

Abstract
PKCε overexpression in FVB/N transgenic mice sensitized skin to ultraviolet radiation (UVR)-induced development of squamous cell carcinomas (SCC) (Wheeler et al., 2004; Wheeler et al., 2005) and suppressed formation of sunburn cells, which are DNA-damaged keratinocytes undergoing apoptosis (Wheeler et al., 2004). Here, we elucidated the mechanisms associated with inhibition of UVR-induced appearance of sunburn cells in PKCε transgenic mice. We found that the inhibition of UVR-induced sunburn cell formation in PKCε transgenic mice may be the result of the inhibition of the expression of Fas, Fas ligand (Fas-L) and the mammalian death adaptor protein termed Fas-associated with death domain (FADD). The adaptor protein FADD is the key component of the death inducing signaling complex of both Fas and tumor necrosis factor receptor 1 (TNF-R1). A decreased expression of epidermal FADD was observed after a single UVR exposure. However, a complete loss of FADD expression was found after four (Monday, Wednesday, Friday and Monday) repeated UVR exposures. FADD transmits apoptotic signals from death receptors to the downstream initiator caspase-8 and connects to the mitochondrial intrinsic apoptotic signal transduction pathway by the cleavage of Bid, a Bcl-2 family member. PKCε-mediated loss of FADD expression inhibited UVR signals to the activation of both extrinsic and intrinsic apoptotic pathways.
Keywords: UVB, PKC, Transgenic mice, Photocarcinogenesis, Apoptosis, FADD
INTRODUCTION
Skin cancer is the most common malignancy in the United States (American Cancer Society, 2008). It is well known that chronic exposure to ultraviolet radiation (UVR) is the major etiological factor in epidermal carcinogenesis. The UV spectrum, a part of the electromagnetic spectrum that lies between visible and X-rays, is divided conventionally into three major categories: UVA (315–400 nm), UVB (280–315 nm) and UVC (190–280 nm). Since stratospheric ozone absorbs most of the radiation below 310nm, UVA and UVB are the most prominent and ubiquitous carcinogenic wavelengths in our natural environment. Squamous cell carcinoma (SCC) and basal cell carcinoma (BCC) are he most common non-melanoma forms of skin cancer (NMSC) (de Gruijl, 1999; Green et al., 1999; Wang et al., 2007; Mukhtar et al., 1999; Morison, 1997). SCC, the second most common skin cancer after BCC, affects more than 200,000 Americans each year. SCC, unlike BCC, invades nearby tissues and metastasizes first to regional lymph nodes and subsequently to distant sites such as the lung and brain (Moller et al., 1979). Although, mortality rate from NMSC is low, skin cancer patients experience mortality from other non-cutaneous cancers (American Cancer Society, 2008). Knowledge about regulatory molecules involved in UVR-induced development of SCC are essential for the rational design of agents for prevention and treatment of SCC. We have reported that protein kinase Cε PKCε, a Ca2+-independent, phospholipid-dependent serine/theronine kinase sensitizes skin to the development of SCC using either chemical tumor promotion by the DMBA-TPA protocol (Jansen et al., 2001; Reddig et al., 2000) or by repeated exposure to UVR (Wheeler et al., 2004; Wheeler et al., 2005). Since the etiology (UVR), pathology (poorly differentiated SCC), and the molecular signatures (p53 mutation) of SCC in PKCε transgenic mice are similar to human SCC (Jansen et al., 2001; Wheeler et al., 2004; Wheeler et al., 2005), PKCε overexpressing mice provide a useful model for investigating the molecular mechanisms and the prevention strategies of SCC.
During studies to find clues about the mechanisms by which PKCε sensitizes skin to UVR carcinogenesis, we found that PKCε overexpression in transgenic mice, as compared with their wild-type littermates, reduced the appearance of sunburn cells. Sunburn cells are DNA-damaged keratinocytes undergoing apoptosis (Hill et al., 1999; Lu et al., 2007; Ziegler et al., 1994). UVR is a complete carcinogen, which both initiates and promotes carcinogenesis. UVR initiates photocarcinogenesis by directly damaging DNA (Berton et al., 1997; de Gruijl et al., 2001; Kunisada et al., 2007), which results in the induction of p53 protein (Berton et at., 1997; Wheeler et al., 2005; Ziegler et al., 1994). The p53 protein transactivates p21WAF1/CIP1 inducing cell cycle arrest to allow DNA repair. If the damage is not repaired, p53-dependent apoptosis is triggered to erase the DNA damage. The p53-dependent apoptosis of UV-damaged normal cells (sunburn cells) is prevented due to p53 mutation. Thus, these mutated cells can clonally expand to form SCC after subsequent UVR exposures. In this context, it is notable that mice deficient in p53 have reduced sunburn cell formation and increased susceptibility to UVR-induced skin carcinogenesis (Li et al., 1998; Ziegler et al., 1994). These findings indicate that apoptosis inhibition may be an important component of the mechanism of UVR-induced skin carcinogenesis.
The Fas pathway is important in eliminating DNA-damaged cells both by augmenting p53-mediated apoptosis (Muller et al., 1998) and by inducing apoptosis when p53 has been mutated (Rossi and Gaidano, 2003). In Fas-mediated apoptosis, the homotrimeric Fas ligand binds to the Fas receptor, inducing it to trimerize within the membrane (Rossi and Gaidano, 2003) (Figure 1). UVR is also able to activate the Fas receptor independently of its ligand by inducing aggregation of the receptor, possibly through disruption of the plasma membrane (Kulms et al., 1998). After the Fas receptor trimerizes, the intracellular death domain of the receptor binds to Fas-associated with death domain (FADD), forming the death-inducing signaling complex (DISC) (Chinnaiyan et al., 1995; Rossi and Gaidano, 2003). FADD then induces the autocatalytic cleavage of initiator caspases 8 or 10, followed by the cleavage of the effector caspases. The executioner caspases cause the cleavage of structural proteins, such as poly(ADP-ribose)polymerases (PARP), leading to membrane blebbing, degradation of nuclear proteins leading to nuclear collapse, fragmentation of nuclear DNA, and finally cell death (Huppertz et al., 1999). FADD is a common adaptor protein in both Fas and TNFR-mediated apoptosis (Gaur and Aggarwal, 2003; Sheikh and Huang, 2003; Sheikh and Huang, 2003; Thorburn, 2004).
Figure 1. PKCε overexpression suppresses UVR-induced apoptosis.
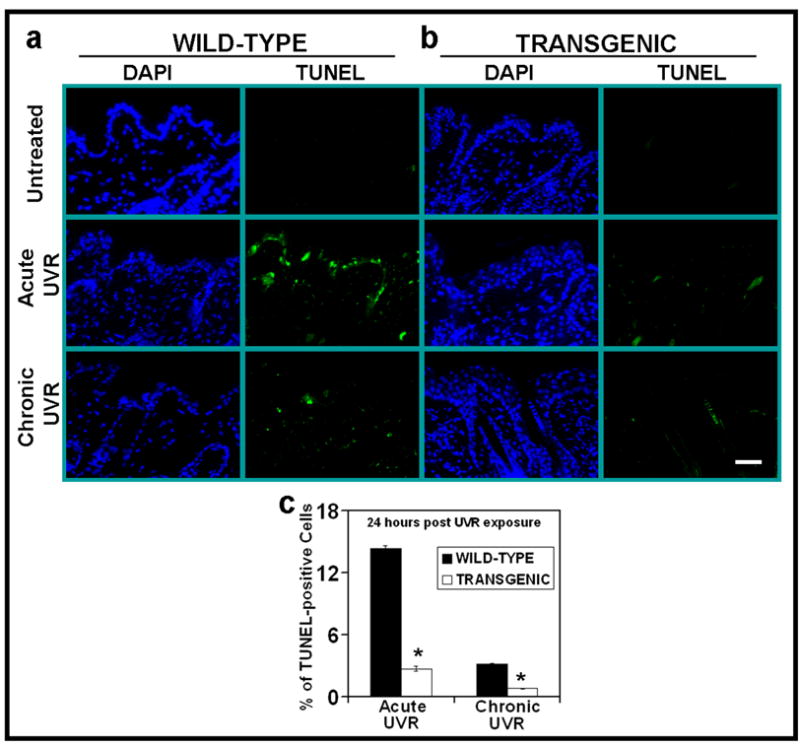
PKCε transgenic mice (line 215) and their wild-type littermates were exposed to acute (single) UVR (4 kJ/m2) or chronic (repeated) UVR (2kJ/m2) four times (Monday, Wednesday, Friday, and Monday). The mice were sacrificed at 24 hours after last UVR exposure. Skin specimens were fixed in 10% neutral buffered formalin and embedded in paraffin. Skin sections of 4-μm thickness were cut and analyzed for the presence of apoptosis cells by TUNEL assay. (a) DAP1 counter stain and TUNNEL assay of WT mice at 0 and 24 hrs post acute or chronic UV exposures (b). DAP1 counter stain and TUNNEL assay of PKCε transgenic mice c: Quantitaion of TUNEL-positive cells 24 hours after acute or chronic UVR exposures. Four mice were sacrificed at each time point, and eight microscopic fields in each mouse were used to calculate average number of TUNEL-positive cells. *, p<0.005. Bars= 50 μm.
In this communication, we determined the effects of PKCε overexpression in transgenic mice on the UVR-induced Fas- and TNFR-mediated apoptotic pathways. We present here for the first time that the inhibition of UVR-induced sunburn cell formation in PKCε transgenic mice may be the result of the inhibition of the expression of the components of Fas/Fas-L (Fas/Fas-L and FADD) and TNFα/TNFR1 (TNFα/TNFR1, FADD)-mediated apoptotic pathways.
RESULTS
PKCε overexpression in the epidermis of FVB/N Transgenic mice suppresses UVR-induced apoptosis
We have shown that UVR exposure in PKCε transgenic mice induced cutaneous damage and the extent of photodamage was proportional to the level of expression of PKCε in transgenic mouse lines (Wheeler et al., 2004; Wheeler et al., 2005). The PKCε transgenic mouse line 224, when exposed to UVR (2kJ/m2 three times weekly) elicited increased SCC multiplicity by 3-fold and decreased tumor latency by 12 weeks (Wheeler et al., 2004). To obtain clues about the mechanisms by which PKCε sensitizes skin to the development of SCC, we found PKCε overexpression suppressed the UVR-induced number of DNA-damaged keratinocytes (sunburn cells) (Wheeler et al., 2004). In these experiments, PKCε transgenic mice and their wild-type littermates were exposed to either single (4kJ/m2) or chronic (2kJ/m2 four times) UVR. Sunburn cells were identified in hemotoxylin and eoison-stained histologic skin sections. The UVR-induced formation of sunburn cells in PKCε transgenic mice was significantly lower than their wild-type littermates at all time points after either single or chronic UVR exposures (Wheeler et al., 2004). In this study, we further determined whether PKCε transgenic mice showed altered susceptibility to apoptosis by measuring TUNEL-positive cells in UVR-exposed PKCε transgenic mouse skin and their wild-type littermates (Figure 1). TUNEL positive cells in PKCε transgenic mice were significantly lower the than wild-type littermates (*p<0.005) (Figure 1a–c) after either acute or chronic UVR treatments.
PKCε overexpression in the epidermis of FVB/N Transgenic mice stimulates UVR-induced TNFR1 protein expression
Tumor necrosis factor (TNF) is a key mediator of inflammation, immunity, and apoptosis (Tracey and Cerami, 1993). Although TNF can signal through two receptors, TNFR1 and TNFR2, the majority of TNF-mediated biological events are mediated via TNFR1 signaling (Chen and Goeddel, 2002; Locksley et al., 2001; MacEwan, 2002; Tartaglia and Goeddel, 1992; Vandenabeele et al., 1995). Furthermore, TNFR1 plays an important role in the induction of several cancers (Arnott et al., 2004; Figueras et al., 2005; Houtenbos et al., 2004; Lind et al., 2004; Wu et al., 2003). TNFα and TNFR1 have been linked to UVR carcinogenesis (Moore et al., 1999; Suganuma et al., 1999; Starcher, 2000; Wheeler et al., 2004; Wheeler et al., 2005; Zhuang et al., 1999). We have previously reported that PKCε transgenic mice were more sensitive than their wild-type littermates to the induction of epidermal TNFα when exposed to UVR (Wheeler et al., 2004). To determine how UVR-induced TNFα may impart sensitivity to the development of SCC in PKCε transgenic mice (Wheeler et al., 2004), we investigated the effects of UVR on the TNFR1-mediated apoptotic signal transduction pathway. In these experiments, mice were exposed to either a single (4kJ/m2) or repeated (2kJ/m2, four times) UVR treatment. TNFR1 expression level in the epidermal extract was analyzed by Western Blot analysis. The results (Figure 2a, c) indicated a biphasic response in the wild type littermates after a single UVR exposure. UVR induced a substantial increase in TNFR1 expression at 1h followed by a decline to basal levels at 12h in wild-type littermates (Figure 2a, c). This was followed by a second phase of TNFR1 expression, which was apparent at 18h and sustained through at least 96h. Similar increases in TNFR1 expression levels were found in PKCε transgenic mice but the expression level of TNFR1 was constitutively activated and the increase in expression was more pronounced at 18 hr and 24 hr post UVR exposure compared to wild-type littermates (Figure 2a, c). As shown in Figure 2b, d, PKCε overexpression also led to the induction of TNFR1 at 1h and 3h after chronic UVR exposures. The effects of UVR on the overexpression of TNFR1 in PKCε transgenic mice were specific because PKCδ transgenic mice, which overexpress PKCδ protein (8-fold) (Reddig et al., 1999), did not affect TNFR1 expression after UVR exposure (Figure 2b).
Figure 2. PKCε overexpression sensitizes skin to UVR-induced TNF-R1 expression.
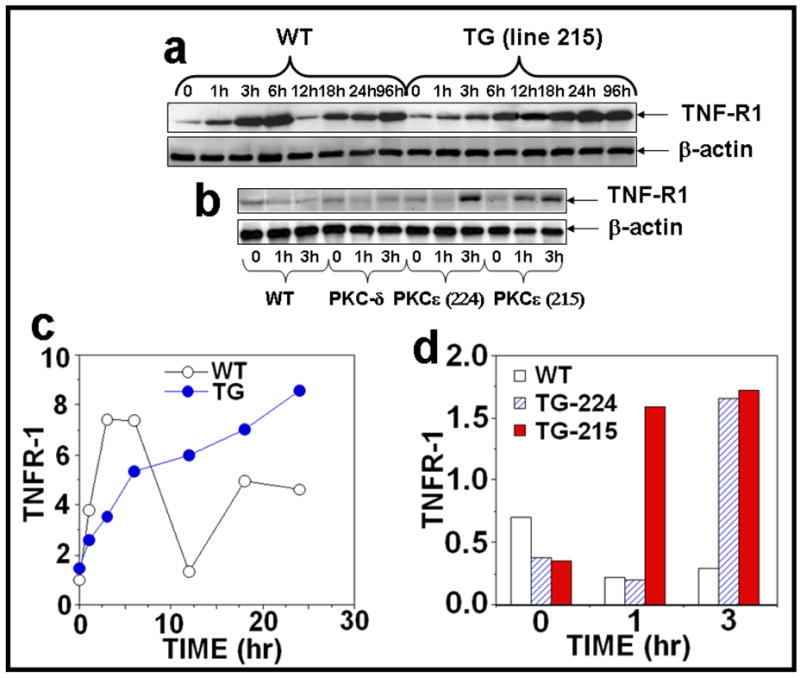
(a): PKCε transgenic mice (line 215) and wild-type littermates (4 mice per group) were exposed to single UVR (4 kJ/m2). The mice were sacrificed at 1, 3, 6, 12, 18, 24, and 96 hours after acute UVR exposure. (b): In a parallel experiment, PKCε (lines 215 and 224), PKCδ transgenic mice and wild-type littermates were exposed to UVR (2kJ/m2) four times (Monday, Wednesday, Friday, and Monday). The mice were sacrificed at 1 and 3 hours post fourth treatment of UVR. Mouse epidermal exrtracts were prepared and TNFR1 and β-actin were assessed by immunoblot analysis as described previously. (c,d): The quantification of proteins (normalized to β-actin).
PKCε overexpression in transgenic mice suppresses the signaling components of the UVR-induced Fas/Fas-L apoptotic pathway
We determined whether the inhibition of UVR-induced sunburn cell formation in PKCε transgenic mice was the result of inhibition of Fas/Fas-L mediated extrinsic pathway of apoptotic signaling. Fas and Fas-L are complimentary receptor-ligand proteins that induce apoptosis in many cell types (Redondo et al., 2002). The Fas/Fas-L pathway has been shown to play an important role in the elimination of UVR-induced DNA-damaged cells (Hill et al., 1999; Hill et al., 1999). In these experiments, PKCε overexpressing transgenic mice and wild-type littermates were either exposed to either a single dose (4kJ/m2) or repeated UVR doses (four times, 2kJ/m2, Monday, Wednesday, Friday, Monday). The expression of Fas/Fas-L was analyzed in the epidermal extract at the indicated times. As shown in Figure 3a, c and d, acute UVR treatment of PKCε transgenic mice resulted in a decrease in the expression of Fas at 6h (Figure 3a, c) and Fas-L at 18h, 24h and 96h (Figure 3a, d) after UVR exposure compared to their wild-type littermates. Our results also showed a reduced expression of Fas (Figure 3b, e) and Fas-L (Figure 3b, g) in PKCε transgenic mice after chronic UVR exposure. However, wild-type littermates had an increase in expression of Fas and Fas-L at 3 hours post chronic UVR exposure (3b, e and g).
Figure 3. PKCε overexpression suppresses UVR-induced Fas and Fas-L expression.
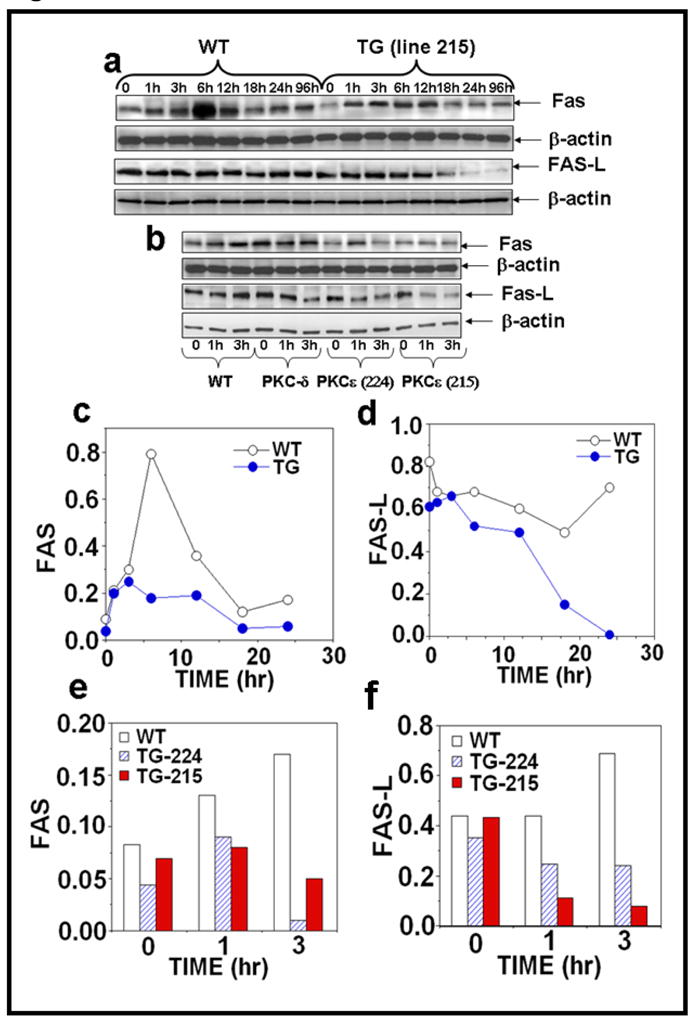
Epidermal extract prepared for Fig. 2 was used to analyze Fas and Fas-L expression. Immunoblot analysis of Fas expression after (a) acute (b) chronic UVR exposed samples and (c–f) the quantification of Fas and Fas-L expression.
The Fas-associated death domain protein (FADD) is essential for death receptor (DR)-induced apoptosis (Gaur and Aggarwal, 2003; Sheikh and Huang, 2003; Sheikh and Huang, 2003; Thorburn, 2004). The expression levels of pFADD and FADD were evaluated by the immunoblot analysis (Fig. 4a, b). pFADD and FADD expression were decreased in PKCε transgenic mice after acute UVR exposure (Figure 4a, c, and d). However, a complete loss of pFADD and FADD expression level was observed in both PKCε transgenic mouse lines (215 and 224) at 1 and 3 hrs after chronic UVR exposure (Figure 4b, f and g).
Figure 4. PKCε overexpression completely suppresses the level of pFADD, FADD and DAP-1 in UVR exposed PKCε transgenic mice.
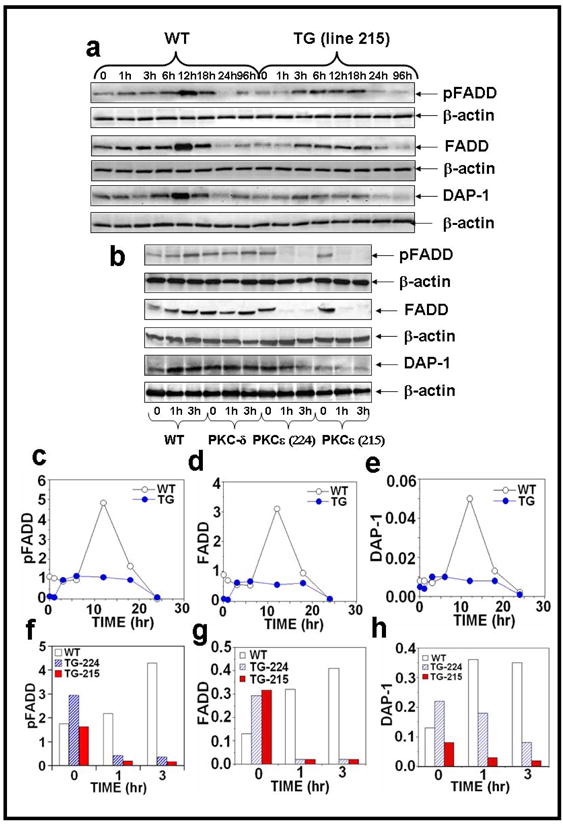
Epidermal extract prepared for Fig. 2 was used to analyze pFADD, FADD and DAP-1 expression. Immunoblot analysis of pFADD, FADD and DAP-1 expression after (a) acute and (b) chronic UVR exposures. (c–h): The quantification of pFADD, FADD and DAP-1 expression.
We also compared UVR-induced expression level of Death associated protein -1 (DAP-1) in PKCε transgenic mice and wild-type littermates. DAP-1 is a 15 kDa, proline rich, cytosolic protein. It has two potential cdk phosphorylation sites. The death domain of DAP-kinase contains all boxes of homology and the conserved amino acids characteristic of analogous domains in other death domain-containing proteins (Feinstein et al., 1995). DAP-1 has a direct involvement in programmed cell death, involving the p55 TNF receptor, the Fas/APO-1 receptor, DR3+5, FADD/MORT-1, RIP, TRADD and RAIDD (Ashkenazi and Dixit, 1998; Daniel et al., 2001; Liou and Liou, 1999). We found that after both acute (Figure 4a, e) and chronic (Figure 4b, h) UVR exposure PKCε transgenic mice have decreased expression of DAP-1. In contrast, wild-type mice have increased expression of DAP-1 6 hours after acute (Figure 4a, e) and both 1 and 3 hours after chronic (Figure 4b, h) UVR exposure.
The extrinsic pathway of apoptosis is initiated via the formation of a DISC, where the ligation of a death receptor, for example, CD95/Fas or TRAIL, facilitates the oligomerization with the adaptor protein FADD. Subsequent recruitment of the initiator caspase-8 concludes the assembly of the DISC and results in activation of caspase-8. We compared the level of UVR-induced caspase-8 cleavage in PKCε transgenic mice to wild-type littermates. Immunoblot analysis of caspase-8 indicated that both acute and chronic UVR exposed wild-type mice showed increased expression of cleaved caspase-8, whereas PKCε overexpression (line 215) resulted in a decrease in cleavage forms of caspase-8 (43- to 41-kDa) after both acute (24h and 96h post-treatment) and chronic (1h and 3h post-treatment) UVR exposure (Figure 5a–g).
Figure 5. Effect of UVR on the activation of caspase-8 in UVR exposed PKCε transgenic mice.

Epidermal extract prepared for Fig. 2 was used to analyze caspase 8 expression. Immunoblot analysis of caspase-8 expression after (a) acute (b) chronic UVR exposed samples. (c–g): The quantification of caspase-8 expression.
Protein Kinase Cε overexpression in transgenic mice suppresses Bid truncation essential to link the extrinsic to intrinsic apoptotic pathway
Intrinsic and extrinsic pathways of apoptosis are connected through caspase-8-mediated processing and activation of the BH3 domain-only death protein Bid. Truncated Bid (tBid) facilitates activation of Bak/Bax, the proapoptotic members of the Bcl-2 family and has important implications for the sensitization of cancer cells to DNA-damaging anticancer drugs (Daniel et al., 2001; Rudner et al., 2005; von Haefen et al., 2004). As shown in Figure 6a, PKCε overexpression inhibited the cleavage of the pro-apoptotic protein from 22 kDa to its truncated form (14 kDa) after chronic UVR exposure at 1h and 3h of treatment (Figure 6a–c). Acute UVR exposure of PKCε transgenic mice did not increase Bax/Bcl-2 ratio (Figures 6d, f). Also, both PKCε transgenic mouse lines exhibited a reduction in Bax/Bcl-2 ratio after chronic UVR treatment (Figure 6e, g).
Figure 6. PKCε overexpression inhibits tBid and Bax/Bcl-2 ratio in UVR exposed PKCε transgenic mice.
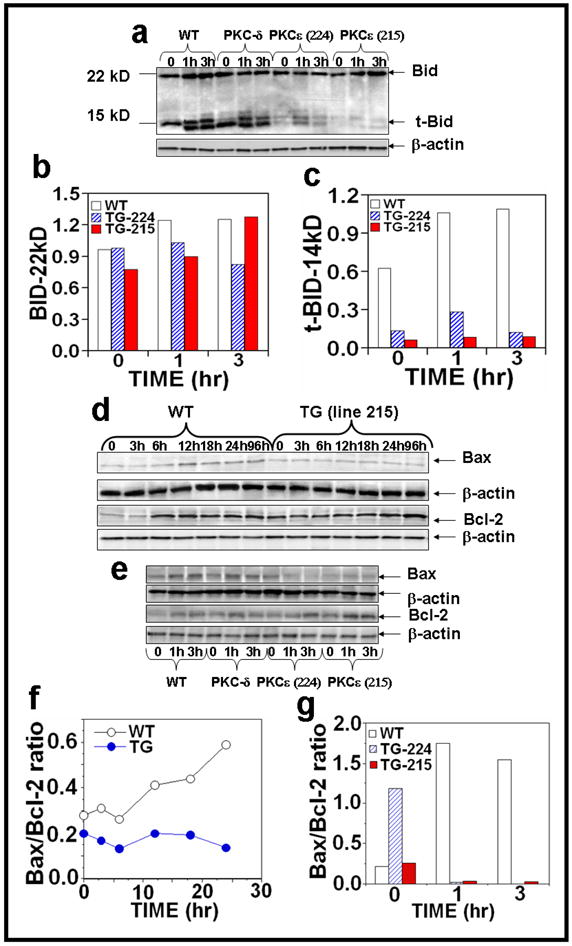
Epidermal extract prepared for Fig. 2 was used to analyze Bid and tBid expression and Bax and Bcl-2 expression after (a,e) chronic and (d) acute UVR exposure. Quantification of (b) Bid and (c) tBid and (f, g) Bax/Bcl-2 ratio after chronic UVR exposure.
Effect of UVR on the activation of caspase-3 in UVR exposed PKCε transgenic mice
Caspase-8 activates the downstream effector caspases-3, which mediate cleavage of a broad range of substrate proteins (Fischer et al., 2007) and initiate DNA fragmentation and cell death. The results indicate that either acute or chronic UVR exposure of PKCε transgenic mice (line 215) resulted in a decrease in the procaspase-3 (Figure 7a–g). In contrast, wild-type mice have an increased expression of the proteolytic cleavage forms of caspase-3 by both acute and chronic (1h and 3h post-treatment) UVR exposures (Figure 7a–g).
Figure 7. Effect of UVR on the activation of caspase-3 in UVR exposed PKCε transgenic mice.

Epidermal extract prepared for Fig. 2 was used to analyze caspase-3 expression. Immunoblot analysis of caspase-3 expression after (a) acute (b) chronic UVR exposure. (c–g): Quantification of caspase-3 expression.
Protein Kinase Cε overexpression suppresses UVR-induced PARP cleavage
Since Poly (ADP-ribose) Polymerase (PARP) cleavage is an indication of the commitment to undergo apoptosis, we determined whether PKCε overexpression inhibits UVR-induced PARP cleavage. In this experiment, PKCε transgenic mice and wild-type littermates were exposed to either acute or chronic UVR, and the epidermal skin extracts were immunoblotted with an antibody specific for PARP. PKCε overexpression inhibited PARP cleavage either after acute (Figs. 8a, c) or chronic (Figs. 8b, d) UVR exposure.
Figure 8. PKCε overexpression inhibits PARP and its cleavage in UVR exposed PKCε transgenic mice.
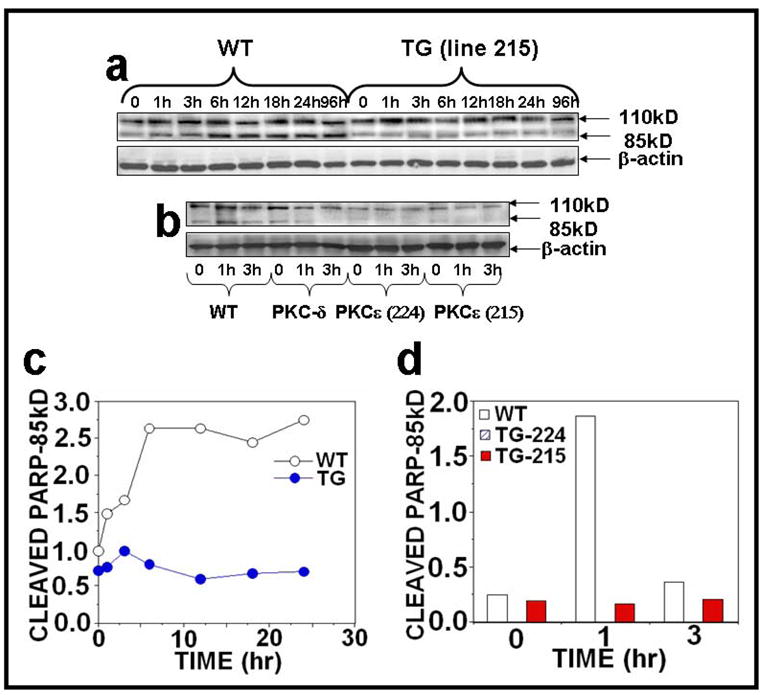
Epidermal extract prepared for Fig. 2 was used to analyze PARP and its cleavage. Immunoblot analysis of PARP and its cleavage product after (a) acute (b) chronic UVR exposure. (c and d): Quantification of PARP and its cleavage product.
We also determined the effects of PKCε overexpression on the expression of other caspase substrates such as Lamin A/C, Lamin B1, and gelsolin after UVR exposures. PKCε overexpression had no effect on Lamin A/C and Lamin B1 but there was a small decrease in the expression of Gelsolin in both acute and chronic UVR exposure (data not shown).
DISCUSSION
The sun’s ultraviolet radiation is the most potent environmental carcinogen and is linked to the development of skin cancer including SCC and BCC (Green et al., 1999; Wang et al., 2007; Morison, 1997; Mukhtar et al., 1999). We have reported that PKCε, which is expressed in both human and mouse epidermis (Wheeler et al., 2005), is an important component of UVR-mediated signal transduction pathways to development of SCC (Wheeler et al., 2004). PKCε overexpression in FVB/N transgenic mice sensitized skin to UVR-induced development of SCC (Wheeler et al., 2004; Wheeler et al., 2005) and suppressed formation of sunburn cells, which are DNA-damaged keratinocytes (Wheeler et al., 2004). Repeated UVR exposures accompanied adecrease in number of apoptotic keratinocytes. However, PKCε overexpression in PKCε transgenic mice attenuated UVR-induced apoptosis after both acute and chronic UVR exposures (Figure 1). This inhibition of apoptosis in DNA-damaged cells (precancerous cells) may contribute to the susceptibility of PKCε transgenic mice to the development of SCC. We now present that PKCε-mediated inhibition of UVR-induced sunburn cell formation may be linked to the ability of PKCε to inhibit both the extrinsic and intrinsic pathways of apoptotic signaling (Rossi and Gaidano, 2003).
UVR is a complete carcinogen, which both initiates and promotes carcinogenesis. UVR initiates carcinogenesis by directly damaging DNA (Balint and Vousden, 2001; Berton et al., 1997; de Gruijl et al., 2001; Li et al., 1998; Muller et al., 1998; Kunisada et al., 2007; Rossie and Gaidano, 2003). DNA-damaged keratinocytes can be eliminated through sunburn cell formation. Sunburn cell formation has been shown to be dependent on Fas-L, a pro-apoptotic protein induced by DNA damage (Ouhtit et al., 2000). Loss of Fas-L expression will inhibit elimination of precancerous cells through sunburn cell formation, a step essential for development of skin cancer. Chronic UVR exposure appears to induce a loss of Fas-L expression and a gain in p53 mutations leading to dysregulation of apoptosis and initiation of skin cancer (Ouhtit et al., 2000). PKCε overexpression suppressed UVR induced apoptosis (Figure 1) and the expression of Fas and Fas-L (Figures 3). A decrease in Fas-L expression was early and more pronounced after chronic than acute UVR exposures (Figure 3a, b, d, and g).
The DISC contains the adaptor protein FADD and caspase-8, which can initiate the process of apoptosis. Fas-L-induced clustering of Fas, FADD, caspase-8 or -10 within the DISC results in autoproteolytic processing of these caspases by induced proximity and in release of processed active proteases. Proper activation of effector caspases by Fas depends on an amplification loop that relies on caspase-8-mediated cleavage of the pro-apoptotic Bcl-2 family member Bid. Truncated Bid initiates release of mitochondrial pro-apoptotic factors cytochrome c and mitochondrial-derived activator of caspases to drive the formation of the caspase-9 activating apoptosome. Active caspase 9 activates the executimer caspase-3 which in turn activates caspase-8 outside the Fas DISC, thereby completing a positive feedback loop. The loss of UVR-induced pFADD and FADD expression in PKCε transgenic mice may block the Fas-induced apoptotic pathway.
The mechanism by which PKCε mediates loss of Fas-L and FADD is not known. It is also unclear how UVR induces the upregulation of the expression of Fas, Fas-L, pFADD and FADD in wild-type mice. In other reports, UVR has been shown to induce the expression of Fas and Fas-L in T cells, where p38 MAPK has been shown to be involved in Fas-L expression in tumor cells (Hsu et al., 1999). The Fas and Fas-L promoters have been studied and the transcription elements NF-AT, NF-kB and AP-1 have been identified in the Fas-L promoter (Kasibhatla et al.,1998; Latinis et al., 1997). Activation of NF-kB and AP-1 has been found to contribute to stress-induced apoptosis via the expression of Fas-L (Kasibhatla et al., 1998; Latinis et al., 1997). The regulation of FADD expression has been shown to occur only at the protein level. Evidence indicates that FADD resides in the nucleus and is shuttled between the nucleus and the cytoplasm (Sheikh and Huang, 2003).
We have reported that UVR-induced levels of epidermal TNFα mRNA and TNFα protein correlate with the level of expression of PKCε protein in PKCε transgenic mouse lines (Wheeler et al., 2004). Also, TNFα knockout mice are resistant to skin carcinogenesis (Moore et al., 1999; Suganuma et al., 1999). We also found that UVR-induced severe cutaneous damage (ulceration, hyperplasia and infiltration of inflammatory cells) in PKCε transgenic mice (line 215) was partially prevented in bigenic PKCε transgenic-TNFα knockout mice (Wheeler et al., 2004). Taken together, it appears that UVR-induced TNFα expression is an important component on PKCε signal transduction pathway to the development of SCC. TNFα has the ability to regulate a vast array of cellular responses including pro-apoptotic, anti-apoptotic, proliferation, and inflammation (Tibbetts et al., 2003). However, it is unclear whether UVR-induced increased expression of TNFα is pro-apoptotic or a proliferating signal in UVR-carcinogenesis. The results presented here indicate that UVR-induced TNFα expression in PKCε transgenic mice is a proliferative signal. TNFα exerts its biological effects by trimerization and binding to two distinct receptors, TNFR1 and TNFR2 (Tartaglia and Goeddel, 1992). TNFR1 is ubiquitously expressed whereas TNFR2 is found predominantly in hematopoietic and endothelial cells (Starcher, 2000; Zhuang et al., 1999). Binding of TNFα induces trimerization of these receptors and then recruits several signaling proteins to the cytoplasmic membrane. FADD is the key component of both Fas and TNFR-mediated apoptosis (Gaur and Aggarwal, 2003; Sheikh and Huang, 2003; Sheikh and Huang, 2003; Thorburn, 2004). The UVR-induced loss of FADD expression in PKCε transgenic mice lends support to the conclusion that UVR-induced level of TNFα is not pro-apoptotic, but rather may contribute to increased cell proliferation of preneoplastic cells.
In summary, PKCε overexpression, which sensitizes skin to UVR-induced carcinogenesis, suppresses UVR-induced sunburn (apoptotic) cell formation (Wheeler et al., 2004). UVR-induced sunburn cell formation involves Fas/Fas-L and TNFα-TNFR1 interactions. FADD is a common adaptor protein for both of these apoptotic pathways (Baud and Karin, 2001; Chinnaiyan et al., 1995; Rossi and Gaidano, 2003; Tibbetts et al., 2003). PKCε attenuated UVR-induced expression of FADD leads to inhibition of both intrinsic and extrinsic apoptotic pathways. These results led us to hypothesize that UVR-induced activated PKCε mediates two potential signals including one that facilitates accumulation of UVR-induced DNA-damaged keratinocytes and the other to promote the proliferation of these DNA-damaged keratinocytes (preneoplastic cells) to form SCC. PKCε exerts these effects through Stat3 activation (Aziz), TNFα expression (Wheeler et al., 2004) and the inhibition of FADD, component of the death inducing signaling complex.
MATERIALS AND METHODS
Materials
The acrylamide, bisacrylamide, sodium dodecyl sulfate (SDS), 0.45 μm supported nitrocellulose membrane, Bio-Rad Protein Assay, and SDS-polyacrylamide gel electrophoresis (PAGE) standards were purchased from Bio-Rad Laboratories (Hercules, CA). The source of antibodies were: TNF-R1, Fas, Fas-L, FADD, Bax, Bcl-2, β-actin (Santa Cruz Biotechnology, Santa Cruz, CA); Caspase-8, caspase-3, Bid, DAP-1 (Cell Signaling Technology, Beverly, MA); PARP (BIOMOL International, L.P., Plymouth Meeting, PA); Anti-Rabbit IgG (H+L) HRP conjugated (MP Biomedicals, Inc., Aurora, Ohio) and Anti-goat IgG (H+L) peroxidase (Roche Diagnostics, Indianapolis, IN). Apoptag plus flurescein in situ apoptosis detection kit S7111 (Chemicon International, Billerica, MA). The ECL Western blotting reagents were purchased from Amersham Life Sciences Inc. (Arlington Heights, IL). FS-40 sunlamps were purchased from National Biological/ETA Systems (Twinsburg, OH). Kodacel filter was purchased from Eastman Kodak Company (Rodchester, NY). FVB/N mice were purchased from Taconic (Germantown, NY).
PKCε Transgenic mice
PKCε transgenic mice were generated as described previously (Wheeler et al., 2004; Wheeler et al., 2005). All animal care protocols were approved by an institutional review board. Transgenic mice were maintained by mating hemizygous transgenic mice with wild-type FVB/N mice. The mice were housed in groups of two to three in plastic bottom cages in light-, humidity-, and temperature-controlled rooms; food and water were available ad libitum. The animals were kept in a normal rhythm of 12h light and 12h dark periods. The transgene was detected by polymerase chain reaction analysis using genomic DNA isolated from one cm tail clips (Wheeler et al., 2004; Wheeler et al., 2005)
UVR source and treatment
The UVR source was Kodacel-filtered FS-40 sunlamps (approximately 60% UV-B and 40% UV-A). Mice were exposed to UVR from a bank of six Kodacel-filtered sunlamps. UVR dose was routinely measured using UVX-radiometer. Mice were used for experimentation at 7–9 weeks of age. The dorsal skin of the mice was shaved 3–4 days before experimentation. Mice were exposed to UVR as indicated in each experiment.
Histology
The tissue to be examined was excised promptly after euthanasia and placed immediately in 10% neutral buffered formalin. The tissue was fixed for 1h in formalin and then embedded in paraffin. Sections of 4μm thickness were cut for hematoxylin and eosin staining.
TUNEL Assay
PKCε transgenic mice (line 215) and wild-type littermates were exposed to single UVR dose (4 kJ/m2). The mice were sacrificed 24 hours after exposure. The tissue to be examined was excised promptly after euthanasia and placed immediately in 10% neutral buffered formalin. The tissue was fixed and then embedded in paraffin. Sections of 4μm thickness were cut for Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP Nick-End Labeling (TUNEL) to detect apoptotic cells in UVR exposed skin tissue. Apoptotic cells were labeledusing Apoptag Plus fluorescein in situ apoptosis detection kit S7111 (Chemicon International, Billerica, MA) according to the manufacturer’s instructions and viewed by fluorescence microscopy. DAPI (0.1 mg/L for 20 min) was used to counterstain all the nuclear DNA. The TUNEL positive cells were identified and scored, based upon the presence of green fluorescent nuclear staining for TUNEL. The sections were scored for a total of at least eight views. Apoptotic cells were expressed as a percentage of total epidermal cells.
Western Blot Analysis
The mice were euthanized at the appropriate time after UVR treatment, the dorsal skin was removed, and the epidermis was scraped off on ice with a razor blade. The epidermis from 4 mice were pooled and placed in immunoprecipitation lysis buffer (50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES]) (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1.5 mM MgCl2, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM phenylmethylsulfonyl fluoride (PMSF), 200 μM Na3VO4, 200 μM NaF and 1 mM ethylene glycol-bis(b-aminoethylether)-N,N,N′,N′-tetra-acetic acid (EGTA]), homogenized in a glass teflon tissue homogenizer, and agitated for 30 min at 4°C. The homogenate was centrifuged at 14, 000 X g for 30 min at 4 °C, the supernatant was used for western blot analysis. 25–35 μg of whole cell lysate was fractionated on 10 or 15% SDS-polyacrylamide gels. The proteins were transferred to 0.45 um of Hybond-P polyvinylidene difluoride (PVDF) transfer membrane (Amersham). The membrane was then incubated with indicated antibodies followed by a horseradish peroxidase secondary antibody, and the detection signal was developed with Amersham’s enhanced chemiluminescence reagent and autoradiography using BioMax film obtained from Kodak Co., (Rochester, NY). The quantitations of Western blots were performed by densitometric analysis using Totollab Nonlinear Dynamic Image analysis software (Nonlinear USA Inc., Durham, NC).
Supplementary Material
Acknowledgments
The work was supported by NIH Grants CA35368 and CA102431.
Abbreviations
- CA
Carcinoma
- DAG
Diacylglycerol
- PKC
Protein Kinase C
- PS
Phosphatidylserine
- TNF
Tumor Necrosis Factor
- TPA
12-O-tetradecanoylphorbol-13-acetate
- UVR
Ultraviolet Radiation
- DMBA
7,12-Dimethylbenzanthracene
- K14
human keratin 14 promoter
- SCC
squamous cell carcinoma
Footnotes
CONFLICT OF INTEREST
None
This work was supported by NIH Grants CA35368 and CA102431.
References
- Anonymous skin cancer; American Cancer Society. Cancer Facts & Figures 2008. Atlanta: American Cancer Society; 2008. [Google Scholar]
- Kulms D, Düssmann H, Pöppelmann B, Ständer S, Schwarz A, Schwarz T. Apoptosis induced by disruption of the actin cytoskeleton is mediated via activation of CD95 (Fas/APO-1) Cell Death Differ. 2002;9:598–608. doi: 10.1038/sj.cdd.4401002. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Balint EE, Vousden KH. Activation and activities of the p53 tumour suppressor protein. Br J Cancer. 2001;85:1813–23. doi: 10.1054/bjoc.2001.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–7. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- Berton TR, Mitchell DL, Fischer SM, Locniskar MF. Epidermal proliferation but not quantity of DNA photodamage is correlated with UV-induced mouse skin carcinogenesis. J Invest Dermatol. 1997;109:340–7. doi: 10.1111/1523-1747.ep12335984. [DOI] [PubMed] [Google Scholar]
- Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–5. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–12. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Daniel PT, Wieder T, Sturm I, Schulze-Osthoff K. The kiss of death: promises and failures of death receptors and ligands in cancer therapy. Leukemia. 2001;15:1022–32. doi: 10.1038/sj.leu.2402169. [DOI] [PubMed] [Google Scholar]
- de Gruijl FR. Skin cancer and solar UV radiation. Eur J Cancer. 1999;35:2003–9. doi: 10.1016/s0959-8049(99)00283-x. [DOI] [PubMed] [Google Scholar]
- de Gruijl FR, van Kranen HJ, Mullenders LH. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J Photochem Photobiol B. 2001;63:19–27. doi: 10.1016/s1011-1344(01)00199-3. [DOI] [PubMed] [Google Scholar]
- Feinstein E, Druck T, Kastury K, Berissi H, Goodart SA, Overhauser J, Kimchi A, Huebner K. Assignment of DAP1 and DAPK--genes that positively mediate programmed cell death triggered by IFN-gamma--to chromosome regions 5p12.2 and 9q34.1, respectively. Genomics. 1995;29:305–7. doi: 10.1006/geno.1995.1255. [DOI] [PubMed] [Google Scholar]
- Figueras M, Busquets S, Carbo N, Almendro V, Argiles JM, Lopez-Soriano FJ. Cancer cachexia results in an increase in TNF-alpha receptor gene expression in both skeletal muscle and adipose tissue. Int J Oncol. 2005;27:855–60. [PubMed] [Google Scholar]
- Fischer U, Janssen K, Schulze-Osthoff K. Does caspase inhibition promote clonogenic tumor growth? Cell Cycle. 2007;6:3048–53. doi: 10.4161/cc.6.24.5118. [DOI] [PubMed] [Google Scholar]
- Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem Pharmacol. 2003;66:1403–8. doi: 10.1016/s0006-2952(03)00490-8. [DOI] [PubMed] [Google Scholar]
- Green A, Whiteman D, Frost C, Battistutta D. Sun exposure, skin cancers and related skin conditions. J Epidemiol. 1999;9:S7–13. doi: 10.2188/jea.9.6sup_7. [DOI] [PubMed] [Google Scholar]
- Hill LL, Ouhtit A, Loughlin SM, Kripke ML, Ananthaswamy HN, Owen-Schaub LB. Fas ligand: a sensor for DNA damage critical in skin cancer etiology. Science. 1999;285:898–900. doi: 10.1126/science.285.5429.898. [DOI] [PubMed] [Google Scholar]
- Hill LL, Shreedhar VK, Kripke ML, Owen-Schaub LB. A critical role for Fas ligand in the active suppression of systemic immune responses by ultraviolet radiation. J Exp Med. 1999;189:1285–94. doi: 10.1084/jem.189.8.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtenbos I, Westers TM, de Gruijl TD, Scheper RJ, Ossenkoppele GJ, van de Loosdrecht AA. TNF-alpha receptor 1 expression on acute myeloid leukemic blasts predicts differentiation into leukemic dendritic cells. Leukemia. 2004;18:1149–53. doi: 10.1038/sj.leu.2403359. [DOI] [PubMed] [Google Scholar]
- Hsu SC, Gavrilin MA, Tsai MH, Han J, Lai MZ. p38 mitogen-activated protein kinase is involved in Fas ligand expression. J Biol Chem. 1999;274:25769–76. doi: 10.1074/jbc.274.36.25769. [DOI] [PubMed] [Google Scholar]
- Huppertz B, Frank HG, Kaufmann P. The apoptosis cascade--morphological and immunohistochemical methods for its visualization. Anat Embryol (Berl) 1999;200:1–18. doi: 10.1007/s004290050254. [DOI] [PubMed] [Google Scholar]
- Jansen AP, Verwiebe EG, Dreckschmidt NE, Wheeler DL, Oberley TD, Verma AK. Protein kinase C-epsilon transgenic mice: a unique model for metastatic squamous cell carcinoma. Cancer Res. 2001;61:808–12. [PubMed] [Google Scholar]
- Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol Cell. 1998;1:543–51. doi: 10.1016/s1097-2765(00)80054-4. [DOI] [PubMed] [Google Scholar]
- Wang LE, Li C, Strom SS, Goldberg LH, Brewster A, Guo Z, Qiao Y, Clayman GL, Lee JJ, El-Naggar AK, Prieto VG, Duvic M, Lippman SM, Weber RS, Kripke ML, Wei Q. Repair capacity for UV light induced DNA damage associated with risk of nonmelanoma skin cancer and tumor progression. Clin Cancer Res. 2007;13:6532–9. doi: 10.1158/1078-0432.CCR-07-0969. [DOI] [PubMed] [Google Scholar]
- Latinis KM, Norian LA, Eliason SL, Koretzky GA. Two NFAT transcription factor binding sites participate in the regulation of CD95 (Fas) ligand expression in activated human T cells. J Biol Chem. 1997;272:31427–34. doi: 10.1074/jbc.272.50.31427. [DOI] [PubMed] [Google Scholar]
- Li G, Tron V, Ho V. Induction of squamous cell carcinoma in p53-deficient mice after ultraviolet irradiation. J Invest Dermatol. 1998;110:72–5. doi: 10.1046/j.1523-1747.1998.00090.x. [DOI] [PubMed] [Google Scholar]
- Lind MH, Rozell B, Wallin RP, van Hogerlinden M, Ljunggren HG, Toftgard R, Sur I. Tumor necrosis factor receptor 1-mediated signaling is required for skin cancer development induced by NF-kappaB inhibition. Proc Natl Acad Sci U S A. 2004;101:4972–7. doi: 10.1073/pnas.0307106101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou ML, Liou HC. The ubiquitin-homology protein, DAP-1, associates with tumor necrosis factor receptor (p60) death domain and induces apoptosis. J Biol Chem. 1999;274:10145–53. doi: 10.1074/jbc.274.15.10145. [DOI] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Lu YP, Nolan B, Lou YR, Peng QY, Wagner GC, Conney AH. Voluntary exercise together with oral caffeine markedly stimulates UVB light-induced apoptosis and decreases tissue fat in SKH-1 mice. Proc Natl Acad Sci USA. 2007;104:12936–41. doi: 10.1073/pnas.0705839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEwan DJ. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–92. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- Moller R, Reymann F, Hou-Jensen K. Metastases in dermatological patients with squamous cell carcinoma. Arch Dermatol. 1979;115:703–5. doi: 10.1001/archderm.1979.04010060011017. [DOI] [PubMed] [Google Scholar]
- Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, Holdsworth H, Turner L, Rollins B, Pasparakis M, Kollias G, Balkwill F. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med. 1999;5:828–31. doi: 10.1038/10552. [DOI] [PubMed] [Google Scholar]
- Morison WL. Sunlight: an environmental toxin for humans. A primer to advise patients. Md Med J. 1997;46:227–30. [PubMed] [Google Scholar]
- Mukhtar H, Forbes PD, Ananthaswamy HN. Photocarcinogenesis--models and mechanisms. Photodermatol Photoimmunol Photomed. 1999;15:91–5. doi: 10.1111/j.1600-0781.1999.tb00065.x. [DOI] [PubMed] [Google Scholar]
- Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998;188:2033–45. doi: 10.1084/jem.188.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisada M, Kumimoto H, Ishizaki K, Sakumi K, Nakabeppu Y, Nishigori C. Narrow-band UVB induces more carcinogenic skin tumors than broad-band UVB through the formation of cyclobutane pyrimidine dimer. J Invest Dermatol. 2007;127:2865–71. doi: 10.1038/sj.jid.5701001. [DOI] [PubMed] [Google Scholar]
- Ouhtit A, Gorny A, Muller HK, Hill LL, Owen-Schaub L, Ananthaswamy HN. Loss of Fas-ligand expression in mouse keratinocytes during UV carcinogenesis. Am J Pathol. 2000;157:1975–81. doi: 10.1016/S0002-9440(10)64836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddig PJ, Dreckschmidt NE, Ahrens H, Simsiman R, Tseng CP, Zou J, Oberley TD, Verma AK. Transgenic mice overexpressing protein kinase Cdelta in the epidermis are resistant to skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1999;59:5710–8. [PubMed] [Google Scholar]
- Reddig PJ, Dreckschmidt NE, Zou J, Bourguignon SE, Oberley TD, Verma AK. Transgenic mice overexpressing protein kinase C epsilon in their epidermis exhibit reduced papilloma burden but enhanced carcinoma formation after tumor promotion. Cancer Res. 2000;60:595–602. [PubMed] [Google Scholar]
- Redondo P, Solano T, BVA, Bauza A, Idoate M. Fas and Fas ligand: expression and soluble circulating levels in cutaneous malignant melanoma. Br J Dermatol. 2002;147:80–6. doi: 10.1046/j.1365-2133.2002.04745.x. [DOI] [PubMed] [Google Scholar]
- Rossi D, Gaidano G. Messengers of cell death: apoptotic signaling in health and disease. Haematologica. 2003;88:212–8. [PubMed] [Google Scholar]
- Rudner J, Jendrossek V, Lauber K, Daniel PT, Wesselborg S, Belka C. Type I and type II reactions in TRAIL-induced apoptosis -- results from dose-response studies. Oncogene. 2005;24:130–40. doi: 10.1038/sj.onc.1208191. [DOI] [PubMed] [Google Scholar]
- Sheikh MS, Huang Y. Death receptor activation complexes: it takes two to activate TNF receptor 1. Cell Cycle. 2003;2:550–2. [PubMed] [Google Scholar]
- Sheikh MS, Huang Y. The FADD is going nuclear. Cell Cycle. 2003;2:346–7. [PubMed] [Google Scholar]
- Starcher B. Role for tumour necrosis factor-alpha receptors in ultraviolet-induced skin tumours. Br J Dermatol. 2000;142:1140–7. doi: 10.1046/j.1365-2133.2000.03539.x. [DOI] [PubMed] [Google Scholar]
- Suganuma M, Okabe S, Marino MW, Sakai A, Sueoka E, Fujiki H. Essential role of tumor necrosis factor alpha (TNF-alpha) in tumor promotion as revealed by TNF-alpha-deficient mice. Cancer Res. 1999;59:4516–8. [PubMed] [Google Scholar]
- Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–3. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–44. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Tibbetts MD, Zheng L, Lenardo MJ. The death effector domain protein family: regulators of cellular homeostasis. Nat Immunol. 2003;4:404–9. doi: 10.1038/ni0503-404. [DOI] [PubMed] [Google Scholar]
- Tracey KJ, Cerami A. Tumor necrosis factor, other cytokines and disease. Annu Rev Cell Biol. 1993;9:317–43. doi: 10.1146/annurev.cb.09.110193.001533. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5:392–9. doi: 10.1016/s0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- von Haefen C, Gillissen B, Hemmati PG, Wendt J, Guner D, Mrozek A, Belka C, Dorken B, Daniel PT. Multidomain Bcl-2 homolog Bax but not Bak mediates synergistic induction of apoptosis by TRAIL and 5-FU through the mitochondrial apoptosis pathway. Oncogene. 2004;23:8320–32. doi: 10.1038/sj.onc.1207971. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Li Y, Verma AK. Protein kinase C epsilon signals ultraviolet light-induced cutaneous damage and development of squamous cell carcinoma possibly through Induction of specific cytokines in a paracrine mechanism. Photochem Photobiol. 2005;81:9–18. doi: 10.1562/2004-08-12-RA-271. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Martin KE, Ness KJ, Li Y, Dreckschmidt NE, Wartman M, Ananthaswamy HN, Mitchell DL, Verma AK. Protein kinase C epsilon is an endogenous photosensitizer that enhances ultraviolet radiation-induced cutaneous damage and development of squamous cell carcinomas. Cancer Res. 2004;64:7756–65. doi: 10.1158/0008-5472.CAN-04-1881. [DOI] [PubMed] [Google Scholar]
- Wu S, Korte A, Gessner R, Henze G, Seeger K. Levels of the soluble, 55-kilodalton isoform of tumor necrosis factor receptor in bone marrow are correlated with the clinical outcome of children with acute lymphoblastic leukemia in first recurrence. Cancer. 2003;98:625–31. doi: 10.1002/cncr.11553. [DOI] [PubMed] [Google Scholar]
- Zhuang L, Wang B, Shinder GA, Shivji GM, Mak TW, Sauder DN. TNF receptor p55 plays a pivotal role in murine keratinocyte apoptosis induced by ultraviolet B irradiation. J Immunol. 1999;162:1440–7. [PubMed] [Google Scholar]
- Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, Brash DE. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–6. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.