

Abstract
The contribution of the NMDA receptors (NMDARs) to synaptic plasticity declines during aging, and the decline is thought to contribute to memory deficits. Here, we demonstrate that an age-related shift in intracellular redox state contributes to the decline in NMDAR responses through Ca2+/calmodulin-dependent protein kinase II (CaMKII). The oxidizing agent xanthine/xanthine oxidase (X/XO) decreased the NMDAR-mediated synaptic responses at hippocampal CA3–CA1 synapses in slices from young (3–8 months) but not aged (20–25 months) rats. Conversely, the reducing agent dithiothreitol (DTT) selectively enhanced NMDAR response to a greater extent in aged hippocampal slices. The enhancement of NMDAR responses facilitated induction of long-term potentiation in aged but not young animals. The DTT-mediated growth in the NMDAR response was not observed for the AMPA receptor-mediated synaptic responses. A similar increase was observed by intracellular application of the membrane-impermeable reducing agent, l-glutathione (l-GSH), through the intracellular recording pipette, indicating that the increased NMDAR response was dependent on intracellular redox state. DTT enhancement of the NMDAR response was dependent on CaMKII activity and was blocked by the CaMKII inhibitor—myristoylated autocamtide-2-related inhibitory peptide (myr-AIP)—but not by inhibition of the activity of protein phosphatases—PP1 and calcineurin (CaN/PP2B) or protein kinase C. CaMKII activity assays established that DTT increased CaMKII activity in CA1 cytosolic extracts in aged but not in young animals. These findings indicate a link between oxidation of CaMKII during aging, a decline in NMDAR responses, and altered synaptic plasticity.
Introduction
The NMDA receptor (NMDAR) is a major source of Ca2+ influx into the postsynaptic neuron during the induction of LTP at hippocampal CA3–CA1 synapses (Bliss and Collingridge, 1993). CA1 region-specific knock-out of the NR1 subunit of the NMDAR abolishes LTP, in addition to impairing spatial memory (Tsien et al., 1996). Aged, memory impaired animals exhibit deficits in LTP induction. Importantly, the NMDAR component of the synaptic response is decreased in aged animals (Barnes et al., 1997; Billard and Rouaud, 2007), suggesting that age-related LTP and memory deficits are due to a decrease in the NMDAR-mediated component of synaptic transmission (Foster, 1999, 2007; Rosenzweig and Barnes, 2003), also referred to here as “NMDAR hypofunction.” Numerous studies indicate that NMDARs contribute less Ca2+ to the induction of LTP in the area CA1 of aged hippocampus, when compared to the young hippocampus (Norris et al., 1998a; Shankar et al., 1998; Boric et al., 2008). However, it is unclear what age-related mechanism underlies the NMDAR hypofunction.
Age-related alterations that may contribute to the NMDAR deficits include altered subunit expression, composition, and splice forms (Magnusson et al., 2005, 2006). However, there is a debate concerning whether NMDAR subunit expression decreases at hippocampal CA3–CA1 synapses (Foster, 2002). In addition, it is possible that functional differences are related to posttranslational modifications associated with oxidation or phosphorylation state rather than number and/or type of receptor subunits (Foster, 2007). Previous research examining the ability of reducing and oxidizing (redox) agents to modulate NMDAR activity in cell cultures and in tissue from neonates suggests that redox state is an important determinant of NMDAR function (Aizenman et al., 1989, 1990; Bernard et al., 1997; Choi and Lipton, 2000; Choi et al., 2001), possibly through oxidation of extracellular cysteine residues on the NMDAR (Lipton et al., 2002). Intracellular signaling molecules that affect NMDAR function are also sensitive to redox state. The aged brain exhibits an increase in oxidative damage (Foster, 2006; Poon et al., 2006), and a decrease in redox buffering capacity (Parihar et al., 2008); however, the effect of redox state on NMDAR function during aging has not been examined.
The current studies confirm that the NMDAR-mediated synaptic potentials are decreased at CA3–CA1 synapses of the aged hippocampus. We now provide evidence that the age-related decline in NMDAR-mediated synaptic responses and impaired induction of LTP in area CA1 of the hippocampus are related to the intracellular redox state. NMDAR responses were modified by redox agents in an age-dependent manner. However, using a combination of extracellular and intracellular recordings with the relatively membrane-impermeable l-glutathione (l-GSH), we found that intracellular redox state mediates that age-dependent shift in NMDAR responses. Furthermore, we demonstrate that the mechanism for the age-dependent redox modulation of NMDARs involves Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity. These findings establish age-related changes in the redox state as a mechanism for the decrease in NMDAR response.
Materials and Methods
Animals.
Procedures involving animals have been reviewed and approved by the Institutional Animal Care and Use Committee and were in accordance with guidelines established by the United States Public Health Service Policy on Human Care and Use of Laboratory Animals. Male Fischer 344 rats, young (3–8 months) and aged (20–25 months), were obtained from National Institute on Aging colony at Harlan Sprague Dawley. All animals were group housed (2 per cage), maintained on a 12:12 h light schedule, and provided ad libitum access to food and water.
Hippocampal slice preparation.
The animals were deeply anesthetized using isoflurane (Webster) and decapitated with a guillotine (MyNeurolab). The brains were rapidly removed and hippocampi were dissected. Hippocampal slices (∼400 μm) were cut parallel to the alvear fibers using a tissue chopper (Mickle Laboratory Engineering). The slices were incubated in a holding chamber (at room temperature) with artificial CSF (ACSF). The ACSF consisted of the following (in mm): NaCl 124, KCl 2, KH2PO4 1.25, MgSO4 2, CaCl2 2, NaHCO3 26, and d-glucose 1. The ACSF was bubbled with 95% O2 and 5% CO2 for 20 min before measurement of pH. The pH was adjusted to 7.4 using 1–2 drops of 10 m NaOH in 4 L of ACSF. At least 30 min before recording, slices were transferred to a standard interface recording chamber (Warner Instrument). The chamber was continuously perfused with oxygenated ACSF at the rate of 2 ml/min, and the temperature was maintained at 30 ± 0.5°C.
Electrophysiological recordings.
Extracellular field EPSPs (fEPSP) from stratum radiatum of the CA1 region of the hippocampus were recorded using glass micropipettes (4–6 MΩ) filled with recording medium (ACSF). Two concentric bipolar stimulating electrodes (outer pole: stainless steel, 200 μm diameter; inner pole: platinum/iridium, 25 μm diameter) (FHC) were localized to the middle of the stratum radiatum to stimulate CA3 inputs onto CA1. Diphasic stimulus pulses of 100 μs duration were delivered by a stimulator (SD9 Stimulator; Grass Instrument) and alternated between the two pathways such that each pathway was activated at 0.033 Hz. The signals were amplified, filtered between 1 Hz and 1 kHz, and stored on a computer disk for off-line analysis. Two cursors were placed to cover the initial descending phase of the waveform and the maximum negative slope (in millivolts per millisecond) of the fEPSP and NMDAR-fEPSP was determined by a computer algorithm that determined the maximum change across a set of 20 consecutively recorded points (20 kHz sampling frequency) between the two cursors. In a few cases, two cursors were placed to cover the entire waveform, and the maximum amplitude (in millivolts) of the NMDAR-fEPSP and NMDAR-mediated intracellular EPSP was determined by a computer algorithm that calculated the valley voltage from zero (for NMDAR-fEPSP) or the maximum voltage from zero (for NMDAR-mediated intracellular EPSP) between the cursors.
For measuring the paired-pulse ratio, paired pulses were delivered through a single stimulating electrode at varying inter pulse intervals. The first pulse was set to elicit 50% of the maximal fEPSP. The various inter pulse intervals between successive pulses were 50 ms, 100 ms, 150 ms, and 200 ms. The ratio of the maximum negative slope of the second pulse to the slope of the first pulse was computed as the paired-pulse ratio. To measure changes in the presynaptic fiber volley (PFV) upon application of dithiothreitol (DTT) and xanthine/xanthine oxidase (X/XO), the average PFV amplitudes from the last 5 min of drug application were normalized to the average PFV amplitude recorded during the baseline.
For induction of LTP, the stimulation intensity was set to elicit 50% of the maximal fEPSP. After stable baseline recording at 0.033 Hz for at least 20 min, high-frequency stimulation (HFS) was delivered to the pathway at 100 Hz for 1 s (100 pulses) at the baseline stimulus intensity, and recorded for 60 min after HFS. A simultaneously recorded control (non-HFS) pathway received the test stimulus but not the HFS. In some cases, the slices were preincubated with the reducing agent DTT for at least 45 min before delivering the HFS. The average fEPSP slope corresponding to the last 5 min from each pathway was used to compare changes in synaptic strength relative to the baseline.
To obtain the NMDAR-mediated field EPSP (NMDAR-fEPSP) of CA3–CA1 synaptic transmission, slices were incubated in ACSF containing low extracellular Mg2+ (0.5 mm), 6,7-dinitroquinoxaline-2,3-dione (DNQX, 30 μm), and picrotoxin (PTX, 10 μm). In each case, the baseline response was collected for at least 10 min before experimental manipulations (drug application). Changes in the transmission properties of the synapses, induced by drug application, were calculated as percentage change from the averaged baseline responses.
To obtain the NMDAR-mediated intracellular synaptic potentials from CA1 pyramidal neurons, slices were incubated in ACSF containing low extracellular Mg2+, DNQX, and PTX as described above. Sharp microelectrodes were pulled from thin walled (1 mm) borosilicate capillary glass using a Flaming/Brown horizontal micropipette puller (Sutter Instruments). Microelectrode tips were filled with 3 m potassium acetate and in some cases included reduced l-GSH (0.7–1.4 mm). The microelectrode resistances ranged from 42 to 55 MΩ and 39 to 50 MΩ for 3 m potassium acetate- and 3 m potassium acetate + l-GSH-containing microelectrodes, respectively. Microelectrodes were visually positioned in the CA1 pyramidal cell layer using a dissecting microscope (SZH10, Optical Elements), and a bipolar stimulating electrode was positioned to stimulate the CA3 afferents onto CA1 pyramidal neurons. On cell entry, positive or negative current was applied to clamp the neuronal membrane potential at −65 mV. Only neurons with a resting membrane potential less than −59 mV, an input resistance >23 MΩ, and an action potential amplitude rising ≥78 mV from the point of spike initiation were included in the analysis. The mean resting membrane potential and holding current was −63 ± 2 mV, −0.07 ± 0.15 nA for the control cells and −63 ± 2 mV, 0.07 ± 0.18 nA for the l-GSH-injected neurons. Inclusion of l-GSH in the pipette had no significant effect on the resting membrane potential, holding current, input resistance, action potential amplitude, or the microelectrode resistance. Diphasic stimulus pulses of 100 μs duration were delivered at 0.033 Hz, and the stimulation intensity was adjusted to elicit an intracellular NMDAR synaptic response, which was below action potential threshold. Baseline response recording began within 3 min after cell entry.
All the drugs were prepared according to the manufacturer's specifications and ultimately dissolved in ACSF before bath application on the slices. 6,7-Dinitroquinoxaline-2,3(1H,4H)-dione (DNQX) (Sigma); 4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl]phenyl isoquinolinesulfonic acid ester (KN-62), okadaic acid (OA) (Tocris Bioscience), and FK-506 (LC Laboratories) were initially dissolved in a small amount of dimethyl sulfoxide (DMSO; Sigma) and diluted in ACSF to a final DMSO concentration of <0.01% and to final DNQX, KN-62, OA, and FK-506 concentrations of 30, 10, 1, and 10 μm, respectively. Picrotoxin (PTX; Tocris Bioscience) and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB; Sigma) were initially dissolved in a small amount of ethanol and diluted in ACSF to a final ethanol concentration of 0.0001% and to final PTX and DTNB concentrations of 10 μm and 0.5 mm, respectively. Xanthine (Calbiochem) was initially dissolved in a small amount of 0.1N NaOH and finally dissolved in ACSF whose pH was readjusted to 7.4 using 10N hydrochloric acid. DTT, dl-2-amino-5-phosphonovaleric acid (AP-5), reduced l-GSH (Sigma), (±)-1-(5-isoquinolinesulfonyl)-2-methylpiperazine dihydrochloride (H-7) (Tocris Bioscience), bisindolylmaleimide I (Bis-I), myristoylated autocamtide-2-related inhibitory peptide (myr-AIP) (Calbiochem), and xanthine oxidase (Roche Diagnostics) were directly dissolved in ACSF. DMSO control studies were interleaved with experiments using DMSO as vehicle.
Measurement of reactive oxygen species levels in the hippocampal slices.
Hippocampal slices were incubated for 30 min in ACSF containing a 10 μm concentration of the reactive oxygen species (ROS) detection reagent 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (c-H2DCFDA; Invitrogen). Slices incubated for 30 min in absence of c-H2DCFDA were used to detect background or autofluorescence. Following incubation, fluorescent images were obtained with an Axiovert 40 CFL fluorescent microscope and Axiocam digital CCD camera (Carl Zeiss). Fluorescence intensity was quantified as follows: fluorescent microscope was used to obtain images under uniform exposure time and intensity. The images were then converted to grayscale images in Adobe Photoshop 5.5; the resulting images were quantified by densitometry analysis using ImageJ software (http://rsbweb.nih.gov/ij). An area of ∼225 μm along the medial–lateral axis and 187.5 μm along the anterior–posterior axis, centered on the CA1 pyramidal neurons was selected for analysis of fluorescence intensity. The mean gray value intensities obtained from the aged and young animals are represented as the mean fluorescence intensities. The mean fluorescence intensity from the dye-exposed slices is normalized to the mean fluorescence intensity obtained from dye-unexposed slices (harvested from the same animal) using the following relationship:
 |
where Fs and Fc are the mean fluorescence intensities obtained from dye-exposed and dye-unexposed slices, respectively.
CaMKII activity.
Hippocampi were isolated from aged F344 rats as described above. CA1 region was separated from the rest of the hippocampus, collected in an Eppendorf tube, flash frozen in liquid nitrogen, and stored at −80°C. The frozen CA1 tissue samples were placed in a Dounce homogenizer containing 1 ml of the homogenization buffer (sucrose, 1 m Tris, pH 7.5, 1 m KCl, protease inhibitor, protein phosphatase 1 inhibitor, protein phosphatase 2 inhibitor, 100 mg/ml sodium butyrate, and 0.1 m phenylmethylsulfonyl fluoride; all prepared in distilled H2O) and homogenized using at least six strokes of the pestle. Homogenates were centrifuged at 7700 × g for 10 min at 4°C. The supernatant (containing the cytosolic fraction) was carefully isolated and stored at −80°C. Protein concentrations of the cytosolic fractions were determined using the BCA assay method (Pierce). CaMKII activity in the cytosolic fraction was measured using the CaMKII assay kit (CycLex). Briefly, uniform amount of cytosolic extracts (protein concentration = 2.0 μg per well) were loaded onto microtiter wells coated with a specific peptide substrate for CaMKII—Syntide-2, along with kinase reaction buffer with or without Ca2+/calmodulin. Purified CaMKII (30 mU per reaction; CycLex) was used as positive control and cytosolic extracts incubated with EGTA + myr-AIP (CaMKII-specific peptide inhibitor) were used as negative control to obtain a measure of comparison for the CaMKII activity in the DTT-treated and untreated cytosolic extracts. CaMKII activity is expressed as spectral absorbance units at 450 nm, normalized to the control.
Statistical analysis.
All statistical analyses were performed using StatView 5.0 (SAS Institute). Student's t tests were used to examine for differences between treatments with significance set at p < 0.05. For tests involving more than one factor, ANOVA was used, followed by Fisher's protected least significant difference (PLSD) post hoc analysis to localize differences. In general only one or two slices per animal were used for a given experimental condition; although, several conditions could be examined using slices from the same animal. Therefore, where stated, n represents the number of slices used in each experiment, unless stated as the number of animals, in which case we used only one slice from each animal. To ensure the reliability of our main effect (increased NMDAR responses following DTT application in aged animals), experiments involving DTT application alone were interleaved with experiments involving kinase/phosphatase inhibitors, DTNB and AP-5, resulting in a relatively large number of slices (n = 16) for this experiment.
Results
Oxidation decreases NMDAR responses in an age-dependent manner
The ROS detection reagent c-H2DCFDA was used to evaluate differential rates of ROS production in hippocampal slices from young and aged animals (Fig. 1A,B). Dye-unexposed slices (aged control: 36.14 ± 4.42%, n = 3 animals; young control: 31.97 ± 3.16%, n = 3 animals) showed no significant difference in fluorescence across the age groups and were used to normalize the fluorescence intensity obtained from c-H2DCFDA-treated slices. Incubation of the hippocampal slices from young and aged animals with c-H2DCFDA for 30 min resulted in significantly (unpaired t test; p < 0.05) enhanced fluorescence in aged animals (242.19 ± 20.96%, n = 3 animals) when compared to young animals (141.61 ± 11.78%, n = 3 animals). To exclude possible ROS outside the cells, aged hippocampal slices were incubated with superoxide dismutase (SOD; 121 U/ml) and catalase (260 U/ml) during the 30 min before imaging. There was no significant difference (p > 0.05) in the c-H2DCFDA fluorescence observed between the SOD/catalase-exposed (292.388 ± 28.04%, n = 3) and unexposed aged hippocampal slices.
Figure 1.

Enhanced ROS production is observed in hippocampal tissue from aged rats. A, Indicated above each image column are the imaging conditions (bright field or green emission) and the exposure time (100 ms) for hippocampal slices that were either untreated or treated (Carboxy-H2DCFDA) with the dye. The rows are images of the CA1 region of young (top row) and aged (second row) hippocampal slices. The lowermost image is a Carboxy-H2DCFDA-treated slice from an aged animal, which was incubated with SOD + catalase. The various layers of hippocampal area CA1—stratum radiatum (s.rad), stratum pyramidale (s.pyr), and stratum oriens (s.or)—are indicated in the last set of images. Scale bars, 50 μm. B, Quantification of the mean fluorescence intensity generated by the oxidation of c-H2DCFDA (c-H2DCFDA Fluorescence) from young (n = 3) (open bar), aged (n = 3), and SOD + catalase-exposed aged (n = 3) (gray bars) hippocampal slices expressed as percentage of fluorescence in untreated (control) slices from the same animal. In this and subsequent figures, error bars represent SEM, asterisks indicate significant difference between the groups indicated, and n.s indicates no significant difference.
The NMDAR-mediated field EPSPs (NMDAR-fEPSPs) were studied to assess changes in the NMDAR response in hippocampal slices from young and aged animals. We confirmed that the NMDAR-mediated synaptic transmission decreases in the area CA1 of the hippocampus during aging. The stimulation-evoked PFV is an indicator of the level of axon activation that gives rise to the NMDAR-fEPSP (Fig. 2A). To examine the relationship between PFV amplitude and the amplitude of the NMDAR-fEPSP across age groups, the PFV amplitude was separated into 0.4 mV bins and plotted against the corresponding NMDAR-fEPSP amplitude obtained from the aged and the young animals (Fig. 2B). An ANOVA revealed that the amplitude of the NMDAR-fEPSP was reduced in the aged animals (n = 6 animals) when compared to young animals (n = 5 animals) (F(1,47) = 27.47, p < 0.0001). The maximum amplitude of the NMDAR-fEPSP was 0.73 ± 0.14 mV and 2.87 ± 0.9 mV in aged and young animals, respectively.
Figure 2.

NMDAR-mediated synaptic potentials (NMDAR-fEPSP) are reduced in area CA1 of the hippocampus during aging. A, Representative traces of NMDAR-fEPSPs obtained at consecutively higher stimulus intensities from the young (left) and aged (right) animals. Open and filled arrows indicate the PFV and NMDAR-fEPSP, respectively. As observed in the traces, the aged animals exhibit a markedly reduced NMDAR-mediated synaptic potential. B, Plot of the mean NMDAR-fEPSP amplitude versus the PFV amplitude (at 0.4 mV binning width). The aged animals (filled circles) (n = 6) exhibited reduced NMDAR-fEPSP when compared to the young animals (open circles) (n = 5).
For subsequent studies, the stimulation intensity was set to evoke a response that was 30% to 50% of the maximal NMDAR-fEPSP, and the slope of the NMDAR-fEPSP was measured before and after pharmacological manipulations. To test the hypothesis that the decrease in the NMDAR response was related to oxidizing conditions X/XO, an enzyme substrate combination that produces two types of ROS—superoxide anion and hydrogen peroxide—was applied to hippocampal slices of young and aged animals. Paired t test revealed that application of X/XO [20 μg/ml/(0.25 U/mg xanthine)] for 60 min, significantly decreased the slope of the NMDAR-fEPSP from the baseline levels in the young (p < 0.01) but not in the aged animals. Furthermore, application of X/XO significantly (F(1,10) = 15.49, p < 0.01) decreased the NMDAR-fEPSP slope to a greater extent in the young animals (66.37 ± 7.04%, n = 5), when compared to the aged animals (96.41 ± 6.14%, n = 7) (Fig. 3A,B). Paired t test on the percentage change in the PFV indicated no effect (p > 0.05) of X/XO (young: 95.16 ± 8.29; aged: 108.23 ± 19.44). The effect of ROS on the NMDAR response of young animals was reversible, such that even in the presence of a higher concentration of xanthine oxidase [X/XO: 20 μg/ml/(1 U/mg xanthine)], which decreased the NMDAR-mediated synaptic response to 54.21 ± 5.79%, the response recovered to 88.11 ± 5.41% of the baseline (n = 5) following a 50 min washout (Fig. 3C).
Figure 3.

The oxidizing agent X/XO decreases NMDAR-mediated synaptic potentials in young animals but not in aged animals. A, Time course of the change in the normalized NMDAR-fEPSP slope in the aged (filled circles) (n = 7) and in the young (open circles) (n = 5) animals following application of X/XO [20 μg/ml/(0.25 U/mg xanthine)] for 60 min. B, Representative traces (average of 5 traces under each condition) illustrating the change in the NMDAR-fEPSP in the young (top) and aged (bottom) animals under control conditions and at the end of a 60 min application of X/XO. Calibration: 20 ms, 0.5 mV. C, Time course of the change in the normalized NMDAR-fEPSP slope in the young animals (n = 5) following application of X/XO [20 μg/ml/(1 U/mg xanthine)] for 30 min and followed by a washout for 50 min. The inset illustrates the average of 5 traces obtained during the indicated time points: baseline (1), under X/XO (2), and upon washout (3). Calibration: 20 ms, 0.5 mV.
Reducing agents enhance NMDAR responses in an age-dependent manner
The age-dependent sensitivity of NMDAR-fEPSP to oxidizing conditions suggests that components of the NMDAR signaling system are initially oxidized to a greater extent in aged animals. Furthermore, the recovery of the NMDAR-fEPSP following washout indicates that young animals possess sufficient antioxidant and/or redox buffering capacity to absorb the excess oxyradical production. To test whether the decline in the NMDAR response in aged animals might be due to the age-dependent increase in the formation of disulfide linkages on the cysteine residues, the reducing agent DTT was applied to hippocampal slices. DTT can reduce the disulfide bonds on proteins into free thiols, thus partially reversing the effect of oxidative stress. Paired t test revealed that DTT significantly enhanced the slope of the NMDAR-fEPSP from the baseline levels in both the aged (p < 0.0001) and young (p < 0.05) animals. However, bath application of DTT (0.7 mm, 45 min) significantly (F(1,19) = 5.49, p < 0.05) increased the slope of the NMDAR-fEPSP to a greater extent in the aged animals (171.38 ± 13.26%, n = 16) when compared to young animals (114.55 ± 4.41%, n = 5) (Fig. 4A). Paired t test on the PFV amplitude before and after application of DTT confirmed no change (p > 0.05) in the PFV amplitude for aged (102.81 ± 3.84) and young (97.08 ± 5.41) animals, indicating that the effect of DTT on the NMDAR-mediated response was not due to changes in excitability and/or the number of axons activated. The enhancement of the NMDAR-fEPSP in the aged animals was maintained (167.32 ± 20.91%, n = 5) following a 45 min washout of DTT (Fig. 4B).
Figure 4.

The reducing agent DTT increases NMDAR-mediated synaptic responses to a greater extent in aged than in the young animals. A, Time course of the change in the normalized NMDAR-fEPSP slope in the aged (filled circles) (n = 16) and young (open circles) (n = 5) animals following application of DTT for 45 min. Inset, Representative traces (average of 5 consecutive traces under each condition) illustrating the change in NMDAR-fEPSP in the presence of DTT in an aged (top) and a young animal. Calibration: 10 ms, 0.5 mV. B, Time course of the change in the normalized NMDAR-fEPSP slope in aged animals (filled circles) (n = 5) upon application of DTT for 45 min followed by a washout for 45 min.
To test whether the NMDAR-fEPSP in aged animals was indeed affected by the redox environment, the oxidizing agent DTNB was applied subsequent to the DTT-mediated enhancement of the NMDAR response in aged animals. Bath application of DTNB (0.5 mm, 45 min) significantly decreased (p < 0.05) the DTT-mediated increase in NMDAR-fEPSP in aged animals, such that the NMDAR-fEPSP slope was 86.49 ± 5.54% (n = 4) of the baseline (Fig. 5A). A repeated-measures ANOVA comparing the mean NMDAR-fEPSP slope corresponding to the last 5 min under each condition indicated a significant (F(2,9) = 6.24, p < 0.05) difference. Post hoc analysis revealed that the NMDAR-fEPSP slope was significantly (p < 0.05) increased under DTT compared to baseline and DTNB (p < 0.05), and no significant difference was observed between the NMDAR-fEPSP slopes in the baseline and under DTNB. Finally, the NMDAR-fEPSP was abolished by application of AP-5 (Fig. 5B), indicating that the response was dependent on NMDARs.
Figure 5.

The effect of DTT on the NMDARs involves cysteine residues. A, Time course of the change in the normalized NMDAR-fEPSP slope in aged animals (n = 4) in response to the bath application of DTT followed by DTNB. The increase in NMDAR-mediated synaptic responses by DTT was decreased by the oxidizing agent DTNB. Error bar in A is indicated for every fourth point, for purposes of clarity. B, Representative traces (average of 5 consecutive traces) of the NMDAR-fEPSP recorded under control conditions (solid black trace) and upon application of 100 μm AP-5 (dashed black trace) for at least 30 min. C, DTT does not affect the AMPAR function of aged animals. Time course of the change in fEPSP upon application of DTT for 45 min in aged hippocampal slices (n = 10). Inset, Representative traces (average of 5 consecutive traces) of the fEPSP recorded under control conditions (black trace) and after application of DTT for 45 min (gray trace). The downward-pointing arrows and cursors indicate the 15 ms time window, where the initial descending phase of the fEPSP is predominantly mediated by AMPARs. Calibration: 20 ms, 0.5 mV.
To further examine the specificity of the DTT effects, slices were bathed in normal ACSF and the AMPA receptor (AMPAR) component was recorded as the initial descending phase of the synaptic response. Application of DTT did not affect the AMPAR component of the synaptic response such that responses were 101.63 ± 2.89% (n = 10) of the baseline after application of DTT for 45 min (Fig. 5C). This confirms previous reports that DTT does not influence AMPAR function (Gozlan et al., 1995; Abele et al., 1998). Together, the results indicate that oxidation of sulfhydryl groups can rapidly regulate responsiveness of NMDARs, and that an age-related reduction in the NMDAR response is linked to the redox environment.
Intracellular redox status contributes to age-related differences in NMDAR synaptic responses
Previous research indicates that l-GSH is a cellular reducing agent that protects hippocampal neurons against oxidative stress (Shin et al., 2005; Shih et al., 2006; Yoneyama et al., 2008). However, l-GSH is relatively membrane impermeable such that exogenous application of l-GSH is not effective in increasing intracellular free thiols when compared to DTT (Mazor et al., 1996; Zou et al., 2001; Susankova et al., 2006). Extracellular application of the reduced form of l-GSH (0.7 mm, 45 min) did not alter NMDAR-fEPSP in the aged animals (104.83 ± 8.39%, n = 6) (Fig. 6A). In contrast to the extracellular application, inclusion of the reduced form of l-GSH (0.7 mm to 1.4 mm) in the intracellular recording pipette significantly (F(1,6) = 6.87, p < 0.05) enhanced the amplitude of the NMDAR-mediated synaptic potentials (203.90 ± 31.38%, n = 5) in single hippocampal CA1 pyramidal neurons from aged hippocampus when compared to age-matched control cells (91.55 ± 22.94%, n = 3), for which l-GSH was not included in the intracellular recording pipette (Fig. 6B). Finally, intracellular delivery of l-GSH in the presence of nifedipine (10 μm) continued to enhance the amplitude of the NMDAR-mediated synaptic potentials (223.54 ± 46.56%, n = 3) (Fig. 6C), suggesting that the DTT effect is not due to differences in L-channel activity. The fact that intracellular delivery of glutathione enhanced the NMDAR response provides strong evidence for intracellular redox status as a mechanism for the age-associated modulation of NMDAR function.
Figure 6.
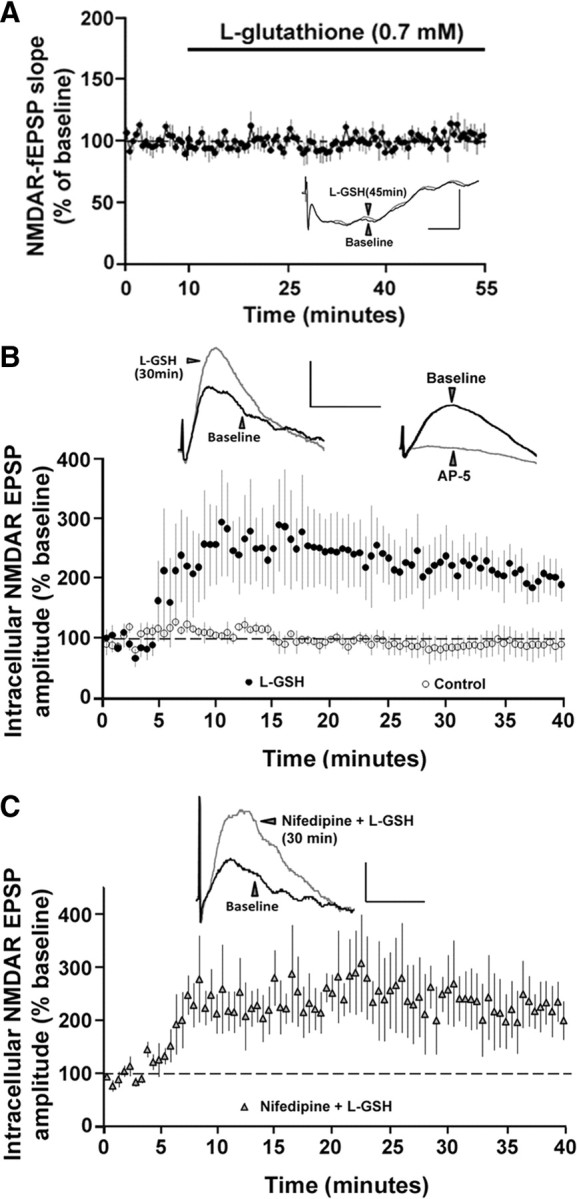
Intracellular, but not extracellular, application of l-GSH enhances NMDAR-mediated synaptic responses in aged animals. A, Time course of the normalized NMDAR-fEPSP slope in the aged animals (n = 6) in response to extracellular application of the l-GSH. Inset, Overlay of the means of five consecutive responses during the baseline (black trace) and 45 min after application of l-GSH (gray trace). Calibration: 20 ms, 0.5 mV. B, Time course of the normalized NMDAR-mediated intracellular EPSP amplitude in the aged animals obtained under control conditions (open circles, n = 3) or with l-GSH in the recording pipette (filled circles, n = 6). Left inset, Overlay of means from five consecutive responses obtained intracellularly during baseline (black trace) and 30 min after impalement (gray trace). Right inset, Overlay of means from five consecutive traces obtained intracellularly during baseline (black trace) and 25 min after application of AP-5 (gray trace). C, Time course of normalized NMDAR-mediated intracellular EPSP amplitude obtained with nifedipine in the ACSF and l-GSH in the recording pipette (filled triangles, n = 3). Inset, Overlay of means from five consecutive responses obtained intracellularly during baseline (black trace) and 30 min after impalement (gray trace). Calibration (in B, C): 40 ms, 2 mV.
Reducing conditions enhance LTP in region CA1 of aged hippocampus
For studies on LTP and paired-pulse facilitation, slices were bathed in normal ACSF to record the AMPAR and NMDAR component of the synaptic response. LTP was induced by a single episode of high-frequency stimulation (HFS; 100 Hz, 1 s). The magnitude of LTP was significantly (F(1,17) = 12.14, p < 0.01) greater in aged hippocampal slices preincubated with DTT for 45 min (136.53 ± 2.77%; n = 10), when compared to the aged controls not exposed to DTT (118.15 ± 4.63%; n = 9) (Fig. 7A,C). In contrast, there was no difference in the levels of HFS-induced LTP between young controls (130.17 ± 8.64%; n = 6) and the young slices exposed to DTT (117.09 ± 12.24%; n = 5) (Fig. 7B,D). Thus, reducing conditions enhance NMDAR responses and the magnitude of LTP only in aged animals. Furthermore, examination of paired pulses delivered at varying interpulse intervals (Δt = 50 ms, 100 ms, 150 ms, 200 ms), under control conditions and 45 min after the bath application of DTT indicated no effect of treatment across the four interpulse intervals (Fig. 7E).
Figure 7.

DTT enhances LTP in hippocampal area CA1 of aged animals. A, Time course for the expression of LTP recorded in hippocampal slices from aged animals. Slices were bathed in control ACSF (n = 9) or ACSF containing DTT (n = 10) for at least 45 min before HFS. Baseline stimulation was applied to a control pathway (open circles) and to a second pathway that received HFS (100 Hz, 1 s) (filled circles). Arrows denote HFS delivery. For purpose of clarity, each point represents the mean of two consecutive responses. B, Time course for the expression of LTP recorded in hippocampal slices from young animals. Slices were bathed in control ACSF (n = 6) (left) or ACSF containing DTT (n = 5) (right) for at least 45 min before HFS. Baseline stimulation was applied to a control pathway (open circles) and to a second pathway that received HFS (100 Hz, 1 s) (filled circles). Arrows denote HFS delivery. C, Distribution of the LTP magnitude for individual slices from aged animals bathed in control ACSF and ACSF + DTT. The rectangular boxes indicate the mean of each group. D, Quantification of the mean percentage change in the fEPSP slope recorded from the control (Cont) and HFS (HFS) pathways from young slices bathed in ACSF or ACSF + DTT. E, Plot of the paired-pulse ratio obtained under control conditions (black circles) and after 45 min bath application of DTT (gray circles) for four interpulse intervals (50, 100, 150, 200 ms). Inset: Responses obtained upon paired pulse stimulation (average of 5 consecutive traces; 50 ms interpulse interval) under control conditions and under DTT application.
CaMKII contributes to DTT-mediated enhancement of NMDAR responses in aged animals
NMDAR function is controlled by several intracellular kinases (Ben-Ari et al., 1992; Westphal et al., 1999; Li et al., 2001). Furthermore, the enzymatic activity of these kinases can be regulated by the reduction and oxidation of the cysteine residues located in their structure (Raynaud et al., 1997; Griendling et al., 2000; Knapp and Klann, 2000). To test whether serine/threonine (Ser/Thr) kinases were involved in the DTT-mediated increase of the NMDAR response, the broad-spectrum and membrane-permeable Ser/Thr kinase inhibitor H-7 was bath applied before and during the application of DTT. In the presence of H-7 (10 μm, 45 min), DTT application failed to produce the robust increase (one-group t test; p > 0.05) in the NMDAR-fEPSP slope (111.86 ± 6.92%, n = 7) from the baseline levels (Fig. 8A). Similarly, bath application of the CaMK inhibitor KN-62 (10 μm, 45 min) (Tokumitsu et al., 1990), before and during the application of DTT, blocked (97.9 ± 7.98%, n = 5) the DTT-mediated increase in NMDAR-fEPSP in aged animals (Fig. 8B). Finally, the specific inhibitor of CaMKII, myr-AIP (5 μm, 60 min), which was bath applied before and during the application of DTT, effectively blocked (104.48 ± 4.29%, n = 4) the DTT-mediated increase in NMDAR-fEPSP in aged animals (Fig. 8C). In contrast, the membrane-permeable PKC inhibitor, Bis-I (500 nm, 45 min) (Knapp and Klann, 2002) failed to block the DTT-mediated increase in the NMDAR-fEPSP (142.58 ± 13.06%, n = 6) (Fig. 8D). An ANOVA comparison of the effect of DTT in the presence and absence of the kinase inhibitors, indicated a significant effect of kinase inhibitors on the DTT effect in aged animals (F(4,33) = 5.85, p < 0.01). Post hoc comparisons indicated that the DTT-mediated increase in the NMDAR response was blocked by H-7, myr-AIP, and KN-62 but not Bis-I (Fig. 8E). Finally, t tests indicated no effect of 0.01% DMSO (96.38 ± 7.64, n = 5) or kinase inhibition per se (H-7: 103.01 ± 4.21%, n = 7; KN-62: 96.23 ± 8.3%, n = 5; myr-AIP: 100.52 ± 4.42%, n = 4; Bis-I: 110.72 ± 11.44%, n = 6) on the baseline NMDAR-fEPSP slope in aged animals.
Figure 8.

CaMKII involvement in the DTT-mediated enhancement of NMDAR synaptic responses in aged animals. A–D, Time course of the change in the normalized NMDAR-fEPSP slope in the aged animals that were incubated with the broad spectrum Ser/Thr kinase inhibitor H-7 dihydrochloride (10 μm, n = 7) (A), the CaMK inhibitor KN-62 (10 μm, n = 5) (B), the specific CaMKII inhibitor myr-AIP (5 μm, n = 4) (C), or the PKC inhibitor Bis-I (500 nm, n = 6) (D) before and during DTT application. E, Quantification of the mean percentage change in the NMDAR-fEPSP slope for aged animals under DTT alone (filled bar), and DTT applied in the presence of H-7, KN-62, myr-AIP, and Bis-I (gray bars). An asterisk indicates a significant difference (p < 0.05) between the increases observed in presence of DTT alone and DTT applied in the presence of H-7, KN-62, myr-AIP, and Bis-I. A pound sign indicates a significant (p < 0.05) increase in the response relative to baseline level of 100%, following DTT application.
The activity of Ser/Thr phosphatases, such as calcineurin (CaN) and protein phosphatase 1 (PP1), is thought to contribute to altered synaptic plasticity during aging (Foster et al., 2001). Furthermore, phosphatase activity decreases NMDAR function (Lieberman and Mody, 1994; Wang et al., 1994). To test whether DTT effects were mediated by CaN, the CaN inhibitor FK-506 (10 μm) (Norris et al., 2010) was bath applied 45 min before and during the application of DTT on aged hippocampal slices. Application of FK-506 per se did not affect the NMDAR-fEPSP slope (109.61 ± 9.16%, n = 5). Importantly, FK-506 failed to block the DTT-mediated increase in the NMDAR response in aged animals such that the NMDAR-fEPSP slope increased to 148.61 ± 16.42% (n = 5) (Fig. 9A,C).
Figure 9.

Calcineurin and PP1 are not involved in the DTT-mediated enhancement of NMDAR synaptic responses in aged animals. A, Time course of the increase in the NMDAR-fEPSP slope in slices from aged animals that were incubated with FK-506 (10 μm), 45 min before and during the application of DTT (n = 5). B, The NMDAR-fEPSP slope exhibited a modest increase (121.46 ± 9.19%) following a 30 min incubation with OA (1 μm) (n = 5). C, Following stabilization of the response in OA, the baseline was recalculated. The figure illustrates the time course for the increase in the renormalized NMDAR-fEPSP slope following application of DTT (n = 5). D, Quantification of the mean percentage change in the NMDAR-fEPSP slope for aged animals under DTT alone (filled bar) and in the presence of FK-506 + DTT, OA + DTT, and OA alone (gray bars).
To examine the role of PP1, the PP1 inhibitor OA (1 μm, 30 min) (Schnabel et al., 2001) was applied before and during application of DTT to aged hippocampal slices. Application of OA significantly increased the NMDAR-fEPSP slope during the baseline recording period (121.46 ± 9.19%, n = 5, p < 0.05). Therefore, a new stable baseline was recorded before the application of DTT. Relative to the new baseline, DTT increased the NMDAR-fEPSP slope (137.89 ± 3.99%, n = 5) in the presence of OA (Fig. 9B,C). An ANOVA comparing the effect of DTT in the presence and absence of the phosphatase inhibitors, indicated no significant difference (F(2,15) = 1.37, p > 0.05) (Fig. 9D).
To determine whether DTT was directly influencing CaMKII activity, cytosolic extracts from CA1 region of the hippocampus from aged and young animals were assayed for CaMKII activity by examining the phosphorylation of the synthetic peptide, syntide-2, in the presence and absence of DTT. Relative to baseline control levels, cytosolic CaMKII activity was significantly enhanced (p < 0.05) in the presence of 0.7 mm (113.11 ± 3.47%, n = 3) and 1.4 mm (120.46 ± 3.14%, n = 3) DTT in the aged CA1 cytosolic extracts (Fig. 10A). Higher levels of DTT (2.8 mm) resulted in a decrease in CaMKII activity, presumably due to denaturation of the enzyme. In contrast to the effect observed in aged animals, DTT had either no effect or decreased CaMKII activity in CA1 cytosolic extracts from young animals (Fig. 10B). In both age groups, the CaMKII activity was inhibited (p < 0.05) by the addition of calcium chelator EGTA (2 mm) and a CaMKII-specific peptide inhibitor myr-AIP (10 μm) (aged: 5.36 ± 2.85%, n = 3; young: 19.99 ± 9.01%, n = 3).
Figure 10.

DTT enhances CaMKII activity in aged but not in young hippocampal CA1 cytosolic extracts. A, CaMKII activity measured from the hippocampal CA1 cytosolic extracts of aged F344 rats. CaMKII activity is represented as percentage of control activity (black bars) in the presence of exogenous calmodulin. CaMKII activity was significantly enhanced in the presence of 0.7 mm and 1.4 mm DTT (gray bars) and was blocked by the addition of EGTA (2 mm) + myr-AIP (10 μm) (white bars). B, Addition of 0.7 mm and 1.4 mm DTT did not increase CaMKII activity in hippocampal CA1 cytosolic extracts of young F344 rats. C, Addition of 0.7 and 1.4 mm DTT decreased the activity of purified CaMKII. An asterisk indicates a significant difference (p < 0.05) from respective controls. Plus and minus represent the presence and absence (respectively) of the indicated component in the reaction mix.
One possibility is that DTT was acting on the CaMKII activity regulator calmodulin (CaM). In this case, the effect of DTT on CaMKII activity may have been reduced by the addition of exogenous and unoxidized CaM. To test this idea the assay was repeated in the absence of exogenously added CaM. The CaMKII activity from aged CA1 cytosolic extract in the presence of 0.7 mm DTT, in the absence of exogenous CaM (112.11 ± 3.91%, n = 3), was not enhanced beyond that observed following addition of exogenous CaM, suggesting that DTT effects were not mediated by reducing cysteines on CaM. Finally, addition of DTT to purified CaMKII (CycLex) decreased CaMKII activity (p < 0.05) (0.7 mm DTT: 86.62 ± 6.04%, n = 3; 1.4 mm DTT: 70.72 ± 18.58%, n = 3) (Fig. 10C), indicating that the DTT effects were specific for CaMKII present in the aged CA1 cytosolic extracts.
Discussion
Intracellular redox state contributes to “NMDAR hypofunction” in the aged hippocampus
Previous work demonstrates that NMDAR responses are reduced in the area CA1 of the hippocampus of aged rat brain (Barnes et al., 1997; Billard and Rouaud, 2007), possibly due to altered receptor function (Cady et al., 2001). We confirmed an age-related decrease in the NMDAR response and demonstrate age-dependent effects of redox modulators on the NMDAR response. Our results reveal a role for intracellular redox state in contributing to the age-related decline in NMDAR-mediated synaptic responses and impaired induction of LTP in area CA1 of the hippocampus. The effects of DTT application were specific for aged animals and not observed for young animals. Furthermore, the DTT-mediated increase in the NMDAR response was not associated with a change in the PFV, was not mimicked for the AMPAR-mediated response (Fig. 5D) and was not blocked by nifedipine, suggesting the effect was not due to a general influence on cell excitability. Finally, the manner in which DTT affects the NMDAR response was specific to the intracellular redox state and the activity of CaMKII. Thus, we establish a mechanism that links general theories of aging (i.e., oxidative stress and redox state) with biological markers of age-related cognitive decline, reduced NMDAR responses, and impaired LTP.
The redox state of the extracellular cysteine residues of the NMDARs have been implicated in regulating the NMDAR function in cell cultures and neonatal animals (Aizenman et al., 1989, 1990; Bernard et al., 1997; Choi and Lipton, 2000; Choi et al., 2001). In contrast, our results are consistent with recent work in hippocampal cell cultures indicating a decrease in the intracellular redox ratio during aging, due in part to a deficit in intracellular GSH (Parihar et al., 2008). Specifically, we observed that intracellular but not extracellular application of l-GSH enhanced the NMDAR response in the aged neurons, indicating that the age-related decrease in NMDAR function is due to a shift in redox state of the intracellular compartment.
CaMKII-dependent mechanism for altered NMDAR responses in aged animals
The DTT-mediated enhancement of NMDAR responses was specific to CaMKII activity. CaMKII inhibitors, myr-AIP and KN-62, blocked the DTT-mediated enhancement of the NMDAR response in aged animals. The DTT effects were not blocked by inhibition of PKC or phosphatases—CaN and PP1. The results point to CaMKII as a critical link between the intracellular redox state and the decrease in the NMDAR response. The role of CaMKII was confirmed by enzyme activity assays, which established that DTT increased CaMKII activity only in CA1 cytosolic extracts of aged animals. DTT did not increase CaMKII activity in samples from young animals or in purified CaMKII.
Oxidation of methionine residues on CaM has been reported to decrease the ability of CaM to activate CaMKII (Robison et al., 2007). Our results are not likely due to CaM methionine oxidation since DTT selectively reduces cysteine residues (Ciorba et al., 1997; Cai and Sesti, 2009; Long et al., 2009). In addition, DTT had equivalent effects in activating CaMKII, regardless of whether exogenous CaM was added to the reaction. The results indicate that oxidation of CaMKII, rather than CaM, underlies the reduction in kinase activity and are consistent with a recent report demonstrating that oxidative stress induced by ischemia results in disulfide linkages on the cysteine residues of CaMKII, which decrease kinase activity (Shetty et al., 2008). While the data provide a link between age-related changes in intracellular redox state, CaMKII activity, and NMDAR function, the exact mechanism through which CaMKII regulates the NMDAR response remains to be determined. In addition to regulating phosphorylation state of proteins, including glutamate receptors, synaptic CaMKII participates in protein–protein interactions with several proteins localized to the dendritic spine, which could ultimately alter NMDAR location and function (Lisman et al., 2002; Robison et al., 2005). Indeed, reduced CaMKII activity is associated with a specific decrease in synaptic NMDARs and decreased LTP (Gardoni et al., 2009).
In addition to a role for CaMKII, we observed that PP1 inhibition resulted in a modest increase in the NMDAR-fEPSP in aged hippocampal neurons. Age differences in the NMDAR response, which depend on kinase/phosphatase activity, are reminiscent of the age-dependent effects of kinase and phosphatase inhibitors on the rapid component of synaptic transmission mediated by AMPARs (Norris et al., 1998b; Hsu et al., 2002; Foster, 2007) and suggest that PP1 activity contributes to a reduction in AMPAR and NMDAR components of synaptic transmission (Foster et al., 2001; Morishita et al., 2005), a characteristic specific to senescent CA1 synapses (Rosenzweig and Barnes, 2003). Our results indicate that a shift in the intracellular redox state during aging may cause or magnify the imbalance in the kinase/phosphatase activity, favoring phosphatases (Foster, 2007).
Finally, NMDAR function is critical to the induction of LTP, and we observed that DTT improved LTP in hippocampal area CA1 in the aged animals. The interaction of NMDARs with CaMKII has been proposed as a model of memory (Lisman et al., 2002) and recent work indicates that disruption of the interaction between CaMKII and NMDAR impairs the induction of LTP and spatial learning (Zhou et al., 2007). We have provided evidence to indicate that an age-related shift in redox state is a biological mechanism that can progressively inhibit NMDAR function in the hippocampus during senescence. Together, the results suggest that age-related changes in the redox state contributes to a decline in CaMKII activity, which ultimately leads to a decline in the NMDAR response. The outcome of such senescent mechanisms is an alteration in the synaptic plasticity at the CA3–CA1 synapses that contributes to the age-related cognitive decline.
Footnotes
This work was supported by the Evelyn F. McKnight Brain Research Foundation, National Institutes of Health Grants AG014979 and MH059891 to T.C.F., and a University of Florida Alumni fellowship to K.B.
References
- Abele R, Lampinen M, Keinänen K, Madden DR. Disulfide bonding and cysteine accessibility in the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor subunit GluRD. Implications for redox modulation of glutamate receptors. J Biol Chem. 1998;273:25132–25138. doi: 10.1074/jbc.273.39.25132. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Lipton SA, Loring RH. Selective modulation of NMDA responses by reduction and oxidation. Neuron. 1989;2:1257–1263. doi: 10.1016/0896-6273(89)90310-3. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Hartnett KA, Reynolds IJ. Oxygen free radicals regulate NMDA receptor function via a redox modulatory site. Neuron. 1990;5:841–846. doi: 10.1016/0896-6273(90)90343-e. [DOI] [PubMed] [Google Scholar]
- Barnes CA, Rao G, Shen J. Age-related decrease in the N-methyl-D-aspartateR-mediated excitatory postsynaptic potential in hippocampal region CA1. Neurobiol Aging. 1997;18:445–452. doi: 10.1016/s0197-4580(97)00044-4. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Aniksztejn L, Bregestovski P. Protein kinase C modulation of NMDA currents: an important link for LTP induction. Trends Neurosci. 1992;15:333–339. doi: 10.1016/0166-2236(92)90049-e. [DOI] [PubMed] [Google Scholar]
- Bernard CL, Hirsch JC, Khazipov R, Ben-Ari Y, Gozlan H. Redox modulation of synaptic responses and plasticity in rat CA1 hippocampal neurons. Exp Brain Res. 1997;113:343–352. doi: 10.1007/BF02450332. [DOI] [PubMed] [Google Scholar]
- Billard JM, Rouaud E. Deficit of NMDA receptor activation in CA1 hippocampal area of aged rats is rescued by D-cycloserine. Eur J Neurosci. 2007;25:2260–2268. doi: 10.1111/j.1460-9568.2007.05488.x. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Boric K, Muñoz P, Gallagher M, Kirkwood A. Potential adaptive function for altered long-term potentiation mechanisms in aging hippocampus. J Neurosci. 2008;28:8034–8039. doi: 10.1523/JNEUROSCI.2036-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady C, Evans MS, Brewer GJ. Age-related differences in NMDA responses in cultured rat hippocampal neurons. Brain Res. 2001;921:1–11. doi: 10.1016/s0006-8993(01)03063-3. [DOI] [PubMed] [Google Scholar]
- Cai SQ, Sesti F. Oxidation of a potassium channel causes progressive sensory function loss during aging. Nat Neurosci. 2009;12:611–617. doi: 10.1038/nn.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YB, Lipton SA. Redox modulation of the NMDA receptor. Cell Mol Life Sci. 2000;57:1535–1541. doi: 10.1007/PL00000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chen HV, Lipton SA. Three pairs of cysteine residues mediate both redox and Zn2+ modulation of the nmda receptor. J Neurosci. 2001;21:392–400. doi: 10.1523/JNEUROSCI.21-02-00392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciorba MA, Heinemann SH, Weissbach H, Brot N, Hoshi T. Modulation of potassium channel function by methionine oxidation and reduction. Proc Natl Acad Sci U S A. 1997;94:9932–9937. doi: 10.1073/pnas.94.18.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC. Involvement of hippocampal synaptic plasticity in age-related memory decline. Brain Res Brain Res Rev. 1999;30:236–249. doi: 10.1016/s0165-0173(99)00017-x. [DOI] [PubMed] [Google Scholar]
- Foster TC. Regulation of synaptic plasticity in memory and memory decline with aging. Prog Brain Res. 2002;138:283–303. doi: 10.1016/S0079-6123(02)38083-X. [DOI] [PubMed] [Google Scholar]
- Foster TC. Biological markers of age-related memory deficits: treatment of senescent physiology. CNS Drugs. 2006;20:153–166. doi: 10.2165/00023210-200620020-00006. [DOI] [PubMed] [Google Scholar]
- Foster TC. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell. 2007;6:319–325. doi: 10.1111/j.1474-9726.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- Foster TC, Sharrow KM, Masse JR, Norris CM, Kumar A. Calcineurin links Ca2+ dysregulation with brain aging. J Neurosci. 2001;21:4066–4073. doi: 10.1523/JNEUROSCI.21-11-04066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Mauceri D, Malinverno M, Polli F, Costa C, Tozzi A, Siliquini S, Picconi B, Cattabeni F, Calabresi P, Di Luca M. Decreased NR2B subunit synaptic levels cause impaired long-term potentiation but not long-term depression. J Neurosci. 2009;29:669–677. doi: 10.1523/JNEUROSCI.3921-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozlan H, Khazipov R, Diabira D, Ben-Ari Y. In CA1 hippocampal neurons, the redox state of NMDA receptors determines LTP expressed by NMDA but not by AMPA receptors. J Neurophysiol. 1995;73:2612–2617. doi: 10.1152/jn.1995.73.6.2612. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- Hsu KS, Huang CC, Liang YC, Wu HM, Chen YL, Lo SW, Ho WC. Alterations in the balance of protein kinase and phosphatase activities and age-related impairments of synaptic transmission and long-term potentiation. Hippocampus. 2002;12:787–802. doi: 10.1002/hipo.10032. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J Biol Chem. 2000;275:24136–24145. doi: 10.1074/jbc.M002043200. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Potentiation of hippocampal synaptic transmission by superoxide requires the oxidative activation of protein kinase C. J Neurosci. 2002;22:674–683. doi: 10.1523/JNEUROSCI.22-03-00674.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BS, Sun MK, Zhang L, Takahashi S, Ma W, Vinade L, Kulkarni AB, Brady RO, Pant HC. Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc Natl Acad Sci U S A. 2001;98:12742–12747. doi: 10.1073/pnas.211428098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca(2+)-dependent phosphatase. Nature. 1994;369:235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, Bankston LA. Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002;25:474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Long LH, Liu J, Liu RL, Wang F, Hu ZL, Xie N, Fu H, Chen JG. Differential effects of methionine and cysteine oxidation on [Ca2+] i in cultured hippocampal neurons. Cell Mol Neurobiol. 2009;29:7–15. doi: 10.1007/s10571-008-9289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson KR, Bai L, Zhao X. The effects of aging on different C-terminal splice forms of the zeta1(NR1) subunit of the N-methyl-d-aspartate receptor in mice. Brain Res Mol Brain Res. 2005;135:141–149. doi: 10.1016/j.molbrainres.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Magnusson KR, Kresge D, Supon J. Differential effects of aging on NMDA receptors in the intermediate versus the dorsal hippocampus. Neurobiol Aging. 2006;27:324–333. doi: 10.1016/j.neurobiolaging.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Mazor D, Golan E, Philip V, Katz M, Jafe A, Ben-Zvi Z, Meyerstein N. Red blood cell permeability to thiol compounds following oxidative stress. Eur J Haematol. 1996;57:241–246. doi: 10.1111/j.1600-0609.1996.tb01370.x. [DOI] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka RC. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC. Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci. 1998a;18:3171–3179. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC. Alterations in the balance of protein kinase/phosphatase activities parallel reduced synaptic strength during aging. J Neurophysiol. 1998b;80:1567–1570. doi: 10.1152/jn.1998.80.3.1567. [DOI] [PubMed] [Google Scholar]
- Norris CM, Blalock EM, Chen KC, Porter NM, Thibault O, Kraner SD, Landfield PW. Hippocampal ‘zipper’ slice studies reveal a necessary role for calcineurin in the increased activity of L-type Ca(2+) channels with aging. Neurobiol Aging. 2010;31:328–338. doi: 10.1016/j.neurobiolaging.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar MS, Kunz EA, Brewer GJ. Age-related decreases in NAD(P)H and glutathione cause redox declines before ATP loss during glutamate treatment of hippocampal neurons. J Neurosci Res. 2008;86:2339–2352. doi: 10.1002/jnr.21679. [DOI] [PubMed] [Google Scholar]
- Poon HF, Calabrese V, Calvani M, Butterfield DA. Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by L-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxid Redox Signal. 2006;8:381–394. doi: 10.1089/ars.2006.8.381. [DOI] [PubMed] [Google Scholar]
- Raynaud F, Evain-Brion D, Gerbaud P, Marciano D, Gorin I, Liapi C, Anderson WB. Oxidative modulation of cyclic AMP-dependent protein kinase in human fibroblasts: possible role in psoriasis. Free Radic Biol Med. 1997;22:623–632. doi: 10.1016/s0891-5849(96)00353-x. [DOI] [PubMed] [Google Scholar]
- Robison AJ, Bass MA, Jiao Y, MacMillan LB, Carmody LC, Bartlett RK, Colbran RJ. Multivalent interactions of calcium/calmodulin-dependent protein kinase II with the postsynaptic density proteins NR2B, densin-180, and alpha-actinin-2. J Biol Chem. 2005;280:35329–35336. doi: 10.1074/jbc.M502191200. [DOI] [PubMed] [Google Scholar]
- Robison AJ, Winder DG, Colbran RJ, Bartlett RK. Oxidation of calmodulin alters activation and regulation of CaMKII. Biochem Biophys Res Commun. 2007;356:97–101. doi: 10.1016/j.bbrc.2007.02.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Barnes CA. Impact of aging on hippocampal function: plasticity, network dynamics, and cognition. Prog Neurobiol. 2003;69:143–179. doi: 10.1016/s0301-0082(02)00126-0. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL. Protein phosphatase inhibitors facilitate DHPG-induced LTD in the CA1 region of the hippocampus. Br J Pharmacol. 2001;132:1095–1101. doi: 10.1038/sj.bjp.0703905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar S, Teyler TJ, Robbins N. Aging differentially alters forms of long-term potentiation in rat hippocampal area CA1. J Neurophysiol. 1998;79:334–341. doi: 10.1152/jn.1998.79.1.334. [DOI] [PubMed] [Google Scholar]
- Shetty PK, Huang FL, Huang KP. Ischemia-elicited oxidative modulation of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2008;283:5389–5401. doi: 10.1074/jbc.M708479200. [DOI] [PubMed] [Google Scholar]
- Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin EJ, Suh SK, Lim YK, Jhoo WK, Hjelle OP, Ottersen OP, Shin CY, Ko KH, Kim WK, Kim DS, Chun W, Ali S, Kim HC. Ascorbate attenuates trimethyltin-induced oxidative burden and neuronal degeneration in the rat hippocampus by maintaining glutathione homeostasis. Neuroscience. 2005;133:715–727. doi: 10.1016/j.neuroscience.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Susankova K, Tousova K, Vyklicky L, Teisinger J, Vlachova V. Reducing and oxidizing agents sensitize heat-activated vanilloid receptor (TRPV1) current. Mol Pharmacol. 2006;70:383–394. doi: 10.1124/mol.106.023069. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Wang LY, Orser BA, Brautigan DL, MacDonald JF. Regulation of NMDA receptors in cultured hippocampal neurons by protein phosphatases 1 and 2A. Nature. 1994;369:230–232. doi: 10.1038/369230a0. [DOI] [PubMed] [Google Scholar]
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Nishiyama N, Shuto M, Sugiyama C, Kawada K, Seko K, Nagashima R, Ogita K. In vivo depletion of endogenous glutathione facilitates trimethyltin-induced neuronal damage in the dentate gyrus of mice by enhancing oxidative stress. Neurochem Int. 2008;52:761–769. doi: 10.1016/j.neuint.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Takahashi E, Li W, Halt A, Wiltgen B, Ehninger D, Li GD, Hell JW, Kennedy MB, Silva AJ. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci. 2007;27:13843–13853. doi: 10.1523/JNEUROSCI.4486-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou CG, Agar NS, Jone GL. Oxidative insult in sheep red blood cells induced by T-butyl hydroperoxide: the roles of glutathione and glutathione peroxidase. Free Radic Res. 2001;34:45–56. doi: 10.1080/10715760100300051. [DOI] [PubMed] [Google Scholar]