

Di-myo-inositol-1,1’-phosphate (DIP), has been identified as a major intracellular solute in several hyperthermophilic organisms. In particular, a number of hyperthermophilic microorganisms (including archaea and the bacterium Thermotoga maritima) accumulate DIP as an osmolyte when grown above 75 °C;[1,2] DIP concentration increases dramatically at temperatures greater than 80 °C.[3] The stereoisomeric nature of naturally occurring DIP has been a matter of discussion in the literature, with reports supporting either the chiral L,L-DIP form, or the meso L,D-isomer (Figure 1). Presently, most of the available data, including biosynthetic,[4] biochemical and bioinformatic[5] studies, support the assignment of the L,D-isomer as a natural product. Importantly, it is also possible that different organisms produce different stereoisomers as a part of their individual metabolic capabilities. Through classical synthesis, van Boom et al. suggested the L,L-isomer could be the naturally occurring form of DIP in Pyrococcus woesei, based on comparison of the optical rotations of natural and synthetic DIP.[6] On the other hand, in 2007 Santos et al. suggested the natural material produced by Archaeoglobus fulgidus is the L,D-isomer based on 13C-labeling and NMR studies.[4] One question of interest, independent of the actual stereochemical assignment for any material of natural origin isolated to date, is whether or not the two stereoisomers are distinct functionally. That is, do the two isomers exhibit differential abilities to exhibit enzyme stabilization properties under extremophilic conditions, when examined with a unique enzyme derived from a common organism? We now address this matter directly through efficient, independent state-of-the-art chemical syntheses of pure samples of known stereochemical identity, and preliminary biochemical studies with an appropriate enzyme at high temperatures.
Figure 1.

Structures of L,L-DIP and L,D-DIP
Previous research from our group has demonstrated that histidine-based peptides can catalyze various enantioselective phosphorylation reactions.[7] Shown in Scheme 1, the triol 2,4,6-tri-O-benzyl-myo-inositol (2) can be subjected to desymmetrization using either of two enantiodivergent catalysts (4 or 5) and diphenyl chlorophosphate to provide phosphates 1 or 3 in good yields and excellent enantioselectivity. We felt that catalysts 4 and 5 could thus be critical tools for the independent synthesis of L,L-DIP and L,D-DIP, enabling unambiguous access to these stereoisomers, and thus allowing direct comparison of their differential prowess as stabilizers of enzyme activity under conditions of high temperature.
Scheme 1.

Desymmetrization of meso-triol 2.
Our retrosynthetic plan was therefore quite straightforward (Scheme 2). In our approach, each inositol would be subjected to phosphorylation in a sequential fashion, allowing for rational control of O−P bond formation at either the 1-position (red) or the 3-position (blue). This plan requires the synthesis of an optically pure, fully-protected inositol-derived phosphoryl chloride (6). Compound 6 could then be coupled to 2, forming the desired phosphate bond under control of chiral catalysts such as 4 or 5.
Scheme 2.

Retrosynthetic analysis of DIP stereoisomers.
Critically, phosphoryl chloride 6 contains a stereogenic P-atom. As a result, catalytic phosphorylation reactions with peptide catalysts 4 and 5 create a fascinating, but complex situation of double asymmetric induction, with stereochemical preferences exerted by catalyst and substrate.[8] Thus, we needed to establish if catalysts 4 and 5 could overcome any inherent substrate control in the stereochemical outcome of the reactions (Scheme 3).
Scheme 3.

Projected peptide-catalyzed coupling reactions.
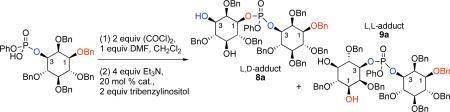
Execution of the synthesis plan began with preparation of phosphoryl chloride 6, and the recognition that it would not likely be accessed in diastereomerically pure form. Furthermore, since myo-inositol phosphate esters with unprotected hydroxyls are prone to undesired cyclizations,[9] we endeavored to alkylate the free hydroxyl groups of 1 (Scheme 4). Conditions that avoid formation of cyclic phosphates were found to be limited to neutral, acidic, or mild basic conditions. In this vein, benzylation according to the conditions of Dudley proved uniquely successful.[10] Hydrolysis to the mono(acid) 7 was then achieved with lithium hydroxide. Preparation of the inositol-based phosphoryl chloride was then achieved using oxalyl chloride and DMF. Within 5 min, chloride 6 was observed as a mixture of diastereomers, (2:1) epimeric at phosphorous, as was revealed in the 31P-NMR spectrum.
Scheme 4.

Conditions: a) 2-benzyloxy-1-methyl-pyridinium triflate, MgO, PhCF3; 73%; b) LiOH, THF/H2O; c) Dowex 50X2-200; quant. over 2 steps; d) 2 equiv (COCl)2, 1 equiv DMF, CH2Cl2, 5 minutes, r.t.; e) 4 equiv Et3N, 1 equiv DMAP, 2 equiv 2, r.t., 12 h, 57%.
We then turned our attention to the identification of efficient conditions for the coupling of 2 and 7. Numerous reaction parameters were evaluated (solvent, base, reagent ratios, temperature, reaction time, order of addition) before settling on addition of phosphoryl chloride 6 to triol 2 under the conditions shown in Scheme 4. These studies were initially conducted with DMAP as an achiral nucleophilic catalyst to define both optimized conditions, and the inherent regioselectivity of the process. The optimized conditions provided a mixture of the two DIP adducts 8a and 9a (3:1) in a combined 57% yield.
Each isomer (8a and 9a) was separated and carried forward to the corresponding isomer of DIP in order to assign stereochemical identity. Notably, reaction of the 1-position of 2 leads to 8a, the precursor to meso L,D-DIP; reaction at the 3-position of 2 leads to 9a, the precursor to optically active L,L-DIP. As shown in Scheme 5, the remaining two hydroxyls of diols 8a and 9a were converted to benzyl ethers to allow for access to meso or optically active material. Optical rotation and 1H NMR data were then used to determine that intermediate 8b was meso ([α]D = 0) and intermediate 9b was optically active ([α]D = +1.2), thus establishing the absolute stereochemistry of products 8a and 9a. Hydrolysis of the phenyl phosphate was then achieved using lithium hydroxide. Exchange to the sodium salt followed by hydrogenolysis yielded each isomer of the DIP product. The final steps were exceptionally clean, enabling isolation of pure DIP with a minimum of late stage purification.
Scheme 5.

Conditions: a) 2-benzyloxy-1-methyl-pyridinium triflate, MgO, PhCF3; 35%; b) LiOH, THF/H2O; Chelex 100 Na form; 69% over 2 steps; c) H2, Pd(OH)2/C, EtOAc/MeOH; 96%; d) 2-benzyloxy-1-methyl-pyridinium triflate, MgO, PhCF3; 27%; e) LiOH, THF/H2O; Chelex 100 Na form; 47% over 2 steps; f) H2, Pd(OH)2/C, EtOAc/MeOH; 82%.
Having established the absolute configurations of products 8a and 9a, a preliminary study of chiral catalysts (e.g., 5 and 4) was undertaken in the coupling reaction to assess if catalyst control could lead to optimized yields of 8a and 9a, respectively (Table 1). Achiral nucleophilic catalysts such as DMAP and NMI (entries 1 and 2), defined the degree of substrate control during the formation of the DIP precursors (entries 1 and 2, ~3-4:1). The simple amino acid catalyst derived from histidine (with the π-nitrogen methylated, Pmh, entry 3) revealed a similar product ratio (3:1, entry 3) as the achiral catalysts. However, catalyst 5 (entry 4) showed a modest reversal (1:1.25) of the inherent selectivity to favor the L,L-isomer (9a) slightly. Catalyst 4 (entry 5), on the other hand, exerted little stereochemical influence and showed similar ratios in comparison to the achiral catalysts. Investigation of 20 additional catalysts[11] led to the identification of a different catalyst (10) that gave substantial selectivity for the L,D-isomer 8a ((13:1; entry 6), and another (11) that more significantly reversed selectivity in favor of the L,L-isomer 9a (1:2.5, entry 7). These results bode well for further study, and amplified instances of improved peptides that could exhibit even higher degrees of catalyst control in these reactions. It is further of note that these differential ratios are achieved in a coupling reaction involving a diastereomeric mixture of phosphoryl chlorides, epimeric at the P-atom, which necessarily contributes to the complexity of these processes. Preparation of diastereomerically pure phosphoryl chlorides of this type is not yet known in the literature to our knowledge.
Table 1.
Examination of catalysts for selective synthesis of 8a and 9a.
 | ||
|---|---|---|
| Entry | Catalyst Sequence | L,D-Adduct:L,L-Adduct |
| 1 | 4,4-Dimethylaminopyridine (DMAP) | 3:1 |
| 2 | N-Methylimidazole (NMI) | 4:1 |
| 3 | Boc-Pmh-OMe | 3:1 |
| 4 | 5: Boc-Pmh-Asn(trt)-His(tBu)-Asp(OtBu)-Ala-OMe | 1:1.25 |
| 5 | 4: Boc-Pmh-Hyp(tBu)-Sp5-Tyr(tBu)-Phe-OMe | 3.6:1 |
| 6 | 10: Boc-D-Pmh-Pro-Aib-Trp(Boc)-Phe-OMe | 13:1 |
| 7 | 11: Boc-Pmh-D-Pro-Aib-D-Phe-D-Phe-OMe | 1:2.5 |
With a robust synthesis of L,L-DIP and L,D-DIP secured, we turned our attention to assessing the differential ability of each isomer to contribute to thermoprotection in the context of a key enzyme found in a unique extremophile. Archaeoglobus fulgidus is one such organism. Therefore, its proteins are good candidates to assess the effect of the solutes on protein thermostability. Previously, it was shown that the inositol monophosphatase (IMPase) from this organism denatures irreversibly when heated above 90 °C.[4, 5, 12] This enzyme thus serves as an interesting case to assess if the DIP anion can reduce the extent of thermal denaturation.
For this study, we examined the effect of KCl and NaCl and the glutamate salts of each cation (Figure 2) in protecting the IMPase activity after heating to 95 °C for 15 min. This treatment in the absence of compatible solutes nearly inactivated the enzyme (residual activity was 3% after this heating). Thermoprotection could be observed at 100 mM added salt, although the effects were much stronger at higher concentrations of the salts. Of particular interest is the observation that the sodium salts were virtually ineffective in this concentration range, while KCl and K+-glutamate were similar and much more effective at protecting IMPase activity.
Figure 2.

Effect of solutes on the residual activity of A. fulgidus inositol monophosphatase, heated to 95 °C for 15 minutes, then cooled and assayed at 85 °C: ▲, KCl; Δ, NaCl; ■, K+-glutamate; □, Na+-glutamate. The inset shows the effect of the sodium salts of L,L- (○) and L,D- (●) DIP at 100 mM, as well as dimethylphosphate (DMP; X) and myo-inositol (◇) compared to the K+-salts solutes (dashed lines) shown in the main figure.
In order to get a measure of what each stereoisomer of the DIP anion might contribute to thermoprotection, we examined the effects of the Na+-salts of DIP (Figure 2, inset). We examined 100 mM of each Na+-DIP on IMPase activity after heating. As a control, 100 mM myo-inositol was also examined for any thermoprotection effects on this enzyme. Surprisingly, both Na+-L,L-DIP and Na+-L,D-DIP were effective in producing a recovery of activity (17% and 11%, respectively). In comparison to the thermoprotection provided by KCl (18% recovery of activity), it is particularly notable the DIP sodium salts offer protection, when NaCl is completely ineffective, further pointing to the thermoprotective properties of the DIP anions. Inositol alone had no effect. In addition, the Na-salt of dimethylphosphate (DMP) offered no thermoprotective capacity, indicating that each DIP anion on its own is unique and has significant thermoprotective effects.
The modest differences in the thermoprotective influence of the two DIP stereoisomers are intriguing. At least in A. fulgidus, as well as in Thermotoga maritima,[13] the natural product appears to be the L,D-isomer. Yet, the fact that the alternative stereoisomer also offers protective effects raises fascinating questions about how the DIP anion stabilizes proteins to thermal denaturation. While the molecular details are not understood, the extended and rigid network of hydroxyl groups on the inositol rings could help to organize the water around the protein and stave off (to some degree) irreversible unfolding.[14] The apparent lack of a strong stereospecificty for these effects with the DIP isomers,[15] elucidated by combined chemical synthesis and biochemical studies, may provide an intriguing and potentially unusual case of chirality in nature more as a matter of biosynthetic convenience, rather than as a matter of evolutionary optimization. Our hope is that these chemical and biochemical approaches will now set the stage for detailed, high precision study of these intriguing phenomena.
Supplementary Material
Acknowledgments
[**] We are grateful to the NIH (GM-068649 to SJM) and to the Department of Energy Physical Biosciences (DE-FG02-91ER20025 to MFR) for support.
Contributor Information
Christina M. Longo, Department of Chemistry, Yale University 225 Prospect Street, New Haven, CT 06520-4900 (USA)
Yang Wei, Department of Chemistry, Boston College Chestnut Hill, MA 02467
Mary F. Roberts, Department of Chemistry, Boston College Chestnut Hill, MA 02467
Scott J. Miller, Department of Chemistry, Yale University 225 Prospect Street, New Haven, CT 06520-4900 (USA)
References
- 1.a Cuilla R, Burggraf S, Stetter KO, Roberts MF. Appl. Environ. Microbiol. 1994;60:3660–3664. doi: 10.1128/aem.60.10.3660-3664.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Martins LO, Santos H. Appl. Environ. Microbiol. 1995;61:3299–3303. doi: 10.1128/aem.61.9.3299-3303.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a Müller V, Spanheimer R, Santos H. Curr. Opin. Microbiol. 2005;8:729–736. doi: 10.1016/j.mib.2005.10.011. [DOI] [PubMed] [Google Scholar]; b Wang YK, Morgan A, Stieglitz K, Stec B, Thompson B, Miller SJ, Roberts MF. Biochemistry. 2006;45:3307–3314. doi: 10.1021/bi052467y. [DOI] [PubMed] [Google Scholar]
- 3.Chen L, Spiliotis ET, Roberts MF. J. Bacteriol. 1998;180:3785–3792. doi: 10.1128/jb.180.15.3785-3792.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodrigues MV, Borges N, Henriques M, Lamosa P, Ventura R, Fernandes C, Empadinhas N, Maycock C, da Costa MS, Santos H. J. Bacteriol. 2007;189:5405–5412. doi: 10.1128/JB.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodionov DA, Kurnasov OV, Stec B, Wang Y, Roberts MF, Osterman AL. Proc. Natl. Acad. Sci. U. S. A. 2007;104:4279–4284. doi: 10.1073/pnas.0609279104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Leeuwen SH, van der Marel GA, Hensel R, van Boom JH. Recl. Trav. Chim. Pays-Bas. 1994;113:335–336. [Google Scholar]
- 7.a Sculimbrene BR, Miller SJ. J. Am. Chem. Soc. 2001;123:10125–10126. doi: 10.1021/ja016779+. [DOI] [PubMed] [Google Scholar]; b Sculimbrene BR, Morgan AJ, Miller SJ. J. Am. Chem. Soc. 2002;124:11653–11656. doi: 10.1021/ja027402m. [DOI] [PubMed] [Google Scholar]; c Sculimbrene BR, Morgan AJ, Miller SJ. Chem. Commun. 2003:1781–1785. doi: 10.1039/b304015c. [DOI] [PubMed] [Google Scholar]
- 8.Masamune S, Choy W, Petersen JS, Sita LR. Angew. Chem. Int. Ed. Engl. 1985;97:1–30. [Google Scholar]
- 9.Billington DC. The Inositol Phosphates: Chemical Synthesis and Biological Significance. VCH; New York: 1993. [Google Scholar]
- 10.Poon KWC, Dudley GB. J. Org. Chem. 2006;71:3923–3927. doi: 10.1021/jo0602773. [DOI] [PubMed] [Google Scholar]
- 11.See SI for details.
- 12.This enzyme is involved in biosynthesis of DIP, removing a phosphate monoester on one of the inositol rings of the DIP precursor to generate the singly charged DIP. It will hydrolyze a wide range of phosphomonoesters and is not specific as to the rest of the molecule. See: Stieglitz KA, Johnson KA, Yang H, Roberts MF, Seaton BA, Head JF, Stec B. J. Biol. Chem. 2002;277:22863–22874. doi: 10.1074/jbc.M201042200.
- 13.Luk H, Roberts MF. Unpublished results.
- 14.For example, two other hydroxylated phosphodiesters have been detected in other hyperthermophiles: α-diglycerophosphate in A. fulgidus under supra-optimal salinities and 1-glyceryl-1-myo-inositylphosphate in Aquifex pyrophilius. See: Gonçalves LG, Huber R, da Costa MS, Santos H H. FEMS Microbiol. Lett. 2003;218:239–244. doi: 10.1111/j.1574-6968.2003.tb11523.x.; Lamosa P, Gonçalves LG, Rodrigues MV, Martins LO, Raven ND, Santos H. Appl. Environ. Microbiol. 2006;72:6169–6173. doi: 10.1128/AEM.00852-06.
- 15.Feng S, Schreiber SL. J. Am. Chem. Soc. 1997;119:10873–10874. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.