [2.8 to 16.7 mM]) exhibits a biphasic kinetic pattern. The first and best-characterized phase is transient (4–8 min). This phase, also coined triggering pathway [12], involves cellular entry of glucose through low-affinity, high capacity, glucose transporters (GLUT)2 [13]; cytoplasmic and mitochondrial glucose metabolism limited by flux through the glucose phosphorylating enzyme glucokinase (reviewed in [14]); increases in intracellular ATP/ADP concentration; closure of ATP-sensitive potassium channels (KATP channels); depolarization of the plasma membrane; opening of voltage-dependent Ca+2 channels (L-type) and release of ‘release-competent’ insulin granules located in the vicinity of these channels. Using capacitance measurements to detect plasma membrane fusion events, it has been estimated that approximately 50 [15] of the approximately 10,000 insulin granules [16] present in each β-cell (i.e., 0.5% of insulin granules/β-cell) are released during this phase. The second, less well-characterized phase (or amplifying pathway [12]) of insulin secretion is sustained (within minutes to hours). Similar to first-phase secretion, it also depends on both glucose metabolism and increases in intracellular calcium but, in addition, involves amplifying signals that are important in recruiting, priming and docking of a ‘highly calcium-sensitive’ pool of intracellular granules [17]. Several metabolic pathways, including pyruvate cycling [18], glutamate metabolism [19], NADH shuttling [20], de novo long-chain acyl CoAs (Lc-CoA) synthesis [21,22] and NADPH production [23,24], have been associated with this phase of insulin release and are believed to support secretion by generating a number of important effectors (e.g., ATP, Lc-CoA, DAG/PKC and NADPH). Recent evidence suggests that these metabolically derived signals are not solely restricted to second-phase secretion but also contribute to first-phase secretion [25]. The importance of these pathways in insulin secretion is further supported by protein expression studies that suggest β-cells are uniquely specialized to maximize the efficiency of aerobic glucose metabolism [26-28]. In addition to these biochemical pathways, a complex cell-to-cell communication network within islets is needed for a coordinated insulin secretory response [29].
Figure 2. Glucose-stimulation secretion coupling.

Aerobic glucose metabolism in β-cells is intimately coupled to insulin secretion. Glucose enters β-cells through high-capacity glucose transporters (GLUT2), is metabolized by the low-affinity (Km ~7–8 mM), rate-limiting, glucose-phosphorylating enzyme (glucokinase or hexokinase IV) and is further oxidized into pyruvate. Reduced NAD (i.e., NADH) generated in glycolysis is transported from the cytosol to the mitochondria via the malate aspartate or glycerol phosphate shuttles, preventing feedback inhibition on glycolytic enzymes and adding mitochondrial-reducing equivalents. Pyruvate is transformed into acetyl-CoA by pyruvate dehydrogenase, and acetyl-CoA is completely oxidized in the citric acid cycle, generating molecules of reduced NAD (i.e., NADH) in the process. Electrons from NADH are donated to the electron-transport chain and a proton gradient is generated across the inner mitochondrial membrane that drives ATP production via oxidative phosphorylation. β-cell pyruvate can also be carboxylated by pyruvate carboxylase to oxaloacetate, adding intermediaries to the citric acid cycle (increasing the efficiency of aerobic glucose metabolism), a process called anaplerosis. Increases in the cytosolic ATP/ADP ratio leads to closure of KATP channels (Kir6.2/SUR1), depolarization of the cell membrane and opening of voltage-sensitive calcium channels. Intracellular calcium entry causes releases of immediately releasable granules in the vincinity of these channels or release of a ‘highly calcium-sensitive’ cytosolic pool of granules. Biochemical effectors also involved in insulin secretion include NADPH generated through pyruvate cycling. Inhibition of fatty acid oxidation by malonyl-CoA has also been shown to play a role in insulin secretion, perhaps by generation of fatty acid-derived effectors such as diacylglycerol.
ER: Endoplasmic reticulum; TCA: Tricarboxylic acid; VDCC: Voltage-dependent calcium channel.
Biochemical pathways linked to hyperinsulinism
The relevance and contribution of these biochemical pathways to the excess insulin secretion observed in adult insulinomas has not been studied but has been well recognized in the setting of congenital hyperinsulinism. The most common form of this disorder results from inactivating mutations (reviewed in [30]) in the genes coding for SUR1 (ABBC8) and Kir6.2 (KCNJ11), both located on chromosome 11 at p15. Less common forms involve activating mutations in the GDH (10q23.3) [31] and the GCK (7p) [32] and inactivating mutations in the short-chain HADH gene (4q) involved in fatty acid oxidation [33,34]. More recently, mutations that result in increased cell surface expression of the monocarboxylase transport protein, MCT-1 (SLC16A1; 1p) [35], a protein responsible for pyruvate and lactate transport into β-cells, were shown to result in inappropriate insulin secretion during exercise, an effect replicated experimentally by intravenous administration of pyruvate [36]. Before the molecular defects underlying the etiology of congenital hyperinsulinism were discovered, histology of pancreatic specimens (i.e., nesidioblastosis) had suggested aberrant growth or development of islets as the basis for the hormone excess [37,38]. This idea was abandoned when nesidioblastosis was revealed to be a nonspecific histologic finding (i.e., present in normal pancreata) and was not found to be associated with an increased rate of β-cell proliferation [39]. While nesidioblastosis as a cause of hypoglycemia has been abandoned for pediatric disorders, it is still invoked as the basis of some hypoglycemic disorders in adults [40-42]. Several controversial issues surround these cases and include an absence of mutations in genes associated with congenital forms of hyperinsulinim, inadequate consideration of factors known to influence islet growth (i.e., morbid obesity and diabetes) or insulin release (i.e., alimentary tract surgery and elevated prandial entero-incretin release), use of a diagnostic test to establish disease never tested for such purpose (i.e., selective arterial calcium stimulation) and inappropriate controls used as the comparator group (i.e., non-BMI-matched subjects with pancreatic cancer). An independent review of these samples did not find an increase in β-cell area or a change in β-cell turnover compared with BMI-matched subjects [43]. In addition, a correlation between β-cell nuclear diameter (i.e., one of the histologic features used to define nesidioblastosis) and obesity in all subjects (i.e., with and without disease) were found. Finally, the presence of islets budding from exocrine ducts, another feature used to diagnose nesidioblastosis, is a common feature of obese and diabetic pancreata [44]. In our experience of more than 35 years, we have not seen a case of hypoglycemia whose etiology could be explained solely on the basis of a pancreatic histology consistent with nesidioblastosis. Clinicians should be aware of these caveats when assessing the risk benefit of pancreatectomy as a treatment option for this disorder.
Determinants of normal β-cell growth in humans
Understanding the molecular events responsible for physiologic β-cell growth is the focus of intense research and beyond the scope of this review [45]. In general, β-cell-mass expansion in humans occurs as a result of normal growth and development [46] or to meet an increased demand for insulin in states such as pregnancy [47,48] or obesity [44,49]. There is accumulating evidence suggesting that postnatal β-cell mass expansion in humans is caused by replication of existing β-cells [46], rather than stem cell differentiation, a process similar to that observed in rodent models [50,51]. Glucose [52-54], prolactin and placental lactogen [47], insulin and IGF-1 [55] and GLP-1 [56] have been proposed as potential circulating physiologic regulators of β-cell replication and growth. Understanding the individual contribution of mitogens to β-cell replication is challenging given the interdependence and multiplicity of cellular pathways triggered by each. Increases in IRS-2 expression, for example, a key downstream protein activated by tyrosine kinase receptors (e.g., insulin and EGF receptors) and directly implicated in β-cell growth and survival [57,58], has been shown to be regulated by both glucose [59] and GLP-1 [60]. The effect of glucose on IRS-2 was shown to be calcium dependent [61], and that of GLP-1; dependent on a coincident calcium and cAMP signal [62]. A number of effectors downstream of IRS-2 [63-70] have been independently associated with rodent β-cell mass expansion. These effectors are involved in insulin biosynthesis, cell-cycle progression, protein synthesis and cell survival pathways (reviewed in [71]).
Insulin processing & significance to insulinoma
As first suggested by Steiner and Oyer [72], insulin is derived from a larger precursor molecule. Preproinsulin is first synthesized in the cytosol and processed in the rough endoplasmic reticulum to form proinsulin (Figure 3) (reviewed in [73]). In β-cell granules [74] and at optimal calcium and pH, proinsulin is cleaved at a pair of dibasic residues (i.e., Arg30–Arg31 and Lys60–Arg61) by two endopeptidases [75,76]. Carboxypeptidase-H subsequently removes the exposed basic residue to give rise to the disulfide-linked two-chain insulin molecule (A- and B-chain) and its connecting peptide (C-peptide) [77]. Incomplete processing of the molecule results in the formation of circulating products that include full-length proinsulin, 31,32 split proinsulin, des-31,32 split proinsulin (i.e., only the B-chain/C-peptide junction is cleaved and exposed basic amino acid are removed), 64,65 split proinsulin or des-64,65 split proinsulin (i.e., only the C-peptide/A-chain connection is disrupted and exposed basic amino acids are removed). It was recognized early that these components, initially termed ‘big insulin’ and re-named proinsulin-like components (PLCs), could be identified in plasma if a gel-filtration step (i.e., molecular-weight separation step) preceded the radioimmunoassay procedure used to quantify insulin [78]. It was later suggested that secretion of these PLCs was elevated in insulin-producing tumors [79], an observation that was confirmed in human plasma samples [78,80-83]. The realization that correct processing of proinsulin, a hallmark of differentiated β-cell function, relied on appropriate sorting of the proinsulin molecule to the ‘regulated’ secretory pathway [84,85] provided a biological basis for this clinical observation. Indeed, proinsulin in insulinoma cells was demonstrated to predominantly enter to the constitutive or ‘unregulated’ secretory pathway usually reserved for plasma membrane proteins [86]. With the advent of more specific, monoclonal, two-site immunometric assays for insulin and a two-site assay that targets both insulin and C-peptide but only labels C-peptide [87], it became possible to directly measure the plasma PLCs and correlate this assay to the older assay used to quantify PLCs from the total immunoreactive insulin [83].
Figure 3. Proinsulin: amino acid sequence, A and B chains; C-peptide and disulfide bonds are shown.

Insulinomas
Incidence & clinical features
Insulinomas are rare neuroendocrine tumors of pancreatic islet cells that retain the ability to produce and secrete insulin. An estimated four insulinoma cases are diagnosed per million person-years [88]. Although rare, insulinomas are the most frequently observed pancreatic neuroendocrine tumors accounting for 70%. Insulinomas are approximately 1.5-times more common in women and can present at any age but, on average, are diagnosed at age 50 years. Patients with multiple endocrine neoplasia (MEN)-1-associated years insulinomas tend to present at a younger age [89]. Insulinomas are usually solitary, small and are equally likely to be found in the pancreatic head or tail. Although insulinoma usually present as solitary tumors, patients presenting with multiple, discreet or 5 mm and above insulin-staining tumors have been described for sporadic [90] and MEN-1-associated insulinoma cases [91]. While the majority of islet cell tumors metastasize (e.g., gastrinoma and glucagonoma), only 10% of insulinomas are malignant on the basis of metastasis [88,92-93]. The clinical course of patients with malignant insulinoma is usually one of prolonged survival [88,94]. Insulin-producing tumors are usually discovered because they cause hypoglycemic symptoms and need to be differentiated from other conditions that cause fasting hypoglycemia (Box 1; reviewed in [95]). These can be divided into three groups: adrenergic, cholinergic and neuroglycopenic. Classic adrenergic symptoms include anxiety, tremulousness, pallor and palpitations. Sweating, tingling, nausea and hunger fall under cholinergic symptoms. Neuroglycopenic symptoms encompass the symptoms of, weakness, fatigue, visual disturbances, confusion, seizures, focal neurological deficit and coma, and denote compromised CNS function due to glucose levels insufficient to meet the brain’s need for energy.
Box 1. Differential diagnosis of fasting hypoglycemia in adults.
-
Drug-induced
– Antidiabetic agents (insulin, sulfonylurea and meglitinides)
– Other (salicylates, quinine derivatives, disopyramide and pentamidine)
– Ethanol
Liver disease
Renal failure
-
Endocrine deficiency
– Cortisol
– Growth hormone
Insulinoma
-
Nonislet tumor
– Mesenchymal tumors
– Hepatocellular carcinoma
– Hematological malignancies
– Other
-
Autoimmune hypoglycemia
– Insulin receptor autoantibodies
– Insulin autoantibodies
Genetics
Familial insulinomas
von Hippel-Lindau disease, MEN-I and tuberous sclerosis complex are three familial disorders that have been associated with the formation of islet tumors that stain for insulin (reviewed in [96]). Of these, only MEN-1 has been associated with tumors that secrete insulin in excess. Insulinomas occurring in the setting of the MEN-1 syndrome (~5–10% of all insulinomas) are associated with the loss of function of menin, the 610-amino acid protein of the tumor-suppressor gene MEN1 located on chromosome 11 at q13. Germline mutation and insulinoma cell-specific deletion that results in biallelic loss of MEN1 leads to the development of insulinomas. Tissue-specific deletion can be recognized by demonstrating loss of heterozygosity at the 11q13 locus in these tumors. The exact molecular mechanism(s) by which functional loss of menin results in tumor formation has/have not been fully elucidated.
Sporadic insulinomas
Less is known about the genetic events linked to sporadic insulinoma development. Monoclonal tumors are hypothesized to arise from less aggressive oligo/polyclonal precursor lesions [97]. Studies using comparative genomic hybridization for genome-wide analysis of pancreatic neuroendocrine tumors (PNETs) suggest a role of genomic instability in tumor initiation and progression [98-101]. These studies have revealed occurrences of nonrandomly distributed genomic alterations in PNETs. Such abnormalities have included regional chromosomal gains (e.g., 4pq, 5q, 7pq, 9q, 12q, 14q, 17pq and 20q) and losses (e.g., 1p, 3p, 6q, 10pq, 11q, Y and X). The type and number of chromosomal anomalies have been shown to correlate with tumor size and malignant potential. The observation that chromosomal losses occur more frequently than gains in PNET has suggested involvement of a tumor suppressor pathway for these tumors. Insulinomas, in keeping with their benign nature, tend to exhibit a lower number of genomic alterations ,with losses in 3p [98] and 6q [99] being especially rare events. The most frequently observed alteration in 62 recently reported sporadic insulinomas was a 9q gain [102]. In this and other studies, malignant insulinomas, exhibited a higher rate of chromosomal alterations than benign insulinomas, suggesting a role for genomic instability in progression of the disease. Loss of heterozygosity studies, using PCR amplificaltion of microsatellite markers, have confirmed and added to the findings of comparative genomic hybridization. Mutations in known oncogenes (e.g., KRAS [12p12.1], MYC [8q24.21], SRC [20q12-q13] or DCC [18q21.3]) and tumor-suppressor genes (e.g., TP53 [17p13.1], CDKN2A4 [9p21], PTEN [10q23.3] or SMAD4 [18q21.1]) common to many gastrointestinal adenocarcinomas, are infrequently observed in benign insulinomas. Finally, single-copy deletion and somatic mutations in the MEN1 gene have been reported in six out of 12 (50%) and two out of 12 (17%) cases of sporadic insulinomas, respectively, suggesting a role for this tumor-suppressor gene in a subset of nonfamilial insulinoma cases [103]. Few studies have evaluated the role of epigenetic modifications on the development of these tumors [104]. Taken together, these studies highlight the complex genetic mechanisms that give rise to insulin-producing tumors. Oncogenesis, as demonstrated by an animal model of insulinoma [104,105], probably involves the coincident occurrence of both proliferative (e.g., c-Myc) and antiapoptotic (e.g., Bcl-XL) signals. These signals may arise from aberrant regulation of signal transduction/growth (e.g., protein kinase B/Akt), hypoxia/angiogenesis (e.g., von Hippel-Lindau disease), cell-cycle progression (e.g., downregulation of cyclin-dependent kinase inhibitors) and/or genome stabilization pathways (e.g., JunD, P53, WRN; extensively reviewed in [96]).
Pathophysiology
Although genetic alterations probably account for tumor initiation and progression, systemic pathophysiology results from the fact that these tumors produce and release insulin inappropriately. Insulinoma cells fail to adequately suppress insulin as plasma glucose concentration falls. The relative insulin excess in relation to glucose leads to hypoglycemia. Clinical, ultrastructural and molecular studies of human insulinomas suggest that defects in insulin biosynthesis, processing, storage and secretion contribute to the pathophysiology of the disease. A total of 95% of patients with insulinomas were shown to have elevated fasting proinsulin plasma levels [83]. This observation suggests a disordered processing of proinsulin and implicates the insulin-secretory pathway in the pathophysiology of the disease [84,86]. Histochemical and ultrastructural studies are consistent with this notion and have shown that human insulinomas contain lower insulin concentration, higher proinsulin concentration and a variable number of atypical-appearing secretory granules when compared with normal β-cells [106]. More recently, contribution of insulin biosynthesis to the pathophysiology of these tumors has been suggested by a report demonstrating overexpression of an insulin mRNA splice variant associated with increased insulin translation efficiency in nine human insulinomas [107]. These findings suggest that insulinoma cells have lost the biological functions that characterise normally differentiated β-cells.
Diagnosis
The most useful and practical approach to establish the diagnosis of insulinoma is the 48 h supervised fast [93]. During the fast, demonstration of whipple’s triad (i.e., biochemical hypoglycemia, symptoms consistent with hypoglycemia and reversal with carbohydrate replacement) and inadequately suppressed insulin levels (>3 μIU/ml) or, in some instances, proinsulin levels (>22 pM) establishes the diagnosis of insulinoma. In a review of 127 cases, whipple’s triad and elevated insulin levels were present in 95% of cases by 48 h [93]. The remaining 5% of cases were diagnosed by 72 h but could have been diagnosed by 48 h based on blood glucose of 50 mg/dl and below (2.7 mM) and insulin levels of 10 μIU/ml and above. Whipple’s triad in the presence of suppressed plasma insulin levels usually favors another etiology (reviewed in [95]). In rare cases, plasma insulin levels in patients harboring an insulinoma may suppress below the 3 μIU/ml cutoff value. Tumors in these patients are presumed to have retained some glucose-sensing ability. Since proinsulin components are no longer detected with newer two-site monoclonal insulin assays, it is possible that these assays are less sensitive at ruling out the presence of insulinoma using a threshold of 3 μIU/ml. In such cases, an elevated fasting plasma proinsulin level (i.e., 22 pmol/l or 0.2 ng/ml on the directly measured assay) predicts the presence of an insulinoma with a sensitivity of 82% and a specificity of 95% [83]. Plasma C-peptide levels and sulfonylurea/meglitinide drug screens are obtained during the fast to exclude a diagnosis of factitious hypoglycemia. At our center we have not studied the value of surrogate markers of peripheral insulin action (i.e., glucagon test and β-hydroxybutyrate levels) in the work-up of adult patients with hypoglycemic disorders [108].
Localization & therapy
Tumor localization followed by surgical enucleation will result in lifelong cure in close to 90% of patients. However, localization of these small tumors (most are <2 cm), represents the major clinical challenge and is a limiting factor to a successful clinical outcome. Blind distal pancreatectomies are not recommended, as they pose considerable risk to patients and are unlikely to result in cure. In a recent review of NIH cases [92], we found that most conventional noninvasive imaging techniques (i.e., trans-abdominal ultrasound, computed tomography and MRI) were poor at localizing small tumors (average tumor size: ~1.8 cm), a result consistent with our previous observations [109] and that of others [110-112]. The Mayo Clinic has reported a much higher success rate with conventional imaging [108]. If conventional imaging studies fail to localize the tumor, invasive preoperative localization procedures selective intra-arterial calcium stimulation (CaStim) should be considered. CaStim can regionalize 80–90% of tumors across pancreatic regions [92,109]. Endoscopic ultrasound has an overall reported sensitivity similar to CaStim [110-114]. Success is skewed towards localization of head tumors. Localization results using radiolabeled somatostatin receptor analog to localize small insulin-producing tumors have been disappointing [114-116]. Despite the high reported success rate of intraoperative localization, it has been our experience that Preoperative tumor localization: influences surgical approach, increases the success of intraoperative localization and increases the likelihood that the initial surgery will result in cure [109]. Before surgery, oral or intravenous carbohydrates are administered to prevent hypoglycemia. Medical therapy, in the form of diazoxide, is reserved for metastatic disease, cases in which the tumor cannot be localized or when surgery is not a therapeutic option. In patients with metastatic disease, we have observed long-term symptomatic control with diazoxide [94]. Cytotoxic, cytoreductive and/or somatostatin analog therapy are seldomly needed in insulinoma. Their usefulness in this setting have been reviewed by others [117,118].
Expert commentary
Insulinomas are small, for the most part, solitary and benign tumors. Although insulinomas retain some of the functional features of normal β-cells (i.e., the capacity to synthesize and secrete insulin), they lack important characteristics of fully differentiated β-cells (i.e., the capacity to process, store and limit insulin secretion to the physiologic glucose range). The exact genetic lesions and molecular mechanisms underlying this difference have not been elucidated and are likely to differ among tumors. As a result of this difference, insulin is released at inappropriate times and results in biochemical and symptomatic hypoglycemia. The diagnosis is made by first excluding other causes of hypoglycemia and, second, by directly observing the coincident occurrence of clinical hypoglycemia and detectable insulin during a 48-h supervised fast. Localization of the tumor is challenging but necessary to ensure optimal outcome for the patient. Successful surgical removal of the tumor results in lifelong cure for the majority of patients.
Five-year view
The molecular events that lead to insulinoma development are still poorly understood and studies to address these issues are needed. Elucidating the underlying basis for aberrant growth and differentiation may shed light on the genetic, molecular and biochemical profile that characterize normally differentiated β-cells. The track-record of promising new noninvasive localization techniques will need to be confirmed in larger series of patients [119,120]. Preoperative localization may become more important with the increasing use of minimally invasive laparoscopic techniques to treat these tumors.
Key issues.
The rate of β-cell insulin secretion is the principal factor determining acute changes in plasma insulin concentration.
The effect of glucose on the rate of β-cell insulin secretion is restricted to the physiologic glucose range.
β-cells are uniquely specialized to respond to ambient fuel, entero-incretin and neural signals to meet an increased demand for insulin.
Lesions in cellular pathways important in glucose-stimulation secretion coupling have been identified as the cause for several forms of congenital hyperinsulinism.
Nesidioblastosis is a nonspecific pancreatic histologic finding.
Nesidioblastosis as the etiological basis for states of insulin excess in adults is controversial.
The processing of proinsulin is characteristic of differentiated β-cell function.
Insulinomas are rare islet tumors that retain the ability to synthesize and release insulin.
Genetic lesions associated with sporadic insulinomas are consistent with the indolent behavior of these tumors.
The diagnosis is made by demonstrating both clinical hypoglycemia and detectable insulin or elevated proinsulin levels during a supervised fast.
Localization of these tumors is difficult with conventional imaging but is necessary to ensure the best possible outcome for patients.
Surgical removal of the tumor leads to resolution of hypoglycemia and prolonged survival.
Footnotes
Financial & competing interests disclosure The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Jean-Marc Guettier, National Institute of Diabetes and Digestive and Kidney Diseases, Building 10-CRC, Room 6–5952, 10 Center Drive, Bethesda, MD 20892–1612, USA, Tel.: +1 301 496 1913, Fax: +1 301 402 0573, guettierj@mail.nih.gov.
Phillip Gorden, National Institute of Diabetes and Digestive and Kidney Diseases, Building 10-CRC, Room 6–5952, 10 Center Drive, Bethesda, MD 20892–1612, USA, Tel.: +1 301 496 1913, Fax: +1 301 402 0573.
References
- 1.Gerich JE, Charles MA, Grodsky GM. Characterization of the effects of arginine and glucose on glucagon and insulin release from the perfused rat pancreas. J Clin Invest. 1974;54(4):833–841. doi: 10.1172/JCI107823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dean PM, Matthews EK. Electrical activity in pancreatic islet cells. Nature. 1968;219(5152):389–390. doi: 10.1038/219389a0. [DOI] [PubMed] [Google Scholar]
- 3.Dean PM, Matthews EK. Glucose-induced electrical activity in pancreatic islet cells. J Physiol. 1970;210(2):255–264. doi: 10.1113/jphysiol.1970.sp009207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature. 1984;311(5983):271–273. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- 5.Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature. 1984;312(5993):446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 6.Schwanstecher M, Loser S, Chudziak F, Panten U. Identification of a 38-kDa high affinity sulfonylurea-binding peptide in insulin-secreting cells and cerebral cortex. J Biol Chem. 1994;269(27):17768–17771. [PubMed] [Google Scholar]
- 7.Clement JP, 4th, Kunjilwar K, Gonzalez G, et al. Association and stoichiometry of K(ATP) channel subunits. Neuron. 1997;18(5):827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 8.Ohara-Imaizumi M, Nakamichi Y, Nishiwaki C, Nagamatsu S. Transduction of MIN6 β cells with TAT-syntaxin SNARE motif inhibits insulin exocytosis in biphasic insulin release in a distinct mechanism analyzed by evanescent wave microscopy. J Biol Chem. 2002;277(52):50805–50811. doi: 10.1074/jbc.M207988200. [DOI] [PubMed] [Google Scholar]
- 9.Ohara-Imaizumi M, Nishiwaki C, Kikuta T, Nagai S, Nakamichi Y, Nagamatsu S. TIRF imaging of docking and fusion of single insulin granule motion in primary rat pancreatic β-cells: different behaviour of granule motion between normal and Goto–Kakizaki diabetic rat β-cells. Biochem J. 2004;381(Pt 1):13–18. doi: 10.1042/BJ20040434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pouli AE, Emmanouilidou E, Zhao C, Wasmeier C, Hutton JC, Rutter GA. Secretory-granule dynamics visualized in vivo with a phogrin-green fluorescent protein chimaera. Biochem J. 1998;333(Pt 1):193–199. doi: 10.1042/bj3330193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varadi A, Ainscow EK, Allan VJ, Rutter GA. Involvement of conventional kinesin in glucose-stimulated secretory granule movements and exocytosis in clonal pancreatic β-cells. J Cell Sci. 2002;115(Pt 21):4177–4189. doi: 10.1242/jcs.00083. [DOI] [PubMed] [Google Scholar]
- 12.Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49(11):1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- 13.Thorens B, Sarkar HK, Kaback HR, Lodish HF. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and β-pancreatic islet cells. Cell. 1988;55(2):281–290. doi: 10.1016/0092-8674(88)90051-7. [DOI] [PubMed] [Google Scholar]
- 14.Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic β-cells and hepatocytes. Diabetes. 1990;39(6):647–652. doi: 10.2337/diab.39.6.647. [DOI] [PubMed] [Google Scholar]
- 15.Barg S, Eliasson L, Renstrom E, Rorsman P. A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse β-cells. Diabetes. 2002;51(Suppl. 1):S74–S82. doi: 10.2337/diabetes.51.2007.s74. [DOI] [PubMed] [Google Scholar]
- 16.Dean PM. Ultrastructural morphometry of the pancreatic-cell. Diabetologia. 1973;9(2):115–119. doi: 10.1007/BF01230690. [DOI] [PubMed] [Google Scholar]
- 17.Pedersen MG, Sherman A. Newcomer insulin secretory granules as a highly calcium-sensitive pool. Proc Natl Acad Sci USA. 2009;106(18):7432–7436. doi: 10.1073/pnas.0901202106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu D, Mulder H, Zhao P, et al. 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS) Proc Natl Acad Sci USA. 2002;99(5):2708–2713. doi: 10.1073/pnas.052005699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maechler P, Wollheim CB. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature. 1999;402(6762):685–689. doi: 10.1038/45280. [DOI] [PubMed] [Google Scholar]
- 20.Eto K, Suga S, Wakui M, et al. NADH shuttle system regulates K(ATP) channel-dependent pathway and steps distal to cytosolic Ca(2+) concentration elevation in glucose-induced insulin secretion. J Biol Chem. 1999;274(36):25386–25392. doi: 10.1074/jbc.274.36.25386. [DOI] [PubMed] [Google Scholar]
- 21.Corkey BE, Glennon MC, Chen KS, Deeney JT, Matschinsky FM, Prentki M. A role for malonyl-CoA in glucose-stimulated insulin secretion from clonal pancreatic β-cells. J Biol Chem. 1989;264(36):21608–21612. [PubMed] [Google Scholar]
- 22.Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J Biol Chem. 1992;267(9):5802–5810. [PubMed] [Google Scholar]
- 23.MacDonald MJ, Fahien LA, Brown LJ, Hasan NM, Buss JD, Kendrick MA. Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am J Physiol Endocrinol Metab. 2005;288(1):E1–E15. doi: 10.1152/ajpendo.00218.2004. [DOI] [PubMed] [Google Scholar]
- 24.Ronnebaum SM, Ilkayeva O, Burgess SC, et al. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J Biol Chem. 2006;281(41):30593–30602. doi: 10.1074/jbc.M511908200. [DOI] [PubMed] [Google Scholar]
- 25.Henquin JC, Nenquin M, Stiernet P, Ahren B. In vivo and in vitro glucose-induced biphasic insulin secretion in the mouse: pattern and role of cytoplasmic Ca2+ and amplification signals in β-cells. Diabetes. 2006;55(2):441–451. doi: 10.2337/diabetes.55.02.06.db05-1051. [DOI] [PubMed] [Google Scholar]
- 26.MacDonald MJ, McKenzie DI, Walker TM, Kaysen JH. Lack of glyconeogenesis in pancreatic islets: expression of gluconeogenic enzyme genes in islets. Horm Metab Res. 1992;24(4):158–160. doi: 10.1055/s-2007-1003284. [DOI] [PubMed] [Google Scholar]
- 27.Sekine N, Cirulli V, Regazzi R, et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic β-cells. Potential role in nutrient sensing. J Biol Chem. 1994;269(7):4895–4902. [PubMed] [Google Scholar]
- 28.MacDonald MJ. Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. J Biol Chem. 1995;270(34):20051–20058. [PubMed] [Google Scholar]
- 29.Bavamian S, Klee P, Britan A, et al. Islet-cell-to-cell communication as basis for normal insulin secretion. Diabetes Obes Metab. 2007;9(Suppl. 2):118–132. doi: 10.1111/j.1463-1326.2007.00780.x. [DOI] [PubMed] [Google Scholar]
- 30.De Leon DD, Stanley CA. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;3(1):57–68. doi: 10.1038/ncpendmet0368. [DOI] [PubMed] [Google Scholar]
- 31.Stanley CA, Lieu YK, Hsu BY, et al. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. 1998;338(19):1352–1357. doi: 10.1056/NEJM199805073381904. [DOI] [PubMed] [Google Scholar]
- 32.Glaser B, Kesavan P, Heyman M, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338(4):226–230. doi: 10.1056/NEJM199801223380404. [DOI] [PubMed] [Google Scholar]
- 33.Clayton PT, Eaton S, Aynsley-Green A, et al. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of β-oxidation in insulin secretion. J Clin Invest. 2001;108(3):457–465. doi: 10.1172/JCI11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molven A, Matre GE, Duran M, et al. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes. 2004;53(1):221–227. doi: 10.2337/diabetes.53.1.221. [DOI] [PubMed] [Google Scholar]
- 35.Otonkoski T, Jiao H, Kaminen-Ahola N, et al. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic β cells. Am J Hum Genet. 2007;81(3):467–474. doi: 10.1086/520960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otonkoski T, Kaminen N, Ustinov J, et al. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes. 2003;52(1):199–204. doi: 10.2337/diabetes.52.1.199. [DOI] [PubMed] [Google Scholar]
- 37.Goossens A, Gepts W, Saudubray JM, et al. Diffuse and focal nesidioblastosis. A clinicopathological study of 24 patients with persistent neonatal hyperinsulinemic hypoglycemia. Am J Surg Pathol. 1989;13(9):766–775. [PubMed] [Google Scholar]
- 38.Yakovac WC, Baker L, Hummeler K. B-cell nesidioblastosis in idiopathic hypoglycemia of infancy. J Pediatr. 1971;79(2):226–231. doi: 10.1016/s0022-3476(71)80105-1. [DOI] [PubMed] [Google Scholar]
- 39.Sempoux C, Guiot Y, Dubois D, et al. Pancreatic B-cell proliferation in persistent hyperinsulinemic hypoglycemia of infancy: an immunohistochemical study of 18 cases. Mod Pathol. 1998;11(5):444–449. [PubMed] [Google Scholar]
- 40.Service FJ, Natt N, Thompson GB, et al. Noninsulinoma pancreatogenous hypoglycemia: a novel syndrome of hyperinsulinemic hypoglycemia in adults independent of mutations in Kir6.2 and SUR1 genes. J Clin Endocrinol Metab. 1999;84(5):1582–1589. doi: 10.1210/jcem.84.5.5645. [DOI] [PubMed] [Google Scholar]
- 41.Patti ME, McMahon G, Mun EC, et al. Severe hypoglycaemia post-gastric bypass requiring partial pancreatectomy: evidence for inappropriate insulin secretion and pancreatic islet hyperplasia. Diabetologia. 2005;48(11):2236–2240. doi: 10.1007/s00125-005-1933-x. [DOI] [PubMed] [Google Scholar]
- 42.Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353(3):249–254. doi: 10.1056/NEJMoa043690. [DOI] [PubMed] [Google Scholar]
- 43.Meier JJ, Butler AE, Galasso R, Butler PC. Hyperinsulinemic hypoglycemia after gastric bypass surgery is not accompanied by islet hyperplasia or increased β-cell turnover. Diabetes Care. 2006;29(7):1554–1559. doi: 10.2337/dc06-0392. [DOI] [PubMed] [Google Scholar]
- 44.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with Type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 45.Bonner-Weir S, Weirr GC. New sources of pancreatic β-cells. Nat Biotechnol. 2005;23(7):857–861. doi: 10.1038/nbt1115. [DOI] [PubMed] [Google Scholar]
- 46.Meier JJ, Butler AE, Saisho Y, et al. β-cell replication is the primary mechanism subserving the postnatal expansion of B-cell mass in humans. Diabetes. 2008;57(6):1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brelje TC, Scharp DW, Lacy PE, et al. Effect of homologous placental lactogens, prolactins, and growth hormones on islet β-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology. 1993;132(2):879–887. doi: 10.1210/endo.132.2.8425500. [DOI] [PubMed] [Google Scholar]
- 48.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. Br J Obstet Gynaecol. 1978;85(11):818–820. doi: 10.1111/j.1471-0528.1978.tb15835.x. [DOI] [PubMed] [Google Scholar]
- 49.Ritzel RA, Butler AE, Rizza RA, Veldhuis JD, Butler PC. Relationship between β-cell mass and fasting blood glucose concentration in humans. Diabetes Care. 2006;29(3):717–718. doi: 10.2337/diacare.29.03.06.dc05-1538. [DOI] [PubMed] [Google Scholar]
- 50.Georgia S, Bhushan A. β-cell replication is the primary mechanism for maintaining postnatal β cell mass. J Clin Invest. 2004;114(7):963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429(6987):41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 52.Alonso LC, Yokoe T, Zhang P, et al. Glucose infusion in mice: a new model to induce β-cell replication. Diabetes. 2007;56(7):1792–1801. doi: 10.2337/db06-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic β-cells in adult rats after short-term glucose infusion. Diabetes. 1989;38(1):49–53. doi: 10.2337/diab.38.1.49. [DOI] [PubMed] [Google Scholar]
- 54.Chick WL. β cell replication in rat pancreatic monolayer cultures. Effects of glucose, tolbutamide, glucocorticoid, growth hormone and glucagon 1. Diabetes. 1973;22(9):687–693. doi: 10.2337/diab.22.9.687. [DOI] [PubMed] [Google Scholar]
- 55.Ueki K, Okada T, Hu J, et al. Total insulin and IGF-I resistance in pancreatic β cells causes overt diabetes. Nat Genet. 2006;38(5):583–588. doi: 10.1038/ng1787. [DOI] [PubMed] [Google Scholar]
- 56.Perfetti R, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology. 2000;141(12):4600–4605. doi: 10.1210/endo.141.12.7806. [DOI] [PubMed] [Google Scholar]
- 57.Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS-2 causes Type 2 diabetes in mice. Nature. 1998;391(6670):900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 58.Kushner JA, Ye J, Schubert M, et al. Pdx1 restores β-cell function in Irs2 knockout mice. J Clin Invest. 2002;109(9):1193–1201. doi: 10.1172/JCI14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Terauchi Y, Takamoto I, Kubota N, et al. Glucokinase and IRS-2 are required for compensatory β-cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest. 2007;117(1):246–257. doi: 10.1172/JCI17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jhala US, Canettieri G, Screaton RA, et al. cAMP promotes pancreatic β-cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003;17(13):1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lingohr MK, Briaud I, Dickson LM, et al. Specific regulation of IRS-2 expression by glucose in rat primary pancreatic islet β-cells. J Biol Chem. 2006;281(23):15884–15892. doi: 10.1074/jbc.M600356200. [DOI] [PubMed] [Google Scholar]
- 62.Screaton RA, Conkright MD, Katoh Y, et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell. 2004;119(1):61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 63.Tuttle RL, Gill NS, Pugh W, et al. Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat Med. 2001;7(10):1133–1137. doi: 10.1038/nm1001-1133. [DOI] [PubMed] [Google Scholar]
- 64.Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet β cell expression of constitutively active Akt1/PKB α induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest. 2001;108(11):1631–1638. doi: 10.1172/JCI13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kitamura T, Nakae J, Kitamura Y, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic β cell growth. J Clin Invest. 2002;110(12):1839–1847. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uchida T, Nakamura T, Hashimoto N, et al. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat Med. 2005;11(2):175–182. doi: 10.1038/nm1187. [DOI] [PubMed] [Google Scholar]
- 67.Tanabe K, Liu Z, Patel S, et al. Genetic deficiency of glycogen synthase kinase-3β corrects diabetes in mouse models of insulin resistance. PLoS Biol. 2008;6(2):e37. doi: 10.1371/journal.pbio.0060037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pende M, Kozma SC, Jaquet M, et al. Hypoinsulinaemia, glucose intolerance and diminished β-cell size in S6K1-deficient mice. Nature. 2000;408(6815):994–997. doi: 10.1038/35050135. [DOI] [PubMed] [Google Scholar]
- 69.Alliouachene S, Tuttle RL, Boumard S, et al. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J Clin Invest. 2008;118(11):3629–3638. doi: 10.1172/JCI35237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnson JD, Bernal-Mizrachi E, Alejandro EU, et al. Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc Natl Acad Sci USA. 2006;103(51):19575–19580. doi: 10.1073/pnas.0604208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elghazi L, Rachdi L, Weiss AJ, Cras-Meneur C, Bernal-Mizrachi E. Regulation of β-cell mass and function by the Akt/protein kinase B signalling pathway. Diabetes Obes Metab. 2007;9(Suppl. 2):147–157. doi: 10.1111/j.1463-1326.2007.00783.x. [DOI] [PubMed] [Google Scholar]
- 72.Steiner DF, Oyer PE. The biosynthesis of insulin and a probable precursor of insulin by a human islet cell adenoma. Proc Natl Acad Sci USA. 1967;57(2):473–480. doi: 10.1073/pnas.57.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rhodes CJ. Diabetes mellitus. In: LeRoith D, Taylor SI, Olefsky JM, editors. A Fundamental and Clinical Text. Lippincott Williams & Wilkins; PA, USA: 2004. pp. 27–50. [Google Scholar]
- 74.Orci L, Ravazzola M, Storch MJ, Anderson RG, Vassalli JD, Perrelet A. Proteolytic maturation of insulin is a post-Golgi event which occurs in acidifying clathrin-coated secretory vesicles. Cell. 1987;49(6):865–868. doi: 10.1016/0092-8674(87)90624-6. [DOI] [PubMed] [Google Scholar]
- 75.Davidson HW, Rhodes CJ, Hutton JC. Intraorganellar calcium and pH control proinsulin cleavage in the pancreatic β cell via two distinct site-specific endopeptidases. Nature. 1988;333(6168):93–96. doi: 10.1038/333093a0. [DOI] [PubMed] [Google Scholar]
- 76.Smeekens SP, Montag AG, Thomas G, et al. Proinsulin processing by the subtilisin-related proprotein convertases furin, PC2, and PC3. Proc Natl Acad Sci USA. 1992;89(18):8822–8826. doi: 10.1073/pnas.89.18.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davidson HW, Hutton JC. The insulin-secretory-granule carboxypeptidase H. Purification and demonstration of involvement in proinsulin processing. Biochem J. 1987;245(2):575–582. doi: 10.1042/bj2450575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roth J, Gorden P, Pastan I. “Big insulin”: a new component of plasma insulin detected by immunoassay. Proc Natl Acad Sci USA. 1968;61(1):138–145. doi: 10.1073/pnas.61.1.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lazarus NR, Tanese T, Gutman R, Recant L. Synthesis and release of proinsulin and insulin by human insulinoma tissue. J Clin Endocrinol Metab. 1970;30(3):273–281. doi: 10.1210/jcem-30-3-273. [DOI] [PubMed] [Google Scholar]
- 80.Gorden P, Sherman B, Roth J. Proinsulin-like component of circulating insulin in the basal state and in patients and hamsters with islet cell tumors. J Clin Invest. 1971;50(10):2113–2122. doi: 10.1172/JCI106705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gutman RA, Lazarus NR, Penhos JC, Fajans S, Recant L. Circulating proinsulin-like material in patients with functioning insulinomas. N Engl J Med. 1971;284(18):1003–1008. doi: 10.1056/NEJM197105062841803. [DOI] [PubMed] [Google Scholar]
- 82.Sherman BM, Pek S, Fajans SS, Floyd JC, Jr, Conn JW. Plasma proinsulin in patients with functioning pancreatic islet cell tumors. J Clin Endocrinol Metab. 1972;35(2):271–280. doi: 10.1210/jcem-35-2-271. [DOI] [PubMed] [Google Scholar]
- 83.Gorden P, Skarulis MC, Roach P, et al. Plasma proinsulin-like component in insulinoma: a 25-year experience. J Clin Endocrinol Metab. 1995;80(10):2884–2887. doi: 10.1210/jcem.80.10.7559869. [DOI] [PubMed] [Google Scholar]
- 84.Halban PA, Irminger JC. Sorting and processing of secretory proteins. Biochem J. 1994;299(Pt 1):1–18. doi: 10.1042/bj2990001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Irminger JC, Vollenweider FM, Neerman-Arbez M, Halban PA. Human proinsulin conversion in the regulated and the constitutive pathways of transfected AtT20 cells. J Biol Chem. 1994;269(3):1756–1762. [PubMed] [Google Scholar]
- 86.Nagamatsu S, Steiner DF. Altered glucose regulation of insulin biosynthesis in insulinoma cells: mouse β TC3 cells secrete insulin-related peptides predominantly via a constitutive pathway. Endocrinology. 1992;130(2):748–754. doi: 10.1210/endo.130.2.1733723. [DOI] [PubMed] [Google Scholar]
- 87.Kao PC, Taylor RL, Service FJ. Proinsulin by immunochemiluminometric assay for the diagnosis of insulinoma. J Clin Endocrinol Metab. 1994;78(5):1048–1051. doi: 10.1210/jcem.78.5.8175958. [DOI] [PubMed] [Google Scholar]
- 88.Service FJ, McMahon MM, O’Brien PC, Ballard DJ. Functioning insulinoma – incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc. 1991;66(7):711–719. doi: 10.1016/s0025-6196(12)62083-7. [DOI] [PubMed] [Google Scholar]
- 89.Marx S, Spiegel AM, Skarulis MC, Doppman JL, Collins FS, Liotta LA. Multiple endocrine neoplasia type 1: clinical and genetic topics. Ann Intern Med. 1998;129(6):484–494. doi: 10.7326/0003-4819-129-6-199809150-00011. [DOI] [PubMed] [Google Scholar]
- 90.Anlauf M, Bauersfeld J, Raffel A, et al. Insulinomatosis: a multicentric insulinoma disease that frequently causes early recurrent hyperinsulinemic hypoglycemia. Am J Surg Pathol. 2009;33(3) doi: 10.1097/PAS.0b013e3181874eca. [DOI] [PubMed] [Google Scholar]
- 91.Anlauf M, Schlenger R, Perren A, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol. 2006;30(5):560–574. doi: 10.1097/01.pas.0000194044.01104.25. [DOI] [PubMed] [Google Scholar]
- 92.Guettier JM, Kam A, Chang R, et al. Localization of insulinomas to regions of the pancreas by intraarterial calcium stimulation: the NIH experience. J Clin Endocrinol Metab. 2009;94(4):1074–1080. doi: 10.1210/jc.2008-1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hirshberg B, Livi A, Bartlett DL, et al. Forty-eight-hour fast: the diagnostic test for insulinoma. J Clin Endocrinol Metab. 2000;85(9):3222–3226. doi: 10.1210/jcem.85.9.6807. [DOI] [PubMed] [Google Scholar]
- 94.Hirshberg B, Cochran C, Skarulis MC, et al. Malignant insulinoma: spectrum of unusual clinical features. Cancer. 2005;104(2):264–272. doi: 10.1002/cncr.21179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guettier JM, Gorden P. Hypoglycemia. Endocrinol Metab Clin North Am. 2006;35(4):753ix–757ix. doi: 10.1016/j.ecl.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 96.Marx SJ, Simonds WF. Hereditary hormone excess: genes, molecular pathways, and syndromes. Endocr Rev. 2005;26(5):615–661. doi: 10.1210/er.2003-0037. [DOI] [PubMed] [Google Scholar]
- 97.Perren A, Roth J, Muletta-Feurer S, et al. Clonal analysis of sporadic pancreatic endocrine tumours. J Pathol. 1998;186(4):363–371. doi: 10.1002/(SICI)1096-9896(199812)186:4<363::AID-PATH197>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 98.Barghorn A, Komminoth P, Bachmann D, et al. Deletion at 3p25.3–p23 is frequently encountered in endocrine pancreatic tumours and is associated with metastatic progression. J Pathol. 2001;194(4):451–458. doi: 10.1002/path.886. [DOI] [PubMed] [Google Scholar]
- 99.Barghorn A, Speel EJ, Farspour B, et al. Putative tumor suppressor loci at 6q22 and 6q23–q24 are involved in the malignant progression of sporadic endocrine pancreatic tumors. Am J Pathol. 2001;158(6):1903–1911. doi: 10.1016/S0002-9440(10)64658-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Speel EJ, Richter J, Moch H, et al. Genetic differences in endocrine pancreatic tumor subtypes detected by comparative genomic hybridization. Am J Pathol. 1999;155(6):1787–1794. doi: 10.1016/S0002-9440(10)65495-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhao J, Moch H, Scheidweiler AF, et al. Genomic imbalances in the progression of endocrine pancreatic tumors. Genes Chromosomes Cancer. 2001;32(4):364–372. doi: 10.1002/gcc.1201. [DOI] [PubMed] [Google Scholar]
- 102.Jonkers YM, Claessen SM, Perren A, et al. Chromosomal instability predicts metastatic disease in patients with insulinomas. Endocr Relat Cancer. 2005;12(2):435–447. doi: 10.1677/erc.1.00960. [DOI] [PubMed] [Google Scholar]
- 103.Zhuang Z, Vortmeyer AO, Pack S, et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res. 1997;57(21):4682–4686. [PubMed] [Google Scholar]
- 104.Arnold CN, Sosnowski A, Schmitt-Graff A, Arnold R, Blum HE. Analysis of molecular pathways in sporadic neuroendocrine tumors of the gastro–entero–pancreatic system. Int J Cancer. 2007;120(10):2157–2164. doi: 10.1002/ijc.22569. [DOI] [PubMed] [Google Scholar]
- 105.Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in β cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109(3):321–334. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 106.Creutzfeldt W, Creutzfeldt C, Frerichs H, Track NS, Arnold R. Histochemistry, ultrastructure and hormone content of human insulinomas. Horm Metab Res. 1976;(Suppl. 6):7–18. [PubMed] [Google Scholar]
- 107.Minn AH, Kayton M, Lorang D, et al. Insulinomas and expression of an insulin splice varient. Lancet. 2004;363(9406):363–373. doi: 10.1016/S0140-6736(04)15438-X. [DOI] [PubMed] [Google Scholar]
- 108.Placzkowski KA, Vella A, Thompson GB, et al. Secular trends in the presentation and management of functioning insulinoma at the Mayo Clinic, 1987–2007. J Clin Endocrinol Metab. 2009;94(4):1069–1073. doi: 10.1210/jc.2008-2031. [DOI] [PubMed] [Google Scholar]
- 109.Brown CK, Bartlett DL, Doppman JL, et al. Intraarterial calcium stimulation and intraoperative ultrasonography in the localization and resection of insulinomas. Surgery. 1997;122(6):1189–1193. doi: 10.1016/s0039-6060(97)90226-9. [DOI] [PubMed] [Google Scholar]
- 110.Pitre J, Soubrane O, Palazzo L, Chapuis Y. Endoscopic ultrasonography for the preoperative localization of insulinomas. Pancreas. 1996;13(1):55–60. doi: 10.1097/00006676-199607000-00007. [DOI] [PubMed] [Google Scholar]
- 111.Schumacher B, Lubke HJ, Frieling T, Strohmeyer G, Starke AA. Prospective study on the detection of insulinomas by endoscopic ultrasonography. Endoscopy. 1996;28(3):273–276. doi: 10.1055/s-2007-1005452. [DOI] [PubMed] [Google Scholar]
- 112.Ardengh JC, Rosenbaum P, Ganc AT, et al. Role of EUS in the preoperative localization of insulinomas compared with spiral CT. Gastrointest Endosc. 2000;51(5):552–555. doi: 10.1016/s0016-5107(00)70288-4. [DOI] [PubMed] [Google Scholar]
- 113.Nesje LB, Varhaug JE, Husebye ES, Odegaard S. Endoscopic ultrasonography for preoperative diagnosis and localization of insulinomas. Scand J Gastroenterol. 2002;37(6):732–737. doi: 10.1080/00365520212501. [DOI] [PubMed] [Google Scholar]
- 114.Zimmer T, Stolzel U, Bader M, et al. Endoscopic ultrasonography and somatostatin receptor scintigraphy in the preoperative localisation of insulinomas and gastrinomas. Gut. 1996;39(4):562–568. doi: 10.1136/gut.39.4.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Krenning EP, Kwekkeboom DJ, Oei HY, et al. Somatostatin-receptor scintigraphy in gastroenteropancreatic tumors. An overview of European results. Ann NY Acad Sci. 1994;733:416–424. doi: 10.1111/j.1749-6632.1994.tb17291.x. [DOI] [PubMed] [Google Scholar]
- 116.van Eyck CH, Bruining HA, Reubi JC, et al. Use of isotope-labeled somatostatin analogs for visualization of islet cell tumors. World J Surg. 1993;17(4):444–447. doi: 10.1007/BF01655102. [DOI] [PubMed] [Google Scholar]
- 117.Oberg K, Astrup L, Eriksson B, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine tumours (including bronchopulmonary and thymic neoplasms). Part I – general overview. Acta Oncol. 2004;43(7):617–625. doi: 10.1080/02841860410018575. [DOI] [PubMed] [Google Scholar]
- 118.Oberg K. Somatostatin analog octreotide LAR in gastro–entero–pancreatic tumors. Expert Rev Anticancer Ther. 2009;9(5):557–566. doi: 10.1586/era.09.26. [DOI] [PubMed] [Google Scholar]
- 119.Kauhanen S, Seppanen M, Minn H, et al. Fluorine-18-l-dihydroxyphenylalanine (18F-DOPA) positron emission tomography as a tool to localize an insulinoma or β-cell hyperplasia in adult patients. J Clin Endocrinol Metab. 2007;92(4):1237–1244. doi: 10.1210/jc.2006-1479. [DOI] [PubMed] [Google Scholar]
- 120.Wild D, Macke H, Christ E, Gloor B, Reubi JC. Glucagon-like peptide 1-receptor scans to localize occult insulinomas. N Engl J Med. 2008;359(7):766–768. doi: 10.1056/NEJMc0802045. [DOI] [PubMed] [Google Scholar]