

GLUT4 is sorted to vesicles whose accumulation beneath and insertion into the plasma membrane are differentially regulated by insulin and selectively affected by insulin resistance.
Abstract
Insulin stimulates glucose transport by recruiting the GLUT4 glucose transporter to the plasma membrane. Here we use total internal reflection fluorescence microscopy to show that two trafficking motifs of GLUT4, a FQQI motif and a TELE-based motif, target GLUT4 to specialized vesicles that accumulate adjacent to the plasma membrane of unstimulated adipocytes. Mutations of these motifs redistributed GLUT4 to transferrin-containing recycling vesicles adjacent to the plasma membrane, and the degree of redistribution correlated with the increases of the GLUT4 mutants in the plasma membrane of basal adipocytes. These results establish that GLUT4 defaults to recycling endosomes when trafficking to specialized vesicles is disrupted, supporting the hypothesis that the specialized vesicles are derived from an endosomal compartment. Insulin stimulates both the accumulation of GLUT4 in the evanescent field and the fraction of this GLUT4 that is inserted into the plasma membrane. Unexpectedly, these two steps are differentially affected by the development of insulin resistance. We ascribe this selective insulin resistance to inherent differences in the sensitivities of GLUT4 vesicle accumulation and insertion into the plasma membrane to insulin. Differences in insulin sensitivities of various processes may be a general mechanism for the development of the physiologically important phenomenon of selective insulin resistance.
INTRODUCTION
Glucose flux into muscle and adipose cells is tightly controlled (Klip, 2009). In unstimulated adipose and muscle cells the insulin-responsive glucose transporter, GLUT4, is predominantly intracellular (∼95%), and its exclusion from the plasma membrane (PM) accounts for the low basal glucose flux into these cells (Huang and Czech, 2007). On insulin stimulation, GLUT4 is redistributed to PM (∼50%), increasing glucose flux into cells. In insulin resistance, insulin does not properly regulate GLUT4 trafficking, thereby contributing to hyperglycemia and the disruption of glucose homeostasis.
GLUT4 trafficking has been extensively studied (Huang and Czech, 2007). Although there remain points of controversy on the details, the emerging picture is that GLUT4 traffics intracellularly between specialized compartments that are accessible to only select proteins and more general endosomal compartments that are accessible to the transferrin receptor (TR; Govers et al., 2004; Karylowski et al., 2004; Dugani and Klip, 2005; Martin et al., 2006). One of the proteins that traffics with GLUT4 through the specialized pathway is the insulin-regulated aminopeptidase (IRAP; Kandror and Pilch, 1994; Keller et al., 1995). In unstimulated adipocytes GLUT4 is rapidly internalized from the PM and slowly recycled back to the cell surface. Slow recycling in unstimulated cells is the predominant kinetic characteristic of the GLUT4 specialized trafficking pathway that distinguishes it from the general endosomal-recycling pathway that traffics the TR back to the PM. Targeting of GLUT4 to the insulin-regulated pathway is required for intracellular retention and this specialized sorting requires information in both the amino and carboxyl cytoplasmic domains of GLUT4. Specifically, an FQQI5-8-based motif in the amino and a TELE498-501-based motif in the carboxyl cytoplasmic domains are required for retention (e.g., Govers et al., 2004; Blot and McGraw, 2008).
Insulin stimulation accelerates recycling and slows endocytosis of GLUT4, resulting in a net accumulation of GLUT4 in the PM (Huang and Czech, 2007). Coincident with the increased recycling is a net redistribution from specialized compartments to endosomes. It is not known if in insulin-stimulated cells GLUT4 traffics to the PM through the rapid general endosomal-recycling pathway or if it traffics to the PM by specialized carriers. The formation and behavior of the carriers that ferry GLUT4 to the PM are sites of insulin regulation, and therefore understanding these processes will further illuminate normal insulin action and will provide a conceptual framework for understanding changes induced by insulin resistance. Although it is known that GLUT4 is not properly translocated to the PM of muscle and adipose from insulin-resistant individuals (e.g., (Maianu et al., 2001); a phenomenon that is recapitulated in model systems of insulin resistance (e.g., Chen et al., 2004; Hoehn et al., 2008), the details of how insulin resistance affects GLUT4 trafficking remain undefined.
Here we use dual-color total internal reflection fluorescence microscopy (TIRFM) to compare wild-type (WT) GLUT4 and GLUT4 mutants whose steady-state distributions between the PM and intracellular compartments are altered. Transferrin (Tf) was used to mark endocytic-recycling vesicles and the insulin-regulated amino peptidase (IRAP) was used to mark the insulin-regulated transport vesicles. Our results establish that in unstimulated adipocytes the GLUT4 mutants are redistributed from IRAP-containing to Tf-containing vesicles in the evanescent field (within 200 nm of PM). The degree of redistribution correlates with the increases of the individual mutants in the PM of unstimulated basal adipocytes. These data support a model in which the GLUT4 mutants with reduced targeting to the specialized insulin-regulated pathway are returned to the PM in rapidly recycling Tf-containing vesicles, thereby accounting, in part, for the increased accumulation of the mutants in the PM of unstimulated adipocytes. We show that insulin does not cause a significant redistribution of GLUT4 to Tf recycling vesicles, strongly supporting the hypothesis that in insulin-stimulated cells, as in basal adipocytes, GLUT4 traffics to the PM in specialized vesicles. Insulin signaling regulates both the accumulation of GLUT4-containing vesicles within the evanescent field as well as the insertion of GLUT4 into the PM. Unexpectedly the accumulation and insertion steps are differentially affected by the development of insulin resistance. In insulin-resistant adipocytes, in which the net redistribution of GLUT4 to the PM is reduced by ∼50%, the insertion of GLUT4 into the PM stimulated by 1 nM insulin is unaffected, whereas the accumulation of GLUT4 in the evanescent field is significantly inhibited. A possible mechanism to explain this “selective” insulin resistance are inherent differences in the insulin sensitivity of GLUT4-containing vesicle accumulation in the evanescent field and GLUT4 insertion into the PM.
MATERIALS AND METHODS
Ligands, Chemicals, and Antibodies
Anti-hemagglutinin (HA) epitope antibody (HA.11) was purified from ascites (Covance, Berkley, CA) on a protein G affinity column (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom). Fluorescent secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Alexa546 was purchased from Invitrogen (Carlsbad, CA) and was conjugated to human Tf (Sigma-Aldrich, St. Louis, MO) according to the manufacturer's instructions. The Akt inhibitor 1/2 (Akti1/2) and phosphatidylinositol 3′-kinase (PI3K) inhibitor (wortmannin) were purchased from Calbiochem (San Diego, CA). Mouse monoclonal anti-TR was a gift from Dr. Fred Maxfield (Weill Cornell Medical College). Rabbit anti-IRAP was a generous gift from Dr. Susanna Keller (University of Virginia).
Cell Culture, Plasmids, and Electroporation
3T3-L1 fibroblasts were cultured, differentiated into adipocytes, and electroporated as previously described (Zeigerer et al., 2002). Studies were performed 1 d after electroporation. The HA-GLUT4-green fluorescent protein (GFP) mutants and TR have been described previously (Zeigerer et al., 2002; Blot and McGraw, 2008). Insulin resistance was induced by incubating adipocytes overnight in DMEM supplemented with 10% FBS and 17 nM insulin. To inhibit Akt or PI3K activity, cells were preincubated for 30 min with 1 μM Akti1/2 or 100 nM wortmannin, respectively, and then incubated with or without 1 nM insulin for 30 min.
Wide-Field Microscopy and TIRFM
TIRFM and epifluorescence (EPI) images were acquired on an Olympus IX 70 (Thornwood, NY) with a 60× 1.45 NA oil-immersion objective using dual-color TIRF imaging with excitation wavelengths at 488 nm for GFP and 532 nm for Cy3. MetaMorph software (Molecular Devices, Sunnyvale, CA) was used for image processing and quantification as described previously (Lampson et al., 2001).
GLUT4 Translocation
Adipocytes incubated in serum-free DMEM medium for 2 h at 37°C in 5% CO2 air were further incubated with or without 1 nM insulin for 30 min. Cells were washed with ice-cold PBS supplemented with Ca2+/Mg2+ and fixed in 3.7% formaldehyde. PM GLUT4 was measured by anti-HA.11 immunofluorescence (IF) of nonpermeabilized cells.
Colocalization
IRAP and HA-GLUT4-GFP.
Adipocytes transiently expressing HA-GLUT4-GFP were fixed and permeabilized with 150 μM saponin, and endogenous IRAP was revealed by indirect IF with a Cy3-labeled secondary antibody. Images were collected in both GFP and Cy3 channels in EPI and TIRF modes. To determine colocalization of the probes in the evanescent field, images in both channels were background-corrected using the median ranking filter of MetaMorph image-processing software. Regions of interest (ROI) were independently identified in the two channels using the internal threshold objects algorithm of MetaMorph and empirically determined low-intensity thresholds. A single threshold value for the red channel was used to process all the red images collected within a given experiment, and a single green channel threshold was used for all the green channel images. The average pixel intensities of the ROI were logged for analysis. The ROI determined in the red channel (IRAP) were transferred to the corresponding image in the green channel, and the green pixel intensities were logged. Similarly the green ROI were transferred to the red channel and the pixel intensities logged. The percent of IRAP ROI that was also positive for GLUT4 (percent colocalization) were calculated as the percent of the red channel ROI that had green channel pixel intensities above the low-intensity threshold of the green channel. Similarly, the GLUT4 vesicles that contained IRAP were determined as the green channel ROI that have red channel pixel intensities above the red channel low-intensity threshold. Images illustrating the steps of the processing are provided in Supplemental Figure 1.
Tf and HA-GLUT4-GFP.
Adipocytes transiently expressing HA-GLUT4-GFP and the human TR were incubated with 5 μg/ml Tf-Alexa546 for at least 2 h at 37°C. The cells were washed with pH 5.0 balanced salt buffer, followed by two washes with neutral pH balanced salt buffer to release Tf-Alexa546 bound to the surface of cells. The cells were incubated for 2 min at 37°C with unlabeled Tf to minimize the number of newly formed endosomes labeled with Tf-Alexa546 in the evanescent field. The quantification of colocalization of HA-GLUT4-GFP and Tf-Alexa546 was determined by the same method as described above for IRAP and HA-GLUT4-GFP.
Accumulation in Evanescent Field and Insertion into the PM
Adipocytes were incubated in serum-free DMEM medium for 2 h. During the final 30 min of this incubation and during the 30-min insulin stimulation, the medium was supplemented with DMSO, 1 μM Akti1/2, or 100 nM wortmannin. Akti1/2 is an inhibitor of Akt 1 and 2 isoforms (Barnett et al., 2005). After fixation, HA-GLUT4-GFP in the PM was revealed by anti-HA.11 IF with Cy3 secondary antibody in unpermeabilized cells. Both EPI and TIRF images were acquired in the Cy3 and GFP channels using dual-color TIRF and EPI microscopy.
To study Tf-containing vesicles, adipocytes were incubated with 5 μg/ml Tf-Alexa546 for at least 2 h at 37°C. During the final 30 min of this incubation and during the 30-min insulin stimulation, the medium was supplemented with DMSO, 1 μM Akti1/2, or 100 nM wortmannin. Surface TR was revealed by IF with Cy2 secondary antibody.
Statistics
Paired Student's t test was applied for the comparisons.
RESULTS
Juxtamembrane Elements of the GLUT4 Retention Pathway Identified in TIRFM
An important aspect of the specialization of GLUT4 behavior is its exclusion from the PM of unstimulated cells. Previous studies have established a correlation between increased GLUT4 localization to endosomes and increased GLUT4 in the PM of basal adipocytes (Martin et al., 1996; Marsh et al., 1998; Blot and McGraw, 2008). For example, mutation of the GLUT4 amino terminal FQQI5-8 motif or the carboxyl terminal TELE498-501-based motif results in an increase of GLUT4 in the PM of unstimulated adipocytes and a shift of GLUT4 from specialized intracellular compartments to Tf-containing endosomal compartments (Marsh et al., 1998; Blot and McGraw, 2008). Those data support a model in which the retention of GLUT4 in unstimulated adipocytes requires targeting to a specialized intracellular trafficking pathway (i.e., diversion from the endosomal system; Figure 1A). Implicit in this model is the hypothesis that the GLUT4 mutants are recycled to the PM through the rapid endosomal-recycling pathway that returns Tf to the PM, resulting in the increased accumulation of GLUT4 mutants at the PM of basal adipocytes.
Figure 1.

GLUT4 retention machinery driven by the FQQI and TELE motifs. (A) GLUT4 retention model. The TR recycling pathway (fast in both basal and insulin-stimulated states) and the GLUT4 specialized pathway (slow in basal and fast in the presence of insulin) contribute to PM GLUT4 distribution. GLUT4 is sorted from the endosomal recycling compartment (ERC) to the GLUT4 specialized vesicles (GSV; retention pathway 2) and an unidentified perinuclear compartment (retention pathway 1) in basal adipocytes. These two retention pathways are controlled by the TELE-based and FQQI motifs, respectively. Mutation of these motifs results in redistribution of GLUT4 to the endosomal compartments and increases PM GLUT4 (Blot and McGraw, 2008). (B) Schematic of the HA-GLUT4-GFP with mutated residues noted in bold type. (C) Surface-to-total distributions in basal adipocytes. WT, wild-type HA-GLUT4-GFP; FA, F5A mutation; EE, EE499,501AA mutation; FA/EE, F5A/EE499,501AA mutation in HA-GLUT4-GFP. The data are averages ± SEM of eight to nine independent experiments normalized to the WT GLUT4 surface-to-total ratio of each experiment. *p < 0.001 compared with WT; #p < 0.001 compared with the FA mutant.
To test this hypothesis, we used TIRFM to examine the effects of the GLUT4 mutations on intracellular compartmentalization in unstimulated adipocytes. In TIRFM, fluorophors within ∼200 nm of the PM are excited, limiting detection of fluorescence to structures that are adjacent to the PM. We reasoned that this method would allow us to determine the effects of the mutations on targeting GLUT4 to transport intermediates ferrying proteins from intracellular compartments to the PM. GLUT4 harboring the F5A mutation of the FQQI motif, the EE499,501AA mutation of the TELE-based motif, or both mutations, F5A/EE499,501AA, were studied (Figure 1B). The behaviors of WT GLUT4 and the different mutants were characterized using HA-GLUT4-GFP, which has an HA epitope inserted in the first exofacial loop and GFP fused to the carboxy terminus of GLUT4 (Lampson et al., 2000). Indirect IF detection of the HA epitope on the surface of nonpermeabilized cells is a measure of the insertion of HA-GLUT4-GFP in the PM and is a quantitative assay of the translocation of GLUT4 to the PM (Zeigerer et al., 2002). As previously reported, GLUT4 mutated in the FQQI or TELE-based motifs accumulated at the PM of basal adipocytes to a greater extent than WT GLUT4, (Figure 1C; Govers et al., 2004; Blot and McGraw, 2008). The different mutants accumulated at the PM to different degrees. The F5A and EE499,501AA mutations increased basal PM GLUT4 by about fourfold and about twofold, respectively, whereas simultaneous mutation of both motifs increased basal PM GLUT4 by about eightfold.
To determine the effects of the mutations on intracellular targeting of GLUT4, we examined the colocalization of the HA-GLUT4-GFP constructs with endogenous IRAP in TIRFM. Adipocytes expressing HA-GLUT4-GFP constructs (WT, F5A, EE499,501AA, or F5A/EE499,501AA) were permeabilized and IRAP localization examined by IF. GFP (GLUT4) and Cy3 (IRAP) images were acquired in a dual-view configuration of the TIRF microscope (Figure 2A). As previously reported, there were a large number (∼350) of WT GLUT4-containing spots (vesicles) in the evanescent field of basal adipocytes (Lizunov et al., 2005; Gonzalez and McGraw, 2006; Bai et al., 2007; Huang et al., 2007). These GLUT4 vesicles are mobile, rapidly moving within the evanescent field, yet rarely fusing with the PM (Lizunov et al., 2005; Huang et al., 2007). Consequently, in basal adipocytes the GLUT4 vesicles detected in TIRFM are not transporting GLUT4 to the PM per se, but rather are vesicular elements of the basal retention mechanism located adjacent to the PM. Past work has established that upon insulin stimulation, these vesicles arrest (dock) and fuse, delivering GLUT4 to the PM (Lizunov et al., 2005; Bai et al., 2007; Huang et al., 2007). As is the case for WT GLUT4, there were abundant GFP-positive puncta in the evanescent field of basal adipocytes expressing the GLUT4 mutants, and all the different GLUT4 constructs had similar number of puncta in the evanescent field.
Figure 2.


Colocalization of the GLUT4 and IRAP in the evanescent field. (A) TIRF images illustrating the distribution of HA-GLUT4-GFP and endogenous IRAP in basal adipocytes. WT, wild-type HA-GLUT4-GFP; FA, F5A mutation; EE, EE499,501AA mutation; FA/EE, F5A/EE499,501AA mutation in HA-GLUT4-GFP. Images are identically scaled and corrected for local background. Cell boundaries are noted by dashed lines. Scale bar, 10 μm. (B) Quantification of GLUT4 colocalization with IRAP in the basal adipocytes. Data are from cells in A. The scatter plots illustrate the intensity distributions, normalized to EPI fluorescence to correct for cell-to-cell variations in expression of the HA-GLUT4-GFP constructs and endogenous IRAP, of ROI picked from the Cy3 channel (IRAP-positive, red circles) or GFP channel (GLUT4-positive, green circles) versus their intensities in the other channel. For data analysis the thresholds used in absolute intensity were the same for each cell type; however, for graphing the data the low-threshold intensity lines shown were scaled to the total expression of the HA-GLUT4-GFP or IRAP of the cells shown. Circles in the quadrant to the right of the green line and above the red line are scored as positive for both probes. The percent of ROI (circles) chosen in the red channel that are positive in the green [GLUT4/IRAP(+)] and vice versa [IRAP/GLUT4(+)] for the data shown in these scatter plots are noted above the plots. (Also see Supplemental Figure 1 for further details on the method). (C) Colocalization of GLUT4 and IRAP from an individual experiment. The percent colocalization is the percent of IRAP vesicles (ROI identified in red channel) whose intensity in the green channel is above the green channel low-intensity threshold. Each symbol is the colocalization determined in an individual cell within the experiment. The lines are the average values. All the data from each individual experiment were collected during one block of time on the microscopy; all the green channel images were processed with the same green channel low-intensity threshold, and all the red images were processed with the same red channel low-intensity threshold. Nine to 19 cells for each construct were analyzed in each individual experiment. (D) The average colocalization (percent of IRAP vesicles positive for GLUT4) measured in three to four independent experiments ± SEM. The data illustrate the reproducibility of the measurements across independent experiments. (E and F) Colocalization of GLUT4 mutants with IRAP normalized to WT GLUT4 colocalization with IRAP. The colocalizations of the different mutants with IRAP determined in individual experiments were normalized to the colocalization of WT GLUT4 measured in the same experiment, illustrating how the mutations affect the WT GLUT4 colocalization. In E the percent of IRAP vesicles (ROI identified in the red channel) that are also positive for GLUT4 are plotted. In F the percent of GLUT4 vesicles (ROI identified in the green channel) that are also positive for IRAP are plotted. The data are averages ± SEM of three to four independent experiments. The p values for paired t test are shown and are the comparison to WT GLUT4. (G) Correlation between the HA-GLUT4-GFP expression of the various constructs and the colocalization of GLUT4 with IRAP. The colocalization of GLUT4 with IRAP in 12–16 cells is plotted as a function of the HA-GLUT4-GFP expression level per cell for each GLUT4 mutant.
Immunofluorescence revealed abundant IRAP-containing vesicles in the evanescent field of basal adipocytes. Data from individual cells expressing the WT or the GLUT4 mutants are shown in Figure 2A. Colocalizations of IRAP and GLUT4 in vesicles adjacent to the PM were quantified as described in detail in supplemental Figure 1. Briefly, IRAP-positive spots above a low-intensity threshold (Figure 2B, red line) were identified as ROI, and the red (IRAP) and green (GLUT4) pixel intensities in these ROI were measured. The ROI with green intensities above the low-intensity threshold (Figure 2B, green line) were scored as IRAP-containing vesicles that were also positive for GLUT4 (Figure 2B). The data were analyzed in reverse, with the ROI chosen in the green channel, and ROI with red intensities above the low-intensity threshold (Figure 2B, red line) were scored as GLUT4-containing vesicles positive for IRAP.
The percent of IRAP vesicles that are also positive for HA-GLUT4-GFP in individual cells from a representative experiment are shown in Figure 2C, and the average data from multiple independent experiments are shown in Figure 2D. About 60% of the IRAP vesicles identified were also positive for WT GLUT4, and the colocalizations of the different GLUT4 mutants with IRAP were all reduced to degrees that paralleled the increases of the individual mutants on the PM of unstimulated adipocytes (Figure 1C). GLUT4 and IRAP have been reported to be better colocalized (>85%) than what we measure here. Past colocalizations of GLUT4 and IRAP have been determined with methods that measure the colocalization of the total cellular pools of IRAP and GLUT4, whereas here we are restricting analysis to structures within the evanescence field. Another consideration is the sensitivity of the assay. To examine this, we measured colocalization between GFP and anti-HA when HA-GLUT4-GFP–expressing cells were permeabilized and stained with anti-HA. We found ∼70% colocalization, suggesting that the 60% value we measured is close to the maximum colocalization measured in the assay. Recently it has been reported that GLUT4 and IRAP colocalize to ∼70% in TIRFM, in line with our measurements (Zhao et al., 2009).
Mutations of the GLUT4 motifs alter the percent colocalizations with IRAP (Figure 2, C and D). The absolute percent colocalization of the two probes will depend on the low-intensity threshold values used to analyze the data. Here we are interested in how mutations in GLUT4 change its localization with IRAP; therefore, the colocalizations of the GLUT4 mutants with IRAP of the individual experiments were normalized to the percent of colocalization of WT GLUT4 and IRAP measured in the same experiment. The averages of the normalized data from multiple experiments are presented in Figure 2, E and F. The largest change was for the F5A/EE499,501AA mutant, whose colocalization with IRAP was reduced by 50% compared with WT GLUT4. Relative to WT GLUT4, the F5A mutation caused a 25% reduction in colocalization with IRAP, whereas the EE499,501AA mutant's colocalization was reduced by ∼10%. The differences in the colocalization of the mutants with IRAP relative to WT GLUT4 followed the same general trends whether the vesicles were identified in the IRAP channel or the GLUT4 channel, although for the F5A mutant, vesicles chosen in the GFP channel had a higher p value than the other comparisons (Figure 2F). The numbers of vesicles quantified were not significantly different among the GLUT4 mutants, and the degree of colocalization did not correlate with the expression levels of the GLUT4 constructs (Figure 2G). Thus, the differences in colocalizations of the mutants are intrinsic to differences in behaviors of the mutants rather than a consequence of differences in the amounts of the mutants expressed. Importantly, previous studies have shown expression of these mutants did not alter IRAP behavior, validating IRAP as a marker of the insulin-regulated retention pathway in adipocytes coexpressing the GLUT4 mutants (Blot and McGraw, 2008). The relative changes in colocalization of the GLUT4 mutants with IRAP parallels the increases of the mutants in the PM of basal adipocytes (Figures 1C and 2, E and F), providing strong support for the hypothesis that these IRAP-containing juxta PM vesicles are indeed elements of the GLUT4 retention machinery.
GLUT4 Mutants Are Redistributed to Tf-containing Vesicles Adjacent to the PM
We next sought to determine whether the decreased targeting of the mutants to IRAP-containing vesicles was paralleled by an increased accumulation in Tf-containing recycling vesicles. To address this question we used a modification of the method discussed above. Adipocytes coexpressing HA-GLUT4-GFP constructs and the human TR were incubated with Tf conjugated to Alexa546 (Tf-Alexa546) for 2 h at 37°C to label endosomes. Cells were incubated for 2 min before fixation in medium with unlabeled Tf. Any endosomes formed from the PM during this 2 min will not be labeled with Tf-Alexa546, reducing the contribution of newly formed Tf-Alexa546–labeled endosomes in the evanescent field.
The localizations of F5A and F5A/EE499,501AA mutants to Tf recycling vesicles were increased relative to WT GLUT4 (Figure 3). These data demonstrate that the reduced targeting of these mutants to the IRAP-containing vesicles correlates with an increased localization to Tf-containing vesicles. The F5A and F5A/EE499,501AA mutations had large effects on the amount of GLUT4 in the PM of basal adipocytes (Figure 1C), providing a link between increased localization to Tf-containing vesicles and increased expression in the PM of basal adipocytes. There was no statistically significant increase of the EE499,501AA mutant in Tf-containing vesicles. This mutation caused the smallest increase in basal PM expression of GLUT4 as well as the smallest decrease in colocalization with IRAP. The sensitivity of the colocalization assay with Tf-labeled endosomes might not be sufficient to detect the change in GLUT4 colocalization induced by the EE499,501AA mutation.
Figure 3.

Colocalization of the GLUT4 and Tf in the evanescent field. (A) Distribution of the HA-GLUT4-GFP and Tf-Alexa546 in TIRFM of basal adipocytes. Data were processed as discussed in Figure 2. Scale bar, 10 μm. (B) Colocalization of GLUT4 and Tf from an individual experiment. The percent colocalization is the percent of Tf vesicles (ROI identified in red channel) whose intensity in the green channel is above the green channel low-intensity threshold. Each symbol is the colocalization determined in an individual cell within the experiment. The lines are the average values. All the data were collected during one block of time on the microscopy; all the green channel images were processed with the same green channel low-intensity threshold, and all the red images were processed with the same red channel low-intensity threshold. Four to20 cells for each construct were analyzed in each individual experiment. (C) The average colocalization (percent of Tf vesicles positive for GLUT4) measured in four to five independent experiments ± SEM. The data illustrate the reproducibility of the measurements across independent experiments. (D and E) Colocalization of GLUT4 mutants with Tf normalized to WT GLUT4 colocalization with Tf. The colocalizations of the different mutants with Tf determined in individual experiments were normalized to the colocalization of WT GLUT4 measured in the same experiment, illustrating how the mutations affect the WT GLUT4 colocalization. (D) The percent of Tf vesicles (ROI identified in the red channel) that are also positive for GLUT4 are plotted. (E) The percent of GLUT4 vesicles (ROI identified in the green channel) that are also positive for Tf are plotted. The data are averages ± SEM of four to five independent experiments. The p values for paired t test are shown and are the comparison to WT GLUT4.
Insulin Controls Both the Accumulation of WT GLUT4 in the Evanescent Field and the Fraction of GLUT4 within the Evanescent Field That Was Inserted into the PM
Insulin signals to GLUT4 in part through activation of Akt downstream of PI3K activation. Insulin regulates GLUT4-vesicle association with the PM at both prefusion and fusion steps, and these steps are distinguished based on sensitivity to Akt inhibition (e.g., Gonzalez and McGraw, 2006). Insulin increases the accumulation of GLUT4 within the evanescent field, an effect that is blocked by Akt inhibition (Gonzalez and McGraw, 2006; Lopez et al., 2009). The fraction of GLUT4 within the evanescent field that is inserted into the PM is also increased by insulin, demonstrating insulin control of vesicle fusion independent of changes in accumulation in the evanescent field (Gonzalez and McGraw, 2006). The effect of insulin on the fraction of GLUT4 within the evanescent field inserted into the PM is not blocked by inhibition of Akt (Gonzalez and McGraw, 2006). Both effects of insulin are blocked by wortmannin inhibition of PI3K (Gonzalez and McGraw, 2006).
Here we use insulin stimulation of the accumulation of GLUT4 in the evanescent field and the fraction of this GLUT4 that is inserted into the PM (“insertion efficiency”) as parameters to further characterize the behaviors of the GLUT4 mutants. We used a two-color TIRFM assay that is a modification of a previously described single color TIRFM method (Gonzalez and McGraw, 2006). Briefly, unstimulated adipocytes and adipocytes treated with insulin for 30 min were fixed (not permeabilized), and the amount of HA-GLUT4-GFP inserted into the PM was revealed by IF with an anti-HA antibody (Cy3; Figure 4A). EPI and TIRF images for both GFP and Cy3 channels were collected. The Cy3EPI/GFPEPI ratio is the standard method used to measure insulin-stimulated translocation of GLUT4 to the PM, and it reflects the fraction of the total cellular HA-GLUT4-GFP that is inserted into the PM (Lampson et al., 2000). The GFPTIRF/GFPEPI is proportional to the fraction of total cellular HA-GLUT4-GFP that is within the evanescent field. The amount in the evanescent field includes GLUT4 inserted in the PM as well as GLUT4 in vesicles adjacent to the PM but not fused with the PM, and an increase in this ratio induced by insulin reflects a net increase of GLUT4 in the evanescent field, without distinguishing between vesicles that have fused with the PM from those that have not. We refer to the GFPTIRF/GFPEPI ratio as “accumulation in evanescent field.” The Cy3TIRF/GFPTIRF ratio is a measure of the fraction of HA-GLUT4-GFP in the evanescent field that is inserted into the PM; therefore, changes in this ratio reflect net changes in the fraction of HA-GLUT4-GFP in the evanescent field that is inserted into the PM. We refer to Cy3TIRF/GFPTIRF as “insertion efficiency.”
Figure 4.

Characterization of GLUT4 accumulation in the evanescent field and fraction of this GLUT4 inserted into the PM. (A) Schematic of the method to measure HA-GLUT4-GFP accumulation in the evanescent field (GFPTIRF/GFPEPI fluorescence ratio) and the fraction of GLUT4 in the evanescent field that is inserted into the PM (αHA Cy3TIRF/GFPTIRF fluorescence ratio). (B–D) Characteristics of HA-GLUT4-GFP translocation (αHA Cy3EPI/GFPEPI fluorescence ratio), the insertion efficiency (fraction of GLUT4 in the evanescent field that is inserted into the PM; αHA Cy3TIRF/GFPTIRF fluorescence ratio) and accumulation in evanescent field (GFPTIRF/GFPEPI fluorescence ratio) in unstimulated and insulin-stimulated adipocytes. Cells were treated with DMSO, 1 μM Akti1/2, or 100 nM wortmannin 30 min before and during the 30-min insulin stimulation. Data are averages ± SEM from six independent experiments normalized to the control in the basal state. *p < 0.05, **p < 0.001 compared with the control in the insulin-stimulated state; #p < 0.05.
Consistent with our previous results, insulin-stimulated GLUT4 translocation to the PM was inhibited by 75% when Akt activation was inhibited with 1 μM of the Akt inhibitor, Akti1/2, whereas inhibition of PI3K with 100 nM wortmannin completely blocked GLUT4 translocation (Figure 4B). Insulin stimulated a threefold increase in the Cy3TIRF/GFPTIRF ratio, demonstrating a threefold increase in the amount of HA-GLUT4-GFP in the evanescent field that is inserted into the PM. The effect of insulin on the amount of HA-GLUT4-GFP in the evanescent field that is inserted into the PM is completely blocked by wortmannin but is inhibited only 48% by Akti1/2 (Figure 4C). Insulin activation of Akt is blocked by Akti1/2 treatment (Supplemental Figure 2). These data show that about half of the effect of the insulin control of GLUT4 in the evanescent field that is inserted into the PM is by an Akt-independent, PI3K-dependent mechanism. Insulin stimulated the accumulation of GLUT4 in the evanescent field by twofold (change in the GFPTIRF/GFPEPI ratio), and this effect of insulin on GLUT4 was completely inhibited by wortmannin and inhibited 76% by Akti1/2 (Figure 4D). Thus, insulin control of GLUT4 accumulation in the evanescent field is more sensitive to Akt inhibition than is insulin control of insertion of GLUT4 into the PM (76 vs. 48% inhibition).
To further characterize this assay, we examined the effect of overexpressing a dominant inhibitory form of the AS160 RabGAP protein AS160-4P (Figure 5). In unstimulated adipocytes AS160 GAP activity is required for the exclusion of GLUT4 from the PM, in part through actions on Rab10 (Sano et al., 2003; Zeigerer et al., 2004; Eguez et al., 2005; Larance et al., 2005; Sano et al., 2007). AS160 controls GLUT4-containing vesicle docking (prefusion step) with the PM (Zeigerer et al., 2004; Jiang et al., 2008). Akt phosphorylates AS160, inactivating the GAP activity and releasing the inhibition on docking. Akt does not phosphorylate the AS160-4P mutant, and therefore AS160-4P inhibits insulin-stimulated GLUT4-containing vesicle docking to the PM. Overexpression of AS160-4P inhibited translocation of GLUT4 by 80%, an effect that is accounted for by a 90% inhibition of insulin-stimulated accumulation of GLUT4 in the evanescent field (Figure 5, A and C). AS160-4P only inhibited the insulin-stimulated increase in the fraction of GLUT4 in the evanescent field that is inserted into the PM by 60% (Figure 5B), consistent with previous results that the effect of AS160-4P is predominantly to inhibit a prefusion step (Jiang et al., 2008). These data show that insulin can increase the insertion of GLUT4 into the PM in the presence of AS160-4P, demonstrating that the assay measures distinct aspects of GLUT4 vesicles trafficking to the PM.
Figure 5.
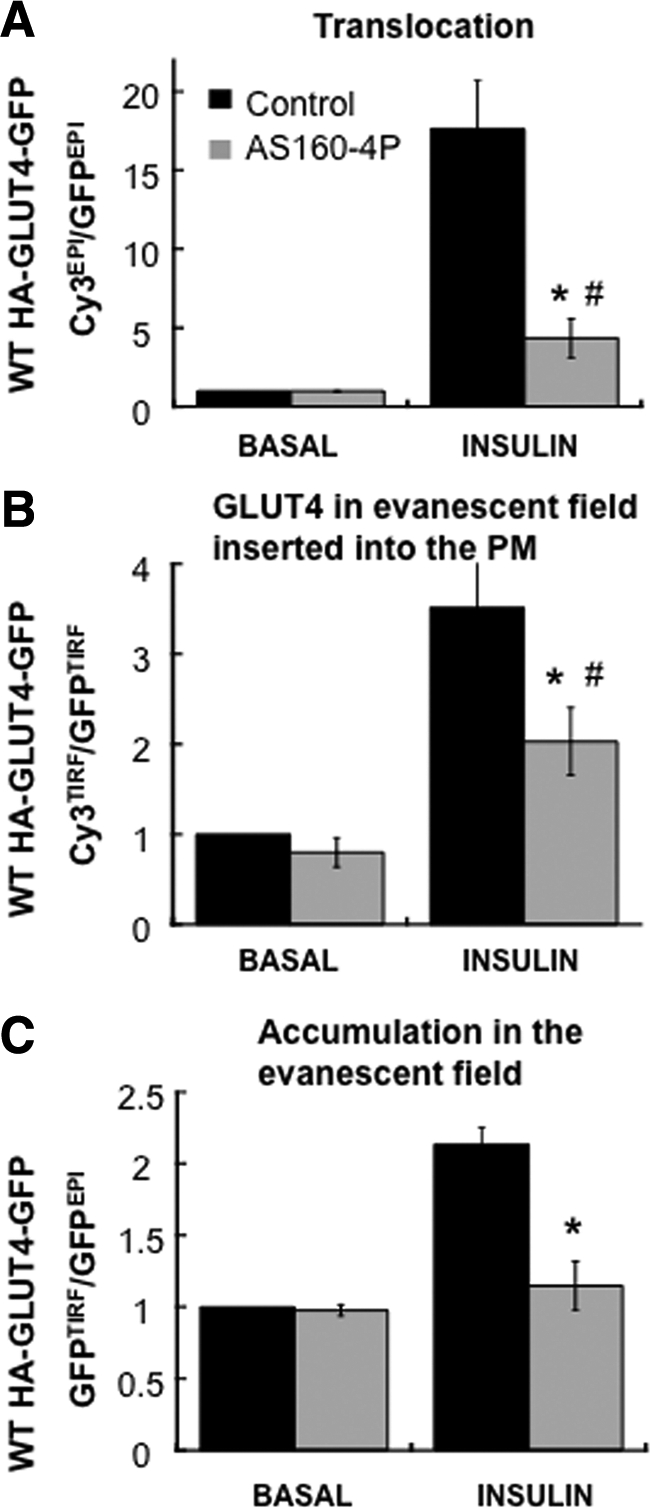
The effects of AS160-4P on GLUT4 accumulation in the evanescent field and the fraction of this GLUT4 inserted into the PM. The effects of AS160-4P on GLUT4 translocation to the PM (αHA Cy3EPI/GFPEPI fluorescence ratio; A), the insertion efficiency (fraction of GLUT4 in the evanescent field that is inserted into the PM (αHA Cy3TIRF/GFPTIRF fluorescence ratio; B), and accumulation in evanescent field (GFPTIRF/GFPEPI fluorescence ratio; C) in unstimulated and insulin-stimulated adipocytes. Adipocytes were transiently transfected with WT HA-GLUT4-GFP and AS160-4P the day before the experiment. The data are the means of four experiments ± SEM; #p < 0.05 compared with basal control; *p < 0.05 compared with insulin-stimulated control.
The Accumulation of F5A/EE499,501AA-GLUT4 in the Evanescent Field and the Fraction of F5A/EE499,501AA-GLUT4 within the Evanescent Field Inserted into the PM Are Not Affected by Insulin
We next examined the behavior of GLUT4 F5A/EE499,501AA mutant in the TIRFM assay. We chose this mutant because its accumulation in the PM of basal adipocytes was most different from WT GLUT4 (Figure 1C). The effects of insulin on this mutant were significantly different from the effects of insulin on WT GLUT4 (Figure 6). First, the effect of Akt inhibition on insulin-stimulated translocation of F5A/EE499,501AA to the PM was smaller than the effect of Akt inhibition on insulin-induced WT GLUT4 translocation (Figures 6A and 4B, respectively). Second, wortmannin inhibition reduced the amount of F5A/EE499,501AA in the PM of unstimulated cells, whereas wortmannin did not have an effect on WT GLUT4 in unstimulated adipocytes (Figures 6A and 4B, respectively). Third, insulin did not increase the fraction of F5A/EE499,501AA-GLUT4 in the evanescent field that is inserted into the PM (Figure 6B). These results support the hypothesis that F5A/EE499,501AA-GLUT4 traffics to the PM in functionally different vesicles than those that ferry WT GLUT4 to the PM. Insulin did increase the accumulation of F5A/EE499,501AA-GLUT4 in the evanescent field, although not to the same degree as the effect on WT GLUT4 (Figure 6C). Furthermore, wortmannin reduced the amount of F5A/EE499,501AA-GLUT4 in the evanescent field of unstimulated adipocytes, indicating that this effect is responsible for the wortmannin-induced decrease in F5A/EE499,501AA-GLUT4 in the PM of basal adipocytes.
Figure 6.

Characterization of FA/EE-GLUT4 mutant accumulation in the evanescent field and fraction of this mutant inserted into the PM. Characteristics of F5A/EE499,501AA HA-GLUT4-GFP translocation (αHA Cy3EPI/GFPEPI fluorescence ratio; A), insertion efficiency (fraction of GLUT4 in the evanescent field that is inserted into the PM; αHA Cy3TIRF/GFPTIRF fluorescence ratio; B), and accumulation in evanescent field (GFPTIRF/GFPEPI fluorescence ratio; C) in unstimulated and insulin-stimulated adipocytes. Cells were treated with DMSO, 1 μM Akti1/2, or 100 nM wortmannin 30 min before and during insulin stimulation. The data are averages ± SEM from four to five independent experiments normalized to the values for WT GLUT4 in the basal state. *p < 0.05 compared with the control in the basal state; #p < 0.05, ##p < 0.001 compared with the control in the insulin-stimulated state; Φp < 0.05.
Because F5A/EE499,501AA-GLUT4 is redistributed to Tf-containing vesicles in the evanescent field, we next examined the behavior of Tf-containing vesicles. Tf-Alexa546 internalized from the medium was used to label the TR recycling vesicles, and an antibody against the extracellular domain of the TR was used to measure insertion of the TR into the PM (Figure 7A). Insulin induced a small increase of TR in the PM of adipocytes, an effect that was inhibited by wortmannin but not Akti1/2 (Figure 7B). Wortmannin reduced the amount of TR in the PM of unstimulated adipocytes, similar to the effect on F5A/EE499,501AA-GLUT4 (Figures 7D and 6C, respectively). In addition, wortmannin reduced the amount of Tf-containing vesicles in the evanescent field of unstimulated cells (Figure 7D). In these respects the behavior of F5A/EE499,501AA-GLUT4 was more like Tf-containing vesicles than WT GLUT4-containing vesicles, consistent with the redistribution of F5A/EE499,501AA-GLUT4 to Tf-containing recycling vesicles in the evanescent field (Figure 3). The wortmannin induced reduction of TR in the PM of unstimulated adipocytes documents a role for PI3K activity in the control of basal TR trafficking, consistent with previous reports (Jess et al., 1996).
Figure 7.

Characterization of Tf-containing vesicle accumulation in the evanescent field and fraction of this TR inserted into the PM. (A) Schematic of the assay. (B–D) Characteristics of TR translocation to the PM (αTR Cy2EPI/Alexa-546EPI fluorescence ratio), the insertion efficiency (fraction of TR in the evanescent field that is inserted into the PM; αTR Cy2TIRF/Alexa-546TIRF fluorescence ratio) and accumulation in evanescent field (Alexa-546TIRF/Alexa-546EPI fluorescence ratio) in unstimulated and insulin-stimulated adipocytes. Cells were treated with DMSO, 1 μM Akti1/2, or 100 nM wortmannin 30 min before and during the 30-min insulin stimulation. The results are averages ± SEM from four independent experiments normalized to the control basal state. *p < 0.05 compared with the control in the basal state; #p < 0.05, ##p < 0.001 compared with the control in the insulin-stimulated state.
We were unable to detect an effect of insulin on the accumulation of Tf-containing vesicles in the evanescent field or a change on the fraction of these vesicles inserted into the PM. Thus, the small change in PM TR induced by insulin did not correlate with measurable changes in the recruitment or fusion of TR recycling vesicles, indicating that the assay is not of sufficient sensitivity to detect the small changes responsible for insulin control of TR in the PM.
GLUT4 Does Not Traffic to PM in TR Recycling Vesicles in Insulin-stimulated Adipocytes
Our data demonstrate the importance of targeting GLUT4 to functionally specialized vesicles in basal retention. In insulin-stimulated cells GLUT4 recycles to the PM with kinetics similar to the TR. The change in GLUT4 behavior could reflect a change in the kinetics of the specialized vesicles in insulin-stimulated cells. Alternatively, GLUT4 could redistribute to endosomes and be returned to the PM in TR recycling vesicles. To provide information on this question we examined, in insulin-stimulated adipocytes, the colocalization of GLUT4 and TR in juxta-PM vesicles (Figure 8). Insulin stimulation did not induce a significant shift in GLUT4 to TR-containing juxta-PM vesicles. Thus, in insulin-stimulated adipocytes GLUT4 does not traffic to the PM in TR recycling vesicles, establishing that GLUT4 is sorted to specialized-recycling vesicles even in stimulated cells.
Figure 8.
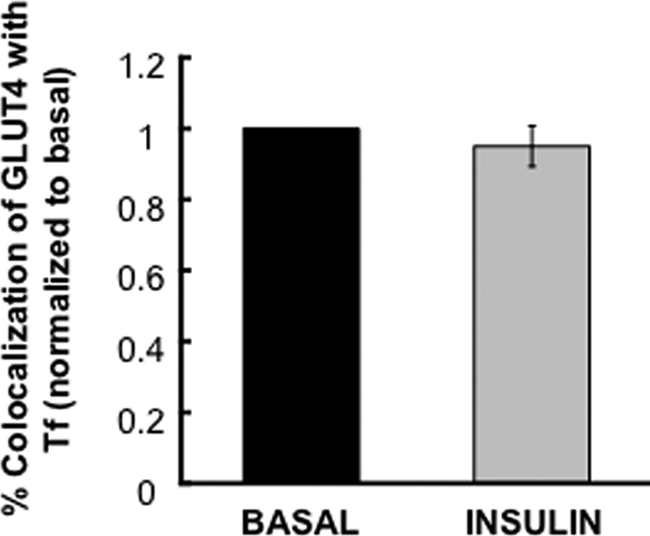
Colocalization of GLUT4 and Tf in the insulin-stimulated state. Colocalization of WT GLUT4 and Tf in the basal and insulin-stimulated states were determined as in Figure 3. The data are averages ± SD from two experiments and are normalized to the basal state.
Effects of Insulin Resistance on the Behavior of GLUT4 Vesicles
The above data demonstrate that GLUT4 is targeted to vesicles whose accumulation in the evanescent field and insertion into the PM are regulated by insulin. These results prompted us to ask how the behaviors of the GLUT4-containing vesicles are affected by insulin resistance. Previous studies have shown that prolonged incubation of adipocytes with high concentrations of insulin induces insulin resistance (Chen et al., 2004; Hoehn et al., 2008). Adipocytes incubated overnight with 17 nM insulin developed insulin resistance characterized by a reduction in the translocation of GLUT4 to the PM when the cells were rechallenged with insulin (Figure 9A). These results establish an approximate 10-fold shift in the insulin dose response for GLUT4 translocation in the insulin-resistant cells (EC50 ∼0.1 nM for control vs. ∼1 nM for insulin-resistant cells). The residual translocation of GLUT4 to the PM in the insulin-resistant cells was blocked by wortmannin, demonstrating that the residual effect of insulin on GLUT4 was dependent on PI3K.
Figure 9.
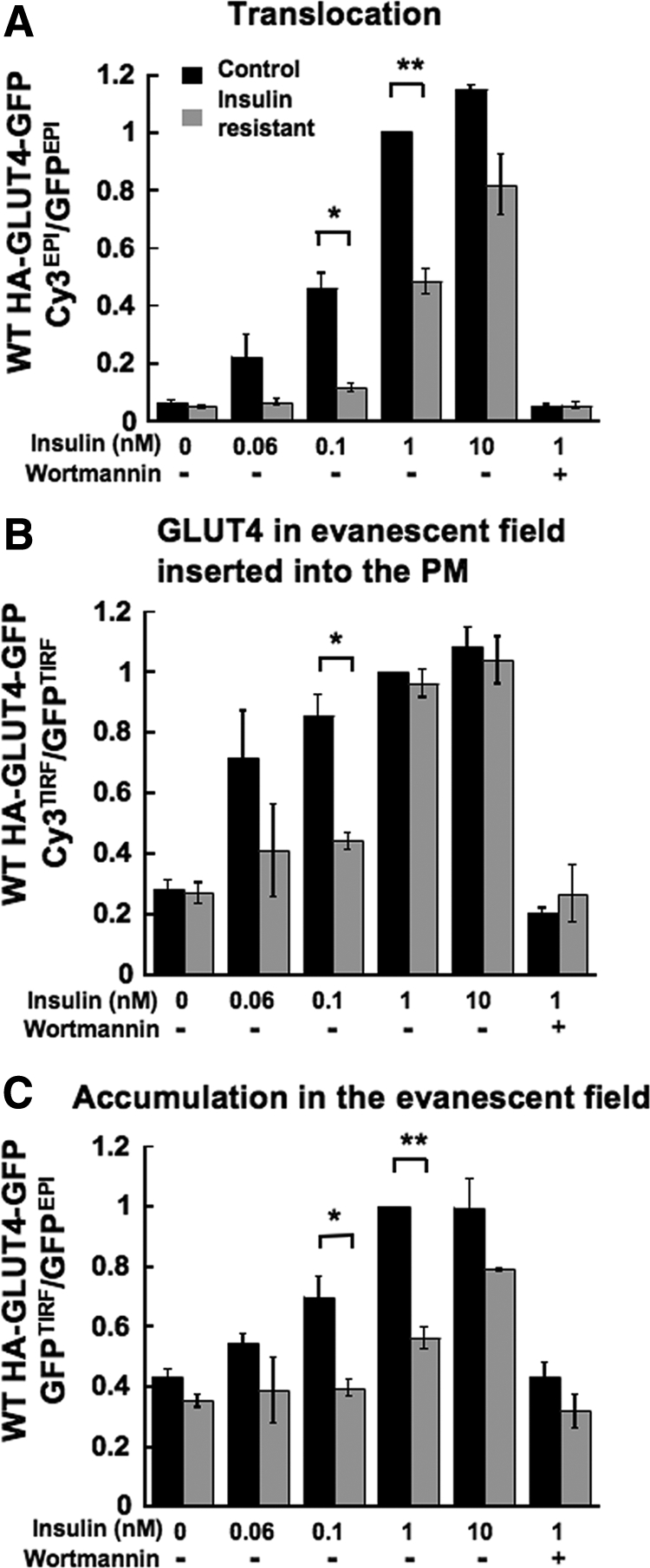
Characterization of GLUT4 accumulation in the evanescent field and fraction of this GLUT4 that is inserted into the PM in insulin-resistant adipocytes. Characteristics of WT HA-GLUT4-GFP translocation (αHA Cy3EPI/GFPEPI fluorescence ratio; A),“insertion efficiency (fraction of GLUT4 in the evanescent field that is inserted into the PM; αHA Cy3TIRF/GFPTIRF fluorescence ratio; B), and accumulation in evanescent field (GFPTIRF/GFPEPI fluorescence ratio; C) in unstimulated and insulin-stimulated control and insulin-resistant adipocytes. 100 nM wortmannin was included as noted. The results are averages ± SEM from three to nine experiments, *p < 0.05; **p < 0.001.
We next determined if the accumulation of GLUT4 in the evanescent field and/or the fraction of this GLUT4 in the evanescent field that is inserted into the PM were affected by insulin resistance. The accumulation of GLUT4 in the evanescent field stimulated by 1 nM insulin was severely blunted in the insulin-resistant cells (Figure 9C). However, unlike the change in recruitment to the PM, the 1 nM insulin-stimulated increase in the amount of GLUT4 in the evanescent field inserted into the PM was unaffected in insulin-resistant adipocytes (Figure 9B). Thus, in the insulin-resistant adipocytes the effects of 1 nM insulin on accumulation in the evanescent field and the fraction of this GLUT4 inserted into the membrane were uncoupled, providing additional support for the hypothesis that these steps are differentially regulated by insulin.
To further explore the differences between accumulation in the evanescent field and insertion into the PM in insulin-resistant adipocytes, we studied cells stimulated with a lower concentration of insulin. The translocation of GLUT4 to the PM stimulated by 0.1 nM insulin was about half that stimulated by 1 or 10 nM insulin (Figure 9A). There was a shift to the right of the insulin dose response for GLUT4-containing vesicle accumulation in the evanescent field in insulin-resistant adipocytes, with an EC50 of the effect in control cells of ∼0.1 nM and an EC50 of greater than 1 nM for the insulin-resistant adipocytes (Figure 9C).
The fraction of GLUT4 in the evanescent field that is inserted into the PM was increased by nearly threefold in cells stimulated with 0.1 nM insulin, similar to the effect of 1 or 10 nM insulin (Figure 9B). The effect of 1 nM insulin on the fraction of GLUT4 in the evanescent field that was inserted into the PM was not affected by insulin resistance, whereas the increase stimulated by 0.1 nM insulin was severely blunted in the insulin-resistant adipocytes (Figure 9B). Thus, although insulin control of GLUT4 insertion into the PM is indeed altered in the insulin-resistant cells, the effect of insulin-resistance is only revealed at insulin concentrations <1 nM. Therefore, these data demonstrate that GLUT4 accumulation in the evanescent field and the fraction of this GLUT4 that is inserted into the PM have different sensitivities to insulin, and as a result of this difference, they are differentially affected in insulin-resistant adipocytes.
One possible explanation for the inhibition of insulin-stimulated recruitment of GLUT4-containing vesicles is a disruption in Akt phosphorylation of AS160. However, there was no detectable reduction in Akt phosphorylation of AS160 in the insulin-resistant cells (Supplemental Figure 3). Therefore, despite normal phosphorylation of AS160, GLUT4-containing vesicle accumulation in the evanescent field is disrupted in the insulin-resistant cells. These results support recent findings that insulin resistance measured by GLUT4 translocation (a functional measure of insulin action) is not accompanied by a significant reduction in AS160 phosphorylation (Hoehn et al., 2008).
DISCUSSION
GLUT4-specialized Vesicles Are Formed from TR-containing Endosomes
Here we use quantitative TIRFM to analyze intracellular trafficking of GLUT4. Past studies have characterized the behavior of GLUT4 using TIRFM (Lizunov et al., 2005; Gonzalez and McGraw, 2006; Bai et al., 2007; Huang et al., 2007). Those results established that GLUT4-containing vesicles adjacent to the PM are elements of the basal retention machinery, emphasizing the importance of understanding the mechanisms that control the biogenesis and targeting of cargo to these vesicles. Here we begin to address some of those questions. We show that mutations that increase GLUT4 in the PM of basal adipocytes also caused an increased colocalization of GLUT4 with TR in vesicles adjacent to the PM and a corresponding decreased colocalization with IRAP-containing vesicles. These findings reveal information on two aspects of the biogenesis and function of these vesicles. One, the altered colocalizations of the GLUT4 mutants establishes that GLUT4 defaults to TR recycling vesicles when its targeting to the specialized vesicles is altered; suggesting that the GLUT4/IRAP-containing specialized vesicles are derived from TR-containing endosomes (as hypothesized in Figure 1A). Previous in vitro studies have shown that GLUT4-containing vesicles can be formed from endosomes (e.g., Shi and Kandror, 2005), but here we provide evidence in studies of intact cells for the formation of specialized GLUT4-containing vesicles from endosomes. Two, correlation between the relative increases of the mutants in TR recycling vesicles and the increased amounts of the mutants in the PM of basal adipocytes, provides compelling evidence that the mutants recycle to the PM in TR recycling vesicles in basal adipocytes. Thus, targeting of GLUT4 from endosomes to specialized vesicles adjacent to the PM, mediated by cytoplasmic GLUT4 motifs, is an essential aspect of proper basal intracellular retention.
In addition to being localized to specialized vesicles adjacent to the PM, GLUT4 also accumulates in a perinuclear compartment that may be an element of the TGN (Figure 1A, retention pathway 1; Govers et al., 2004; Blot and McGraw, 2008). Cycling of GLUT4 between this compartment and endosomes, which is determined by the FQQI motif, is important for basal retention (Blot and McGraw, 2008). Although it is likely that the majority of the GLUT4- and IRAP-containing vesicles in the evanescent field are the specialized vesicles noted in retention pathway 2 (Figure 1A), we cannot eliminate the possibility that some of the GLUT4- and IRAP-containing vesicles in the evanescent field are trafficking between the perinuclear compartment and endosomal compartments (retention pathway 1). Regardless, our data demonstrate that mutations that cause an increase of GLUT4 in TR recycling vesicles result in an increase of GLUT4 in the PM of basal adipocytes.
In Insulin-stimulated Adipocytes GLUT4 Does Not Traffic to the PM in TR Recycling Vesicles
Within 15 min of insulin stimulation, the GLUT4 distribution between the PM and interior of cells achieves a new steady state in which PM GLUT4 is increased by ∼10-fold. This increased steady-state level of PM GLUT4 supports increased glucose transport and a corresponding decrease in blood glucose. It is of considerable importance to understand how elevated PM GLUT4 is maintained in the insulin-stimulated steady state. The increases of the GLUT4 mutants in the PM of basal adipocytes, particularly the eightfold increase of the GLUT4 mutated in both the FQQI and TELE-based motifs, raised the possibility that recycling of GLUT4 in TR recycling vesicles is involved in maintaining elevated levels of PM GLUT4. However, insulin did not alter the colocalization of GLUT4 and TR, demonstrating that GLUT4 is segregated from the TR recycling vesicles in the presence of insulin. Consequently, GLUT4 traffics by a specialized pathway in both basal and insulin-stimulated adipocytes.
Differential Regulation in the Accumulation of GLUT4 Adjacent to the PM and Its Insertion into the PM
We had previously concluded, based on different sensitivities to Akt inhibition, that the accumulation of GLUT4 in the evanescent field and the fraction of the GLUT4 in the evanescent field that is inserted into the PM are differentially regulated by insulin (Gonzalez and McGraw, 2006). A similar conclusion has been reached for control of GLUT4-containing vesicle trafficking in muscle cells (Randhawa et al., 2008). The results reported here, using a dual-color TIRFM assay, are in agreement with those previous conclusions. Moreover, we show that the control of GLUT4 accumulation in the evanescent field and the control of the fraction of GLUT4 in the evanescent field that is inserted into the PM have different sensitivities to insulin. The effect of 1 nM insulin on the accumulation of GLUT4 in the evanescent field was blocked in insulin-resistant adipocytes, whereas stimulation of GLUT4 insertion into the PM was not affected. The different sensitivities of these two steps in insulin-resistant adipocytes provide independent and compelling support for the hypothesis that GLUT4-containing vesicle accumulation and insertion into the PM are differentially controlled by insulin.
Selective Insulin Resistance in the Regulation of GLUT4 Translocation
The differential effect of insulin resistance on GLUT4 accumulation in the evanescent field and insertion of this GLUT4 into the PM is an example of selective insulin resistance, a state when one insulin-regulated process is resistant to the effect of insulin, whereas another retains sensitivity (Brown and Goldstein, 2008). In type 2 diabetes the liver displays characteristics of selective insulin resistance with the gluconeogenic program resistant to the inhibitory effects of insulin, whereas the stimulatory effect of insulin on lipogenesis is intact. The mechanism underlying this difference is not known. Our results suggest inherent differences in insulin sensitivities as a possible means for developing selective insulin resistance. The accumulation in the evanescent field and insertion into the PM of GLUT4-containing vesicles are controlled by insulin, yet in insulin-resistant adipocytes GLUT4 accumulation stimulated by 1 nM insulin was blocked, whereas insertion into the PM fusion was unaffected. These results establish selective insulin resistance in two steps of insulin signaling to GLUT4. In control adipocytes, 0.1 nM insulin had the same stimulatory effect on GLUT4 insertion into the PM as 1 nM insulin, whereas GLUT4 accumulation in the evanescent field stimulated by 0.1 nM insulin was less than that stimulated by 1 nM insulin. We propose that this difference in sensitivity accounts for the selective insulin resistance. We do not know whether differences in the inherent sensitivity to insulin contribute to other examples of selective insulin resistance, but our findings establish the importance of considering how differences in the EC50s might effect how these different processes are altered in insulin resistance.
Mechanisms That Control GLUT4-containing Vesicle Accumulation and Insertion into the PM
How insulin controls the fusion of GLUT4-containing vesicles is not known. Phospholipase D (PLD) has been shown to be involved in the control of GLUT4 vesicle fusion (Huang et al., 2005). Thus, one possibility is that the stimulation of insertion measured in the TIRFM assay reflects control of PLD activity. Insulin may also exert control of vesicle fusion through PLD-independent actions. There is precedent for the control of GLUT4 vesicle fusion via control of SNARE protein function, and this remains one possible site of insulin action (Watson and Pessin, 2007). In addition, recent studies suggest that control of actin is important for promoting GLUT4 translocation to the PM, perhaps by regulation of GLUT4-containing vesicle fusion (Chen et al., 2006; McCarthy et al., 2006; Brozinick et al., 2007; Lopez et al., 2009).
A number of steps could contribute to the increased accumulation of GLUT4 in the evanescent field. These include formation of GLUT4-containing vesicles, the movement of vesicles to the PM, and the docking of vesicles at the PM, as well as the fusion of these vesicles to the PM. Changes in one or all of these steps would contribute to the increased recruitment. Of particular interest is the role of AS160. Past studies have shown that AS160 is a negative regulator of GLUT4 vesicle recruitment/docking (Zeigerer et al., 2004; Jiang et al., 2008). Here we show that although insulin-stimulated accumulation of GLUT4-containing vesicles is inhibited in the insulin-resistant cells, insulin-stimulated phosphorylation of AS160 (mechanism for inhibition of AS160 GAP activity) is not inhibited. Our results extend previous studies that demonstrated reduced GLUT4 translocation in insulin-resistant adipocytes was not accompanied by a detectable change in AS160 phosphorylation (Hoehn et al., 2008). Possible explanations for the uncoupling of AS160 phosphorylation from GLUT4 translocation in the insulin-resistant adipocytes include the following: the deregulation of AS160 function at a different level (e.g., AS160 localization), a change in a step preceding that controlled by AS160, or the effect of insulin resistance reflecting the development of a “dominant” activity that blocks recruitment, as may be the case if a negative feedback loop is up-regulated as a consequence of insulin resistance. The use of methods to interrogate the individual steps controlling GLUT4 redistribution to the PM will provide novel insights into insulin action as well as how control of GLUT4 trafficking is disrupted by the development of insulin resistance.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Sameer Mohammed, Dorothee Molle, and Salihah Dick for discussions; Gus Lienhard (Dartmouth University) for insightful suggestions; Fred Maxfield for advice on data analysis; and David Iaea for expert technical assistance. This work was supported by National Institutes of Health Grants DK52852 and DK69982 (T.E.M.) and an American Diabetes Association mentor-based postdoctoral fellowship (I. J.).
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-08-0751) on February 24, 2010.
REFERENCES
- Bai L., Wang Y., Fan J., Chen Y., Ji W., Qu A., Xu P., James D. E., Xu T. Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab. 2007;5:47–57. doi: 10.1016/j.cmet.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Barnett S. F., et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J. 2005;385:399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blot V., McGraw T. E. Molecular mechanisms controlling GLUT4 intracellular retention. Mol. Biol. Cell. 2008;19:3477–3487. doi: 10.1091/mbc.E08-03-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. S., Goldstein J. L. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7:95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Brozinick J. T., Jr, Berkemeier B. A., Elmendorf J. S. “Actin”g on GLUT4, membrane and cytoskeletal components of insulin action. Curr. Diabet. Rev. 2007;3:111–122. doi: 10.2174/157339907780598199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Liu P., Pattar G. R., Tackett L., Bhonagiri P., Strawbridge A. B., Elmendorf J. S. Chromium activates glucose transporter 4 trafficking and enhances insulin-stimulated glucose transport in 3T3-L1 adipocytes via a cholesterol-dependent mechanism. Mol. Endocrinol. 2006;20:857–870. doi: 10.1210/me.2005-0255. [DOI] [PubMed] [Google Scholar]
- Chen G., Raman P., Bhonagiri P., Strawbridge A. B., Pattar G. R., Elmendorf J. S. Protective effect of phosphatidylinositol 4,5-bisphosphate against cortical filamentous actin loss and insulin resistance induced by sustained exposure of 3T3-L1 adipocytes to insulin. J. Biol. Chem. 2004;279:39705–39709. doi: 10.1074/jbc.C400171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugani C. B., Klip A. Glucose transporter 4, cycling, compartments and controversies. EMBO Rep. 2005;6:1137–1142. doi: 10.1038/sj.embor.7400584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguez L., Lee A., Chavez J. A., Miinea C. P., Kane S., Lienhard G. E., McGraw T. E. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Gonzalez E., McGraw T. E. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol. Biol. Cell. 2006;17:4484–4493. doi: 10.1091/mbc.E06-07-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govers R., Coster A. C., James D. E. Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol. Cell. Biol. 2004;24:6456–6466. doi: 10.1128/MCB.24.14.6456-6466.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehn K. L., Hohnen-Behrens C., Cederberg A., Wu L. E., Turner N., Yuasa T., Ebina Y., James D. E. IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 2008;7:421–433. doi: 10.1016/j.cmet.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P., Altshuller Y. M., Hou J. C., Pessin J. E., Frohman M. A. Insulin-stimulated plasma membrane fusion of Glut4 glucose transporter-containing vesicles is regulated by phospholipase D1. Mol. Biol. Cell. 2005;16:2614–2623. doi: 10.1091/mbc.E04-12-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Czech M. P. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Huang S., Lifshitz L. M., Jones C., Bellve K. D., Standley C., Fonseca S., Corvera S., Fogarty K. E., Czech M. P. Insulin stimulates membrane fusion and GLUT4 accumulation in clathrin coats on adipocyte plasma membranes. Mol. Cell. Biol. 2007;27:3456–3469. doi: 10.1128/MCB.01719-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jess T. J., Belham C. M., Thomson F. J., Scott P. H., Plevin R. J., Gould G. W. Phosphatidylinositol 3′-kinase, but not p70 ribosomal S6 kinase, is involved in membrane protein recycling: wortmannin inhibits glucose transport and downregulates cell-surface transferrin receptor numbers independently of any effect on fluid-phase endocytosis in fibroblasts. Cell Signal. 1996;8:297–304. doi: 10.1016/0898-6568(96)00054-x. [DOI] [PubMed] [Google Scholar]
- Jiang L., Fan J., Bai L., Wang Y., Chen Y., Yang L., Chen L., Xu T. Direct quantification of fusion rate reveals a distal role for AS160 in insulin-stimulated fusion of GLUT4 storage vesicles. J. Biol. Chem. 2008;283:8508–8516. doi: 10.1074/jbc.M708688200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandror K. V., Pilch P. F. gp160, a tissue-specific marker for insulin-activated glucose transport. Proc. Natl. Acad. Sci. USA. 1994;91:8017–8021. doi: 10.1073/pnas.91.17.8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karylowski O., Zeigerer A., Cohen A., McGraw T. E. GLUT4 is retained by an intracellular cycle of vesicle formation and fusion with endosomes. Mol. Biol. Cell. 2004;15:870–882. doi: 10.1091/mbc.E03-07-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S. R., Scott H. M., Mastick C. C., Aebersold R., Lienhard G. E. Cloning and characterization of a novel insulin-regulated membrane aminopeptidase from Glut4 vesicles [published erratum appears in J. Biol. Chem. 1995 Dec 15; 270(50):30236] J. Biol. Chem. 1995;270:23612–23618. doi: 10.1074/jbc.270.40.23612. [DOI] [PubMed] [Google Scholar]
- Klip A. The many ways to regulate glucose transporter 4. Appl. Physiol. Nutr. Metab. 2009;34:481–487. doi: 10.1139/H09-047. [DOI] [PubMed] [Google Scholar]
- Lampson M. A., Racz A., Cushman S. W., McGraw T. E. Demonstration of insulin-responsive trafficking of GLUT4 and vpTR in fibroblasts. J. Cell Sci. 2000;113(Pt 22):4065–4076. doi: 10.1242/jcs.113.22.4065. [DOI] [PubMed] [Google Scholar]
- Lampson M. A., Schmoranzer J., Zeigerer A., Simon S. M., McGraw T. E. Insulin-regulated release from the endosomal recycling compartment is regulated by budding of specialized vesicles. Mol. Biol. Cell. 2001;12:3489–3501. doi: 10.1091/mbc.12.11.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larance M., et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J. Biol. Chem. 2005;280:37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- Lizunov V. A., Matsumoto H., Zimmerberg J., Cushman S. W., Frolov V. A. Insulin stimulates the halting, tethering, and fusion of mobile GLUT4 vesicles in rat adipose cells. J. Cell Biol. 2005;169:481–489. doi: 10.1083/jcb.200412069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J. A., Burchfield J. G., Blair D. H., Mele K., Ng Y., Vallotton P., James D. E., Hughes W. E. Identification of a distal GLUT4 trafficking event controlled by actin polymerization. Mol. Biol. Cell. 2009;17:3918–3929. doi: 10.1091/mbc.E09-03-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maianu L., Keller S. R., Garvey W. T. Adipocytes exhibit abnormal subcellular distribution and translocation of vesicles containing glucose transporter 4 and insulin-regulated aminopeptidase in type 2 diabetes mellitus: implications regarding defects in vesicle trafficking. J. Clin. Endocrinol. Metab. 2001;86:5450–5456. doi: 10.1210/jcem.86.11.8053. [DOI] [PubMed] [Google Scholar]
- Marsh B. J., Martin S., Melvin D. R., Martin L. B., Alm R. A., Gould G. W., James D. E. Mutational analysis of the carboxy-terminal phosphorylation site of GLUT-4 in 3T3-L1 adipocytes. Am. J. Physiol. 1998;275:E412–E422. doi: 10.1152/ajpendo.1998.275.3.E412. [DOI] [PubMed] [Google Scholar]
- Martin O. J., Lee A., McGraw T. E. GLUT4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulin-modulated bipartite dynamic mechanism. J. Biol. Chem. 2006;281:484–490. doi: 10.1074/jbc.M505944200. [DOI] [PubMed] [Google Scholar]
- Martin S., Tellam J., Livingstone C., Slot J. W., Gould G. W., James D. E. The glucose transporter (GLUT-4) and vesicle-associated membrane protein-2 (VAMP-2) are segregated from recycling endosomes in insulin-sensitive cells. J. Cell Biol. 1996;134:625–635. doi: 10.1083/jcb.134.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy A. M., Spisak K. O., Brozinick J. T., Elmendorf J. S. Loss of cortical actin filaments in insulin-resistant skeletal muscle cells impairs GLUT4 vesicle trafficking and glucose transport. Am. J. Physiol. Cell Physiol. 2006;291:C860–C868. doi: 10.1152/ajpcell.00107.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa V. K., Ishikura S., Talior-Volodarsky I., Cheng A. W., Patel N., Hartwig J. H., Klip A. GLUT4 vesicle recruitment and fusion are differentially regulated by Rac, AS160, and Rab8A in muscle cells. J. Biol. Chem. 2008;283:27208–27219. doi: 10.1074/jbc.M804282200. [DOI] [PubMed] [Google Scholar]
- Sano H., Eguez L., Teruel M., Fukuda M., Chuang T., Chavez J., Lienhard G., McGraw T. Rab10 is a target of the insulin regulated AS160 rabGAP protein required for insulin-stimulated translocation of GLUT4 to the plasma membrane of adipocytes. Cell Metab. 2007;5:293–303. doi: 10.1016/j.cmet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Sano H., Kane S., Sano E., Miinea C. P., Asara J. M., Lane W. S., Garner C. W., Lienhard G. E. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- Shi J., Kandror K. V. Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes. Dev. Cell. 2005;9:99–108. doi: 10.1016/j.devcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Watson R. T., Pessin J. E. GLUT4 translocation: the last 200 nanometers. Cell Signal. 2007;19:2209–2217. doi: 10.1016/j.cellsig.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Zeigerer A., Lampson M., Karylowski O., Sabatini D., Adesnik M., Ren M., McGraw T. GLUT4 retention in adipocytes requires two intracellular insulin-regulated transport steps. Mol. Biol. Cell. 2002;13:p2421–p2435. doi: 10.1091/mbc.E02-02-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigerer A., McBrayer M. K., McGraw T. E. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol. Biol. Cell. 2004;15:4406–4415. doi: 10.1091/mbc.E04-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P., Yang L., Lopez J. A., Fan J., Burchfield J. G., Bai L., Hong W., Xu T., James D. E. Variations in the requirement for v-SNAREs in GLUT4 trafficking in adipocytes. J. Cell Sci. 2009;122:3472–3480. doi: 10.1242/jcs.047449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.