

The PPR family protein LRPPRC is implicated in the French Canadian variant of Leigh syndrome, a fatal neurodegenerative disease. LRPPRC functions in posttranscriptional mitochondrial gene expression as part of an RNP complex with SLIRP, a stem-loop RNA-binding protein, to regulate the stability and handling of mature mRNAs.
Abstract
Mutations in LRPPRC are responsible for the French Canadian variant of Leigh syndrome (LSFC), a neurodegenerative disorder caused by a tissue-specific deficiency in cytochrome c oxidase (COX). To investigate the pathogenic mechanism of disease, we studied LRPPRC function in LSFC and control fibroblasts. The level of mutated LRPPRC is reduced in LSFC cells, and this results in decreased steady-state levels of most mitochondrial mRNAs, but not rRNAs or tRNAs, a phenotype that can be reproduced by siRNA-mediated knockdown of LRPPRC in control cells. Processing of the primary transcripts appears normal. The resultant defect in mitochondrial protein synthesis in LSFC cells disproportionately affects the COX subunits, leading to an isolated COX assembly defect. Further knockdown of LRPPRC produces a generalized assembly defect in all oxidative phosphorylation complexes containing mtDNA-encoded subunits, due to a severe decrease in all mitochondrial mRNAs. LRPPRC exists in a high-molecular-weight complex, and it coimmunoprecipitates with SLIRP, a stem-loop RNA-binding protein. Although this interaction does not depend on mitochondrial mRNA, both proteins show reduced stability in its absence. These results implicate LRPPRC in posttranscriptional mitochondrial gene expression as part of a ribonucleoprotein complex that regulates the stability and handling of mature mRNAs.
INTRODUCTION
The French Canadian variant of Leigh syndrome (LSFC) is an early onset, and usually fatal, neurodegenerative disorder caused by mutations in LRPPRC, a leucine-rich protein of the pentatricopeptide repeat family (Mootha et al., 2003). LSFC is a founder effect disease in Quebec, and most patients are homozygous for a single missense mutation predicting a A354V substitution in the protein. The disease is characterized by a tissue-specific decrease in cytochrome c oxidase (COX) activity in which the brain and liver are particularly affected, but the heart, skeletal muscle, and kidneys are relatively spared (Merante et al., 1993); however, the precise role of LRPPRC in the disease mechanism remains unclear. The pentatricopeptide repeat family of proteins is characterized by the presence of a tandemly repeated, degenerate, 35-amino acid motif, which is predicted to form an array of α-helical structures (Schmitz-Linneweber and Small, 2008). This family contains more than 400 members in land plants, and nearly all proteins that have been investigated localize to mitochondria or chloroplasts where they have sequence-specific roles in RNA stability, editing, splicing, processing, or translational control (Schmitz-Linneweber and Small, 2008).
Although the majority of LRPPRC protein localizes to the mitochondrial compartment, it was in fact first identified as a protein present in nuclear ribonucleoprotein (mRNP) complexes (Mili et al., 2001) and only later was shown to bind polyadenylated mitochondrial RNAs, with a preference for polypyrimidine tracts in in vitro assays (Mili and Pinol-Roma, 2003). Mutational analysis showed that the RNA binding activity mapped to the C-terminal region of the protein, in which no known canonical RNA-binding motifs are present and which contains only two of the predicted 11 PPR motifs (Mili and Pinol-Roma, 2003). LRPPRC has also been shown to form a complex with PGC-1α, regulating the expression of genes whose products are involved in gluconeogenesis and mitochondrial respiration, (Cooper et al., 2006) and to act as a modulator of PGC-1 coactivator pathways in brown fat differentiation (Cooper et al., 2008). It has further been associated with the eIF4E-dependent nuclear mRNA export pathway by binding to specific mRNAs (Topisirovic et al., 2009). Whether these nuclear functions of LRPPRC are affected in LSFC patients remains unknown.
PET309, the yeast homologue of LRPPRC is a mitochondrial translational activator that binds specifically to the 5′UTR of the COX1 mRNA, an activity that is essential for the stabilization and efficient translation of this mRNA (Manthey and McEwen, 1995; Naithani et al., 2003). Because mammalian mitochondrial mRNAs do not contain significant 5′UTRs (Montoya et al., 1981), the interaction of LRPPRC with polyadenylated mRNAs in mammals (Mili and Pinol-Roma, 2003) must involve a different mechanism. Mutational analysis of PET309 demonstrated that the PPR motifs themselves are not necessary for the stability of the COX1 mRNA, but that they are essential for its translational activation activity, perhaps by binding the COX1 mRNA in an extended single-stranded conformation (Tavares-Carreon et al., 2008).
A previous report suggested that the mutant form of LRPPRC was not imported correctly into mitochondria in LSFC cells and that it caused a specific decrease in the synthesis of the COX I and COX III polypeptides (but not COX II; Xu et al., 2004), which correlated with a specific decreases in the steady-state levels of both the COX I and COX III mRNAs. However, studies in cells depleted of LRPPRC by siRNA-mediated knockdown have demonstrated that other mitochondrial mRNAs are also decreased (Cooper et al., 2006; Cooper et al., 2008). To investigate the function of LRPPRC in detail, we studied its function in several patient cell lines; used siRNA to knock it down in control and patient cells, and performed immunoprecipitation (IP) experiments and mass spectrometry analysis to identify interacting protein partners.
MATERIALS AND METHODS
Cell Culture and Small Interfering RNA Transfection
Control and patient fibroblasts were grown in high-glucose DMEM supplemented with 10% FBS, at 37°C and 5% CO2. LSFC fibroblasts were obtained from the LSFC Consortium Biobank (Université du Québec, Chicoutimi, QC, Canada) and from the fibroblast cell bank at Hôpital Ste. Justine (Montreal). All reported LSFC patients, except one, are homozygous for a common C1119T transition (A354V). One patient is a compound heterozygote for the common mutation in LRPPRC and an 8-base pair deletion in exon 35. Fibroblasts were immortalized by transduction with a retroviral vector expressing the HPV-16 E7 gene and with another one expressing the catalytic component of human telomerase (htert; Yao and Shoubridge, 1999).
For the knockdown experiments, two Stealth RNA interference (RNAi) duplexes constructs for LRPPRC (Invitrogen, Carlsbad, CA) were used (Stealth1173: AAA UGG AUG UCU GUC UGA UAG UGA U and Stealth3017: AAA UAA UCC CGC CUA ACU UGC GUU A). Stealth RNAi duplexes or the fluorescent oligo control Block-iT Alexa FluorRed (Invitrogen) were transiently transfected at a final concentration of 10 nM using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer's specifications. Transfection was repeated on day 3, and cells were harvested and analyzed on day 6.
COX Activity Measurements
A spectrophotometric assay of whole cell extracts was used to measure the enzymatic activity of COX in control and patient fibroblasts (Capaldi et al., 1995; Antonicka et al., 2003). COX activity was normalized to the activity of citrate synthase and specific COX activity was calculated after measuring the protein concentration of the samples by the Bradford assay.
Blue-Native PAGE and Immunoblotting
Mitoplasts were prepared from fibroblasts by treatment with 0.8 mg of digitonin per milligram of protein and then solubilized with 1% lauryl maltoside, after which samples containing 20 μg solubilized protein were run in the first dimension on 6–15% polyacrylamide gradient gels as described in detail elsewhere (Leary and Sasarman, 2009). For the second-dimension analysis, Blue-Native (BN)-PAGE/SDS-PAGE was carried out as detailed previously (Antonicka et al., 2003). Immunodetection in the first and second dimensions was performed with monoclonal antibodies against structural subunits of complexes I-V (Molecular Probes, Eugene, OR), with the exception of the ND1 subunit of complex I, for which a polyclonal antibody was used (a kind gift from A. Lombes, INSERM, Paris, France).
For immunoblotting, whole cells or mitochondria were extracted in 1.5% lauryl maltoside/PBS, and 10 or 20 μg of protein per sample were loaded and run on 12 or 15% polyacrylamide gels, then transferred to a nitrocellulose membrane, and used for the detection of COX I, COX II, COX IV, or CoII-70 kDa with monoclonal antibodies from Molecular Probes, or with a mAb against porin (Sigma, St. Louis, MO). LRPPRC polyclonal antibodies were prepared by immunizing rabbits with peptides of 22 amino acids corresponding to the sequence CEPPESFEFYAQQLRKLRENSS (antibody 295–313; Zymed Laboratories, San Francisco, CA), and used for immunoblotting at a dilution of 1:3000. A polyclonal antibody against SLIRP (sRA-stem loop interacting RNA-binding protein) was purchased from Abcam (Cambridge, MA) and used at a dilution of 1:1000.
35S-Labeling of Mitochondrial Translation Products
Pulse-labeling of mitochondrial translation products in control and patient fibroblasts was performed with a 200 μCi/ml [35S]methionine/cysteine mix (PerkinElmer-Cetus, Woodbridge, ON, Canada), in DMEM lacking methionine and cysteine and containing 100 μg/ml of a cytoplasmic translation inhibitor (emetine or anisomycin), for 60 min, followed by chasing of the label, as described in detail elsewhere (Leary and Sasarman, 2009).
Immunofluorescence Analysis
Cultured control and patient fibroblasts were fixed at 50% confluency in 4% paraformaldehyde and then incubated with the polyclonal anti-LRPPRC antibody (dilution 1:500) or with the monoclonal anti-cytochrome c antibody (dilution 1:500). The appropriate anti-species secondary antibodies coupled with Alexa fluorochromes (Invitrogen) were subsequently used at a dilution of 1:1000. Images were obtained on a confocal microscope.
Northern Blotting and Northern-PAGE Analysis
Northern blotting was carried out essentially as described previously (Weraarpachai et al., 2009). For Northern-PAGE analysis of mitochondrial (mt) tRNAs, 5 μg total RNA from control and patient fibroblasts were run on a 10% polyacrylamide gel containing 7 M urea, followed by transfer to Hybond N+ membrane (GE Healthcare, Mississauga, ON, Canada). Prehybridization and hybridization were carried out in EXPRESSHyb solution (Clontech, Palo Alto, CA) according to the manufacturer's instructions. The oligonucleotides used for the generation of the 32P-labeled probes by the end-labeling technique had the following sequences: 5′tggtattctcgcacggactacaac3′ for mt tRNAGlu; 5′tggctaggactatgagaatcgaac3′ for mt tRNAGln; 5′tggtcactgtaaagaggtgttggt3′ for mt tRNALys, 5′tggcagaaattaagtattgcaact3′ for mt tRNATrp, 5′tggtcagagcggtcaagttaagtt3′ for mt tRNAVal, and 5′tggtggttccctgaccgggaatcg3′ for cytoplasmic tRNAcyt Glu.
Mitochondrial Isolation for Immunoblotting and Immunoprecipitation Experiments
Control fibroblasts, 143B human osteosarcoma cells or 143B cells lacking mtDNA (rho0) were resuspended in ice-cold SET buffer (250 mM sucrose, 2 mM EDTA, 10 mM Tris-HCl, pH 7.4) containing protease inhibitors (complete cocktail tablets from Roche, Laval, QC, Canada), and homogenized with 10 strokes of a prechilled, zero-clearance homogenizer (Kimble/Kontes, Vineland, NJ). Samples were then centrifuged twice at 650 × g for 10 min at 4°C, and mitochondria were pelleted from the supernatant by centrifugation at 12,000 × g for 20 min at 4°C, followed by one wash in cold SET buffer. Mitochondrial fractions were then extracted either in lauryl maltoside or in taurodeoxycholate, as described in the sections on immunoblotting and IP, respectively.
Size Exclusion Chromatography
Soluble proteins from mitochondrial extracts were fractionated on a Tricorn Superdex 200 10/30 HR column (GE Healthcare) as described (Kaufman et al., 2007), and the elution profile of LRPPRC was determined by immunoblot analysis of the fractions with the polyclonal anti-LRPPRC antibody described above.
IP Experiments
For each IP reaction, mitochondrial protein isolated from control fibroblasts (350 μg) or from 143B rho0 cells (450 μg) was extracted in 100 μl extraction buffer (50 mM HEPES buffer, pH 7.6, 150 mM NaCl) and 1% taurodeoxycholate in the presence of complete protease inhibitors (Roche), on ice, for 45 min with occasional vortexing. The extract was subsequently centrifuged at 25,000 × g at 4°C, for 40 min, and the supernatant was used to immunoprecipitate LRPPRC or SLIRP with the antibodies described earlier. Immunoprecipitation was performed with Dynabeads protein A (Invitrogen) according to the manufacturer's instructions (version no. 004), except that the incubation of the antibody with the beads and the incubation of the extract with antibody cross-linked to the beads were both carried out overnight. The IP fractions were then analyzed by immunoblotting, and the eluates were sent for mass spectrometry analysis (Orbitrap, Thermo Scientific, Waltham, MA) at the Institut de Recherches Cliniques de Montréal. Because our starting material was isolated mitochondria, we did not expect to identify nonmitochondrial proteins previously reported to interact with LRPPRC by this analysis.
To isolate RNA after the IP reaction, the beads were washed five times with extraction buffer and then incubated for 30 min at 37°C with DNase I, followed by a 30-min incubation at 55°C with proteinase K in the presence of 0.1% SDS. Samples were then supplemented with EDTA (5 mM), SDS (to 1%), and 2 μg of yeast tRNA and incubated for 15 min at room temperature. The magnetic beads were then discarded, and phenol-chloroform extraction of RNA was subsequently performed. The resulting RNA was treated again with DNase I, followed by a second round of phenol-chloroform extraction. RT-PCR experiments were performed using OneStep RT-PCR (QIAGEN, Chatsworth, CA) with primers specific for COX I and COX II.
RESULTS
COX Assembly Defect in LSFC Fibroblasts
We first measured the enzymatic activity of COX in immortalized fibroblasts from eight LSFC patients and four controls. COX activity in patient cells was decreased to 36 ± 7% of control values (range 28–45%), similar to previous reports (Merante et al., 1993). Analysis of the oxidative phosphorylation (OXPHOS) complexes by BN-PAGE showed a specific defect in COX assembly in LSFC fibroblasts (Figure 1A). Immunoblot analysis showed a marked decrease in the steady-state levels of the mtDNA-encoded subunits COX I and COX II and a smaller decrease in the levels of the nuclear-encoded COX IV in patient fibroblasts (Figure 1B). The steady-state level of mutant LRPPRC protein in patient fibroblasts was reduced to <30% of control levels (Figure 1B), as shown previously (Xu et al., 2004).
Figure 1.

The COX assembly defect in LSFC fibroblasts is accompanied by a decreased synthesis of several mitochondria-encoded polypeptides. Control and LSFC fibroblasts were analyzed by BN-PAGE (A) and by Western blotting (B). In A, each of the five OXPHOS complexes (Co I–V) was visualized with a subunit-specific antibody. In B, the blots were incubated with antibodies against the proteins indicated at the right of the panel. The 70-kDa subunit of complex II was used as a loading control. (C) Control and LSFC fibroblasts were pulse-labeled with [35S]methionine and cysteine in the presence of anisomycin, an inhibitor of cytoplasmic protein synthesis, and subsequently were chased for 10 min (PULSE) or overnight (CHASE). Fifty micrograms of total protein were run on a 15–20% polyacrylamide gradient gel. The seven subunits of complex I (ND), one subunit of complex III (cyt b), three subunits of complex IV (COX), and two subunits of complex V (ATP) are indicated at the left of the figure. (D) Quantification of the synthesis of individual mitochondria-encoded polypeptides in fibroblasts from eight LSFC patients (four fibroblast lines in triplicate; four lines as a single experiment).
Defect in Mitochondrial Protein Synthesis in LSFC Fibroblasts
To test whether the observed reduction in steady-state levels of mitochondrial-encoded COX subunits was a consequence of a decrease in their synthesis or stability, we pulse-labeled the mitochondrial translation products in control and LSFC fibroblasts with [35S]methionine/cysteine and chased the label either for 10 min (pulse experiment) or overnight (chase experiment). The pulse experiment can detect defects in protein synthesis, whereas the chase experiment reports on the stability of the newly synthesized polypeptides. A decrease in the synthesis of all three mitochondrial COX subunits, particularly COX I and COX II, was observed in LSFC fibroblasts (Figure 1C). In some patients (e.g., patient 1 in Figure 1C), a defect in the synthesis of some of the other mtDNA-encoded polypeptides was also observed, but it was less severe than that seen in the COX subunits. Quantification of the defect in the rate of synthesis of all mtDNA-encoded polypeptides in eight LSFC fibroblast lines is summarized in Figure 1D. These results are contrary to those in a previous study in which a specific defect in the synthesis of COX I and COX III was reported (Xu et al., 2004). Interestingly, COX II appeared to be preferentially stabilized in the chase in LSFC cells. A similar pattern of a defect in the synthesis of COX II coupled with its stabilization was previously reported in a patient with a mutation in SCO2, encoding a COX assembly factor, in which the levels of mutant SCO2 protein are severely reduced (Leary et al., 2009). However, we found control levels of SCO2 protein in LSFC fibroblasts (data not shown).
Mutant LRPPRC Localizes to Mitochondria in LSFC Fibroblasts
We next investigated whether the mutation in the LRPPRC protein interferes with its import into mitochondria, resulting in its accumulation in the cytoplasm, as was previously suggested (Xu et al., 2004). Immunofluorescence analysis in control and LSFC fibroblasts using antibodies against LRPPRC and cytochrome c showed that the protein perfectly colocalized with mitochondria in both control and LSFC cells. It could not be detected outside the mitochondrial compartment in LSFC cells, indicating that the mutant protein is correctly imported (Figure 2), and not mistargeted. The level of the protein is, however, reduced, and it is distributed unevenly in a fragmented mitochondrial network in LSFC fibroblasts. We cannot rule out the possibility that the mutation in LRPPRC interferes with the efficiency of import and that this contributes to the decreased steady-state level in mitochondria, but we consider it unlikely as the mutation does not alter the putative mitochondrial targeting sequence.
Figure 2.

Localization of mutant LRPPRC to mitochondria in LSFC fibroblasts. Fibroblasts from control and one LSFC patient were grown on coverslips and incubated with antibodies against LRPPRC and against cytochrome c followed by incubation with secondary antibodies coupled to red and green fluorescent dyes, respectively. The overlay of these two images demonstrates the mitochondrial localization of mutant LRPPRC in patient fibroblasts and confirms the reduction in mutant LRPPRC levels in LSFC fibroblasts.
The Steady-State Levels of Mitochondrial mRNAs Are Decreased in LSFC Fibroblasts
Given the postulated role of LRPPRC in RNA binding, the mitochondrial translation defect in LSFC cells is likely to be a consequence of a decrease in the levels of mitochondrial mRNAs. To investigate this, Northern blotting analysis was carried out for the three COX subunits and for several Complex I and Complex V subunits in four LSFC lines (Figure 3). We found a global reduction in the levels of all tested mitochondrial mRNAs except ND3 and ND6 (ND6 is not shown in the figure). The levels of the two mitochondrial rRNAs were normal in LSFC cells (Figure 3; see also Figure 5), as well as five different mitochondrial tRNAs spanning the mtDNA genome (see Figure 6), suggesting a specific role for LRPPRC in the binding/stabilization of mitochondrial mRNAs. Mature mitochondrial mRNAs are generated by the processing of large polycistronic transcripts, which are transcribed from the light and heavy strands of mtDNA. To test whether the decrease in mitochondrial mRNAs in LSFC cells could be the result of altered processing of the polycistronic mitochondrial transcripts, we used multiple primer pairs spanning the mitochondrial genome. This permitted us to search for the presence of increased levels of unprocessed transcripts, but we did not detect a difference between control and LSFC cell lines (data not shown), suggesting that LRPPRC function is limited to mature mRNAs.
Figure 3.

Reduction in the steady-state levels of mitochondrial mRNAs in LSFC fibroblasts. (A) Northern blot analysis was carried out with total RNA extracted from control and LSFC fibroblasts. Hybridization was performed with probes specific for the mitochondrial mRNAs encoding the three COX subunits, four of the complex I subunits (ND), the bicistronic mRNA encoding the complex V subunits (ATP6/8) and, as a loading control, with a probe for the 12S mitochondrial rRNA. (B) Quantification of mitochondrial mRNA levels in fibroblasts from four LSFC patients normalized to 12S RNA levels (3–8 replicates per mRNA).
Figure 5.

Generalized decrease in mitochondrial translation and in the levels of mitochondrial mRNAs in control and LSFC fibroblasts after LRPPRC knockdown. Fibroblasts from one control and one LSFC patient were transiently transfected either with one of two different siRNA constructs specific to LRPPRC (S1173 and S3017) or with a fluorescent control siRNA (Alexa). On day 6, transfected and untransfected cells were either pulse-labeled with [35S]methionine and cysteine (A) or total RNA was extracted and analyzed by Northern blot (B). (A) The seven subunits of complex I (ND), one subunit of complex III (cyt b), three subunits of complex IV (COX), and two subunits of complex V (ATP) are indicated at the left of the figure. (B) Hybridization was performed with probes specific for the mitochondrial mRNAs encoding the three COX subunits, the ND1 subunit of complex I, and the bicistronic mRNA encoding the complex V subunits (ATP6/8) and with probes for the 12S and 16S mitochondrial rRNAs.
Figure 6.
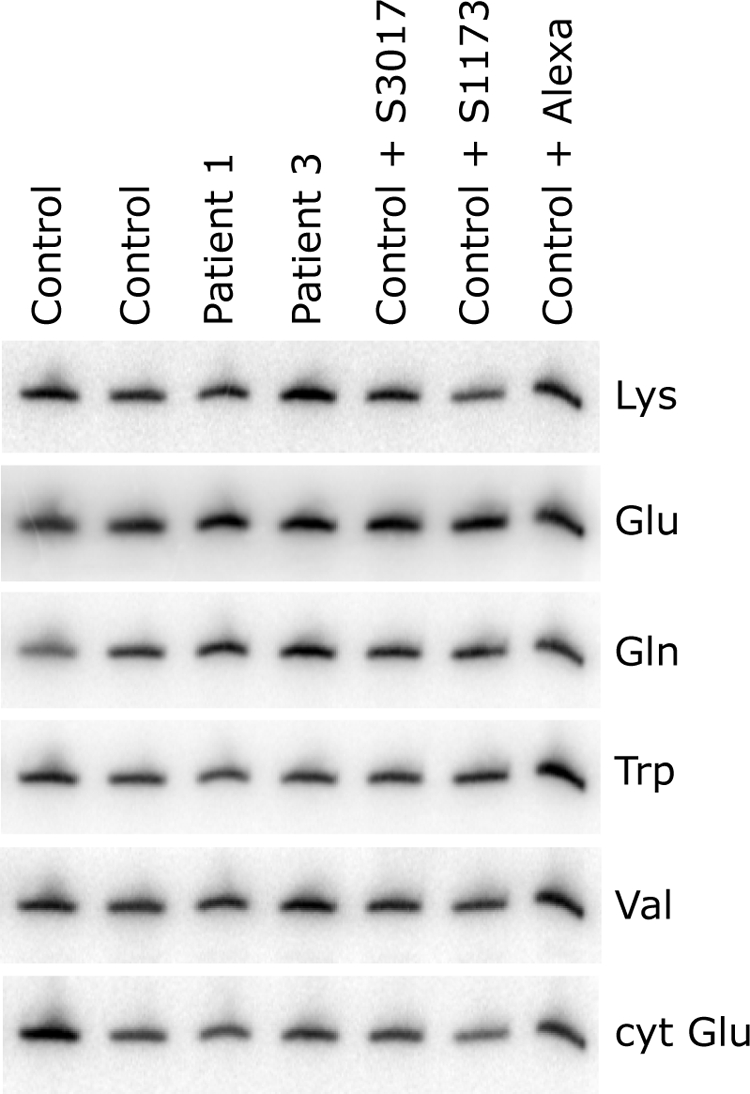
Normal levels of mitochondrial tRNAs in fibroblasts from LSFC patients and in fibroblasts in which LRPPRC was knocked down. Total RNA was extracted from control cells transiently transfected either with one of two different siRNA constructs specific to LRPPRC (S1173 and S3017) or with a fluorescent control siRNA (Alexa) and from untransfected controls and LSFC patients, after which 5 μg total RNA/sample were run on a 10% polyacrylamide gel containing 7 M urea. After transfer to membrane, hybridization was performed with oligonucleotide probes complementary to the mitochondrial tRNAs for Lys, Glu, Gln, Trp, and Val and, as a loading control, the cytoplasmic tRNA for Glu (cyt Glu), as indicated at the right of the figure.
Steady-State Level of LRPPRC Determines the Extent of the OXPHOS Defect
The molecular defects in LSFC fibroblasts could result from an impaired function of the mutant LRPPRC protein, or from a decrease in its steady-state level. To distinguish between these two possibilities, we knocked down LRPPRC in control and patient fibroblasts using two different small interfering RNA (siRNA; Stealth) constructs. In control cells in which LRPPRC was reduced by 70% (Stealth S3017, Figure 4B), similar to LSFC cells, steady-state levels of the COX subunits (Figure 4B), mitochondrial mRNA levels (Figure 5B), mitochondrial protein synthesis (Figure 5A), and assembly of the OXPHOS complexes (Figure 4A) were indistinguishable from those in LSFC fibroblasts. When the levels of LRPPRC were reduced to <10% of normal in either control or LSFC cells (Stealth S1173, Figure 4B), the reduction in the steady-state levels of mitochondrial mRNAs (Figure 5B), translation products (Figure 5A), and assembled OXPHOS complexes (Figure 4A) was severe and generalized. The levels of mitochondrial rRNAs (Figure 5B) or tRNAs (Figure 6) were not affected in any of the knockdown experiments. We conclude that the phenotype in LSFC fibroblasts is a consequence of the reduced levels of LRPPRC protein caused by the mutation and that the nature and extent of the OXPHOS deficiency varies as a function of the level of LRPPRC.
Figure 4.

Decreased assembly of the OXPHOS complexes in control and LSFC fibroblasts after knockdown of LRPPRC. Control and LSFC fibroblasts were transiently transfected with one of two different siRNA constructs specific to LRPPRC (S1173 and S3017) or with a fluorescent control siRNA (Alexa) and analyzed by BN-PAGE (A) and SDS-PAGE (B).
LRPPRC Interacts with SLIRP (Steroid Receptor RNA Activator-Stem Loop Interacting RNA-binding Protein) as Part of a High-Molecular-Weight Complex
To learn more about the mechanism of action of LRPPRC, we searched for interacting protein partners of LRPPRC. We first performed size-exclusion chromatography and detected LRPPRC predominantly in a high-molecular-weight complex of approximately 300 kDa (Figure 7A), suggesting that LRPPRC might interact with other protein partners. To investigate this, we carried out IP experiments with an antibody specific to human LRPPRC, followed by mass spectrometry analysis. The highest scoring protein that coimmunoprecipitated with LRPPRC was SLIRP, a protein with a molecular weight of approximately 12 kDa that was recently shown to be involved in mitochondrial RNA homeostasis (Baughman et al., 2009) and that was first described as a nuclear receptor corepressor (Hatchell et al., 2006). To validate our findings, we repeated the IP with the anti-LRPPRC antibody and performed the reciprocal IP, using an anti-SLIRP antibody. As shown in Figure 7B, in both IP reactions, LRPPRC and SLIRP are coordinately depleted in the unbound fractions and recovered in the eluates, confirming an interaction between these two proteins.
Figure 7.

LRPPRC exists in a higher molecular weight complex and interacts with SLIRP. (A) Mitochondria from control fibroblasts were separated by size exclusion and the individual fractions were analyzed by SDS-PAGE. The molecular weights of the individual fractions were calculated from the elution profile of a set of standards. (B) Coimmunoprecipitation of LRPPRC and SLIRP. Mitochondria from control fibroblasts were extracted with 1% sodium deoxycholate, and the extract (preclear fraction) was incubated overnight with naked beads to reduce nonspecific binding to the beads during the subsequent IP reaction. The cleared extract (input fraction) was divided among naked beads (control IP), beads cross-linked with anti-LRPPRC antibody (LRPPRC IP), and beads cross-linked with anti-SLIRP antibody (SLIRP IP). “Unbound” refers to the fractions collected after the IP reactions were allowed to proceed overnight. Subsequently, the beads were washed (wash fractions) and eluted with acidic glycine (eluate fractions). Individual fractions were then analyzed by immunoblotting with antibodies against LRPPRC and SLIRP. (C) The LRPPRC–SLIRP complex contains mitochondrial mRNAs. Immunoprecipitation from control mitochondrial extracts was performed with the antibody against LRPPRC as described in B, and RNA was then extracted from the immunoprecipitate (pellet) and unbound (supernatant) fractions and was used to amplify COX I and COX II mRNA sequences by RT-PCR. Mitochondrial mRNA, but not mtDNA, was specifically found in the LRPPRC immunoprecipitate. (D) LRPPRC and SLIRP are detected as part of the same high-molecular-weight complex on 2D-PAGE. Extracts of control and LSFC fibroblasts were run on a nondenaturing gel (BN-PAGE) after which the individual lanes were excised and run on a second, denaturing gel (SDS-PAGE). Antibodies against the subunits of all five OXPHOS complexes, LRPPRC, and SLIRP were used, as indicated at the right of the gel. In the upper middle panel analysis of three subunits of COX shows reduced levels in LSFC cells. The lower middle panel was probed for the indicated complexes after stripping. The COX I signal is the residual signal from the upper middle blot. The sizes of the LRPPRC–SLIRP-containing complexes are indicated at the top of the figure.
To confirm that the protein complex containing LRPPRC and SLIRP also contains mitochondrial mRNAs, we performed an IP experiment with the anti-LRPPRC antibody and extracted RNA from the immunoprecipitate (pellet) and from the fraction recovered after the IP reaction was complete (supernatant). Using RT-PCR, we then looked for the presence of two different mitochondrial mRNAs, COX I and COX II, after treatment with DNase. As shown in Figure 7C, COX I and COX II sequences were amplified by RT-PCR from the anti-LRPPRC immunoprecipitate, but not from the control.
To further verify our conclusions, control and LSFC fibroblast extracts were run on a nondenaturing BN-PAGE, after which individual lanes were excised and run in a second, denaturing dimension (SDS-PAGE). This analysis allows the separation, in the second dimension, of proteins that run as part of a complex in the first, nondenaturing dimension. In both the control and the LSFC patient, SLIRP and LRPPRC are predominantly detected as components of a complex of approximately 250 kDa, slightly larger than the COX dimer at 230 kDa (Figure 7D). Lower amounts of LRPPRC and SLIRP are found in higher molecular weight complexes of about 500 kDa and 1 MDa, which may represent oligomers of the 250 kDa complex. This analysis further shows that the levels of SLIRP are reduced in the LSFC patient, consistent with a codependent interaction between SLIRP and LRPPRC. In fact, immunoblot analysis of two LSFC patients along with two patients with uncharacterized COX deficiencies, but which have reduced levels of LRPPRC, shows that all four patients contain low endogenous levels of SLIRP (Figure 8A). These results provide strong evidence for an interaction between LRPPRC and SLIRP. To test whether the levels of LRPPRC and SLIRP depended on the presence of mRNA, we examined the steady-state levels of the proteins in 143B rho0 cells, which do not contain any mtDNA and therefore no mitochondrial mRNAs (Figure 8B). The levels of both proteins, and especially SLIRP, were markedly reduced in these cells, and subcellular fractionation experiments showed that the remaining protein was enriched in the mitochondrial fraction. To investigate whether mitochondrial mRNAs are necessary for the LRPPRC–SLIRP interaction, we immunoprecipitated SLIRP in 143B rho0 cells and tested for the presence of LRPPRC. All of the residual SLIRP bound to the anti-SLIRP antibody and coimmunoprecipitated with LRPPRC (Figure 8C), confirming that the interaction between the two proteins does not depend on mRNA.
Figure 8.

LRPPRC and SLIRP are interdependent and show reduced stability in the absence of mitochondrial mRNA. (A and B) Western blot analyses of SLIRP and LRPPRC in patients with reduced endogenous levels of LRPPRC (A) and human 143B rho0 cells (whole cells or mitochondrial fractions as indicated; B) show coordinate decreases in the levels of SLIRP and LRPPRC. Antibodies against the 70-kDa subunit of complex II and against porin were used as loading controls. The lanes labeled COX patients represent patients with uncharacterized COX deficiencies. (C) Coimmunoprecipitation of LRPPRC and SLIRP from mitochondrial extracts of rho0 cells. Mitochondria from human rho0 cells were extracted and used for IP reactions with the anti-SLIRP antibody as described in Figure 7B. The individual fractions identified at the top of the gel were analyzed by immunoblotting with the anti-LRPPRC and anti-SLIRP antibodies.
DISCUSSION
This study demonstrates that LRPPRC and SLIRP interact in a high-molecular-weight complex of at least 250 kDa, that, when disrupted, results in a decrease in the steady-state levels of all mitochondrial mRNAs. Both proteins are known to bind RNA, and we were able to detect mitochondrial mRNAs in the immunoprecipitated complex. Because we found no evidence for mitochondrial mRNA processing defects in LSFC fibroblasts, it is reasonable to propose that both are part of an mRNP complex that regulates the stability and handling of mature mRNAs. Supporting this conclusion, SLIRP was recently identified in a screen for OXPHOS regulators and shown to be important for mitochondrial mRNA stability (Baughman et al., 2009). The stability of both LRPPRC and SLIRP depends on the presence of mRNA, as demonstrated by the marked reduction in both proteins in rho0 cells. This is consistent with a previous report showing a reduction in LRPPRC in response to RNAi-mediated knockdown of SLIRP, and a decrease in SLIRP in cells treated with ethidium bromide (Baughman et al., 2009).
Interestingly, both LRPPRC and SLIRP were originally characterized as nuclear RNA-binding proteins, although immunofluorescence and subcellular fractionation studies clearly show that the vast majority of both proteins is targeted to the mitochondrial compartment (Mili and Pinol-Roma, 2003; Xu et al., 2004; Hatchell et al., 2006; this study). The molecular basis for the dual targeting of these proteins remains unknown. Although this is the only study to date in which an interacting protein partner has been reported for mitochondrial LRPPRC, a stable physical interaction has been reported between nuclear LRPPRC and the translation initiation factor 4E (eIF4E), which modulates the export of eIF4E-sensitive mRNAs from the nucleus (Topisirovic et al., 2009) and also with the coactivator PGC-1α (Cooper et al., 2008). LRPPRC contains at least two binding sites for eIF4E, defined by a YxxxxLΦ motif, where x is any amino acid and Φ is any hydrophobic residue (Topisirovic et al., 2009). The domains promoting the interaction between LRPPRC and SLIRP remain to be characterized.
Recent studies on the nuclear function of LRPPRC suggest that it could be part of a pathway that coordinates the expression of nuclear-encoded mitochondrial proteins, with some evidence that it binds to the promoters of these genes, such as that coding for the uncoupling protein UCP1 (Cooper et al., 2008). This model would predict that large decreases in LRPPRC, like that we produced with one of the siRNAs, would have dramatic consequences on the mitochondrial transcriptome. However, we failed to find any evidence of this using RNA expression arrays (Illumina; Ambion, Austin, TX; data not shown) to analyze patient and control cells with different levels of LRPPRC knockdown. In particular, we saw no obvious coordinate changes in expression of PGC-1α targets (data not shown). This may of course reflect the fact that we only investigated skin fibroblasts, and it does not rule out such a role for LRPPRC in other cell types, such as brown fat (Cooper et al., 2008). It is not known whether the nuclear functions of LRPPRC are significantly perturbed by the mutations found in LSFC patients.
SLIRP was originally identified in a screen for proteins that interact with steroid receptor RNA activator (SRA), which is an RNA species that acts as a nuclear receptor coactivator (Hatchell et al., 2006). SLIRP binds to a substructure on SRA and acts as a corepressor of nuclear receptor transactivation, an activity that depends on an intact RRM RNA-binding motif (Hatchell et al., 2006). Unexpectedly, this activity depends on the presence of an intact mitochondrial targeting signal peptide in SLIRP, suggesting that the mitochondrial localization of SLIRP is crucial to its nuclear corepressor activity. Although there are reports of nuclear hormone receptors in mitochondria (Colley and Leedman, 2009), it is unclear how, or if, this is important in the transactivation function of these receptors.
A previous report of LRPPRC function in LSFC cells (Xu et al., 2004) is clearly at odds with the results we present here. Although we confirm that the missense mutation in LRPPRC results in reduced levels of the mutant protein, it appears to be targeted normally to the mitochondrial compartment. Our results indicate that most mRNAs are decreased in LSFC cells, consistent with results reported in Cooper et al. (2008) and that the synthesis of several of the mtDNA polypeptides is decreased. However, the translation of the COX subunit mRNAs, and particularly COX II, is disproportionately affected in patient cells, and this presumably accounts for the specific decrease in the assembly of COX and the enzymatic deficiency in LSFC patients. The synthesis of the polypeptide labeled as COX II in (Xu et al., 2004) is not affected in their mitochondrial translation assay, but the labeling pattern in their experiment is not similar to any published analysis of mammalian mitochondrial translation.
It is not yet clear why the steady-state levels of the COX mRNAs are apparently more sensitive than most to decreases in LRPPRC in LSFC cell lines or why their translation is disproportionately decreased. It could relate to differences in the regulation of the posttranscriptional processing or translation of individual mRNAs. In yeast, a family of translational activator proteins exists to promote specific translation of individual mRNAs by binding to sequences in the 5′UTR (Naithani et al., 2003). These proteins do not share any homology, and in fact PET309 is the only PPR protein among them. Mammalian mitochondrial mRNAs lack significant 5′UTRs, so other mechanisms must exist to promote their translation. We recently identified TACO1 as a specific translational activator of COX I, implying that the handling of individual mitochondrial mRNAs in mammals may also require a set of unique, and mRNA-specific proteins (Weraarpachai et al., 2009). How or whether these proteins interact with the LRPPRC–SLIRP RNP complex is unknown. We did not identify TACO1 in the IP experiments we performed in this study, and conversely we have not been able to find any evidence for the presence of either LRPPRC or SLIRP in TACO1 immunoprecipitates (unpublished data). Nevertheless, the interactions of translational activator proteins with RNP complexes could be weak or transient, so these results do not rule out that possibility.
Most PPR proteins have sequence-specific RNA binding activity, but how these proteins recognize their specific targets is essentially unknown. Other than LRPPRC, six additional mammalian PPR proteins have been identified, all of which localize to mitochondria (Davies et al., 2009), and at least three of these are involved in specific mitochondrial posttranscriptional processing or translation events. PTCD1 associates with tRNA leucine and acts as a negative modulator of translation by reducing the abundance of the leucine tRNAs (Rackham et al., 2009), PTCD2 appears to be important in the maturation of the cytochrome b mRNA (Xu et al., 2008), and PTCD3 binds 12S rRNA, an interaction that is necessary for efficient mitochondrial protein synthesis (Davies et al., 2009). LRPPRC appears to be more promiscuous in its choice of RNA-binding partners as it apparently binds most, if not all, 13 mitochondrial mRNAs, in addition to a subclass of nuclear mRNAs. Further studies will be required to determine the RNA recognition specificities for LRPPRC and to determine how the mitochondrial mRNP complex interacts with the mitochondrial translation apparatus.
ACKNOWLEDGMENTS
This research was supported in part by grants from L'Association de l'Acidose Lactique and the Canadian Institutes for Health Research (CIHR). We thank Dr. Grant Mitchell (Université de Montréal) for providing LSFC cell lines from the fibroblast cell bank at Hôpital Ste. Justine, Montreal. E.A.S. is an International Scholar of the Howard Hughes Medical Institute (HHMI). The LSFC Consortium is a group of scientists and clinicians dedicated to investigating the causes of and developing treatments for LSFC. The members of the consortium are, in alphabetical order: Bruce Allen, Yan Burelle, Guy Charron, Lise Coderre, Christine DesRosiers, Catherine Laprise, Charles Morin, John Rioux, and Eric A. Shoubridge.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-01-0047) on March 3, 2010.
REFERENCES
- Antonicka H., Leary S. C., Guercin G. H., Agar J. N., Horvath R., Kennaway N. G., Harding C. O., Jaksch M., Shoubridge E. A. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003;12:2693–2702. doi: 10.1093/hmg/ddg284. [DOI] [PubMed] [Google Scholar]
- Baughman J. M., Nilsson R., Gohil V. M., Arlow D. H., Gauhar Z., Mootha V. K. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009;5:e1000590. doi: 10.1371/journal.pgen.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldi R. A., Marusich M. F., Taanman J. W. Mammalian cytochrome-c oxidase: characterization of enzyme and immunological detection of subunits in tissue extracts and whole cells. Methods Enzymol. 1995;260:117–132. doi: 10.1016/0076-6879(95)60134-1. [DOI] [PubMed] [Google Scholar]
- Colley S. M., Leedman P. J. SRA and its binding partners: an expanding role for RNA-binding coregulators in nuclear receptor-mediated gene regulation. Crit. Rev. Biochem. Mol. Biol. 2009;44:25–33. doi: 10.1080/10409230802661719. [DOI] [PubMed] [Google Scholar]
- Cooper M. P., Qu L., Rohas L. M., Lin J., Yang W., Erdjument-Bromage H., Tempst P., Spiegelman B. M. Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC-1α/LRP130 complex. Genes Dev. 2006;20:2996–3009. doi: 10.1101/gad.1483906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper M. P., Uldry M., Kajimura S., Arany Z., Spiegelman B. M. Modulation of PGC-1 coactivator pathways in brown fat differentiation through LRP130. J. Biol. Chem. 2008;283:31960–31967. doi: 10.1074/jbc.M805431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S. M., Rackham O., Shearwood A. M., Hamilton K. L., Narsai R., Whelan J., Filipovska A. Pentatricopeptide repeat domain protein 3 associates with the mitochondrial small ribosomal subunit and regulates translation. FEBS Lett. 2009;583:1853–1858. doi: 10.1016/j.febslet.2009.04.048. [DOI] [PubMed] [Google Scholar]
- Hatchell E. C., et al. SLIRP, a small SRA binding protein, is a nuclear receptor corepressor. Mol. Cell. 2006;22:657–668. doi: 10.1016/j.molcel.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Kaufman B. A., Durisic N., Mativetsky J. M., Costantino S., Hancock M. A., Grutter P., Shoubridge E. A. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol. Biol. Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary S. C., Sasarman F. Oxidative phosphorylation: synthesis of mitochondrially encoded proteins and assembly of individual structural subunits into functional holoenzyme complexes. Methods Mol. Biol. 2009;554:143–162. doi: 10.1007/978-1-59745-521-3_10. [DOI] [PubMed] [Google Scholar]
- Leary S. C., Sasarman F., Nishimura T., Shoubridge E. A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009;18:2230–2240. doi: 10.1093/hmg/ddp158. [DOI] [PubMed] [Google Scholar]
- Manthey G. M., McEwen J. E. The product of the nuclear gene PET309 is required for translation of mature mRNA and stability or production of intron-containing RNAs derived from the mitochondrial COX1 locus of Saccharomyces cerevisiae. EMBO J. 1995;14:4031–4043. doi: 10.1002/j.1460-2075.1995.tb00074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merante F., Petrova-Benedict R., MacKay N., Mitchell G., Lambert M., Morin C., De Braekeleer M., Laframboise R., Gagne R., Robinson B. H. A biochemically distinct form of cytochrome oxidase (COX) deficiency in the Saguenay-Lac-Saint-Jean region of Quebec. Am J. Hum. Genet. 1993;53:481–487. [PMC free article] [PubMed] [Google Scholar]
- Mili S., Pinol-Roma S. LRP130, a pentatricopeptide motif protein with a noncanonical RNA-binding domain, is bound in vivo to mitochondrial and nuclear RNAs. Mol. Cell. Biol. 2003;23:4972–4982. doi: 10.1128/MCB.23.14.4972-4982.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mili S., Shu H. J., Zhao Y., Pinol-Roma S. Distinct RNP complexes of shuttling hnRNP proteins with pre-mRNA and mRNA: candidate intermediates in formation and export of mRNA. Mol. Cell. Biol. 2001;21:7307–7319. doi: 10.1128/MCB.21.21.7307-7319.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya J., Ojala D., Attardi G. Distinctive features of the 5′-terminal sequences of the human mitochondrial mRNAs. Nature. 1981;290:465–470. doi: 10.1038/290465a0. [DOI] [PubMed] [Google Scholar]
- Mootha V. K., et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naithani S., Saracco S. A., Butler C. A., Fox T. D. Interactions among COX1, COX2, and COX3 mRNA-specific translational activator proteins on the inner surface of the mitochondrial inner membrane of Saccharomyces cerevisiae. Mol. Biol. Cell. 2003;14:324–333. doi: 10.1091/mbc.E02-08-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham O., Davies S. M., Shearwood A. M., Hamilton K. L., Whelan J., Filipovska A. Pentatricopeptide repeat domain protein 1 lowers the levels of mitochondrial leucine tRNAs in cells. Nucleic Acids Res. 2009;37:5859–5867. doi: 10.1093/nar/gkp627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz-Linneweber C., Small I. Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci. 2008;13:663–670. doi: 10.1016/j.tplants.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Tavares-Carreon F., Camacho-Villasana Y., Zamudio-Ochoa A., Shingu-Vazquez M., Torres-Larios A., Perez-Martinez X. The pentatricopeptide repeats present in Pet309 are necessary for translation but not for stability of the mitochondrial COX1 mRNA in yeast. J. Biol. Chem. 2008;283:1472–1479. doi: 10.1074/jbc.M708437200. [DOI] [PubMed] [Google Scholar]
- Topisirovic I., Siddiqui N., Lapointe V. L., Trost M., Thibault P., Bangeranye C., Pinol-Roma S., Borden K. L. Molecular dissection of the eukaryotic initiation factor 4E (eIF4E) export-competent RNP. EMBO J. 2009;28:1087–1098. doi: 10.1038/emboj.2009.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weraarpachai W., et al. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009;41:833–837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- Xu F., Ackerley C., Maj M. C., Addis J. B., Levandovskiy V., Lee J., Mackay N., Cameron J. M., Robinson B. H. Disruption of a mitochondrial RNA-binding protein gene results in decreased cytochrome b expression and a marked reduction in ubiquinol-cytochrome c reductase activity in mouse heart mitochondria. Biochem. J. 2008;416:15–26. doi: 10.1042/BJ20080847. [DOI] [PubMed] [Google Scholar]
- Xu F., Morin C., Mitchell G., Ackerley C., Robinson B. H. The role of the LRPPRC (leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III mRNA. Biochem. J. 2004;382:331–336. doi: 10.1042/BJ20040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Shoubridge E. A. Expression and functional analysis of SURF1 in Leigh syndrome patients with cytochrome c oxidase deficiency. Hum. Mol. Genet. 1999;8:2541–2549. doi: 10.1093/hmg/8.13.2541. [DOI] [PubMed] [Google Scholar]