

In this study, the authors demonstrate a role and a mechanism for h3/acidic calponin in regulating ERK1/2 activity and REF52.2 fibroblast motility, likely due to a regulation of ERK1/2 mediated l-caldesmon phosphorylation.
Abstract
Migration of fibroblasts is important in wound healing. Here, we demonstrate a role and a mechanism for h3/acidic calponin (aCaP, CNN3) in REF52.2 cell motility, a fibroblast line rich in actin filaments. We show that the actin-binding protein h3/acidic calponin associates with stress fibers in the absence of stimulation but is targeted to the cell cortex and podosome-like structures after stimulation with a phorbol ester, phorbol-12,13-dibutyrate (PDBu). By coimmunoprecipitation and colocalization, we show that extracellular signal-regulated kinase (ERK)1/2 and protein kinase C (PKC)α constitutively associate with h3/acidic calponin and are cotargeted with h3/acidic calponin in the presence of PDBu. This targeting can be blocked by a PKC inhibitor but does not require phosphorylation of h3/acidic calponin at the PKC sites S175 or T184. Knockdown of h3/acidic calponin results in a loss of PDBu-mediated ERK1/2 targeting, whereas PKCα targeting is unaffected. Caldesmon is an actin-binding protein that regulates actomyosin interactions and is a known substrate of ERK1/2. Both ERK1/2 activity and nonmuscle l-caldesmon phosphorylation are blocked by h3/acidic calponin knockdown. Furthermore, h3/acidic calponin knockdown inhibits REF52.2 migration in an in vitro wound healing assay. Our findings are consistent with a model whereby h3/acidic calponin controls fibroblast migration by regulation of ERK1/2-mediated l-caldesmon phosphorylation.
INTRODUCTION
The cytoskeleton of a nonmuscle cell controls cell shape and motility but can also play an important role in many signaling pathways (Carpenter, 2000). Integrin receptors for example connect the extracellular matrix to mitogen-activated protein kinase (MAPK) cascades, thereby leading to alterations in gene expression and actin organization (Chu et al., 2000).
Extracellular signal-regulated kinase (ERK)1/2 is a member of the MAPK family. It is a serine/threonine kinase and is activated by a mitogen-activated protein (MAP)-kinase/ERK kinase (MEK). Activated ERK1/2 molecules form homodimers that are able to translocate into the nucleus, phosphorylate transcription factors such as Myc, and thereby regulate gene expression (Khokhlatchev et al., 1998; Lewis et al., 1998). In undifferentiated cells, MAPK signaling mainly promotes cell growth, but several indications exist that this cascade also controls cytoskeleton remodelling and thereby shape, contractility, and motility (English et al., 1999; Hai and Gu, 2006).
The protein kinase C (PKC) family consists of at least 12 phospholipid-dependent serine/threonine protein kinases, whose activity is controlled by both the interaction of cofactors as well as by its phosphorylation status. For release of the autoinhibitory pseudosubstrate domain from the substrate binding site, all PKCs require phosphatidylserine as cofactor. For the binding of lipids and for activation, PKCs must additionally be phosphorylated at three highly conserved sites in the catalytic domain (Newton, 1997). Depending on their subcellular localization, expression status, and substrate specificity, members of the PKC family are implicated in a variety of cellular processes, including proliferation, differentiation, apoptosis, cell motility, and cytoskeleton rearrangements (Toker, 1998).
The actin-associated protein calponin (CaP) has also been proposed to have both cytoskeletal and signaling functions, although in differing settings, and by different investigators (Winder and Walsh, 1990a; Shirinsky et al., 1992; Menice et al., 1997; Leinweber et al., 1999, 2000). CaP was first isolated from gizzard smooth muscle cells as h1CaP (CNN1, basic CaP) and was shown to inhibit in vitro actomyosin ATPase activity and thus, by implication, contractility (Winder and Walsh, 1990b). To date, there are three known isoforms of calponin that are encoded by different genes. Basic or h1CaP is restricted in its expression to differentiated smooth muscle (Takahashi et al., 1988), whereas h2CaP (CNN2, neutral CaP) (Strasser et al., 1993) and h3/acidic CaP (CNN3, aCaP) (Applegate et al., 1994) are more widely expressed yet far less well studied. All CaP proteins consist of a conserved CaP homology domain located in their N terminus and three CaP like repeats referred to as CLIK motifs (Mezgueldi et al., 1995; Mino et al., 1998; Burgstaller et al., 2002), whereas the very C terminus is a highly variable region. The h3/acidic isoform of CaP contains a C-terminal domain that is strongly acidic (Applegate et al., 1994).
Only the basic isoform of CaP has been investigated in any detail mechanistically and yet its function in smooth muscle remains highly controversial. Some authors (Takahashi et al., 1986; Allen and Walsh, 1994) have suggested that it functions in a manner similar to that of troponin in striated muscle by directly inhibiting actomyosin interactions. Others (Menice et al., 1997; Leinweber et al., 1999; Je et al., 2001) have suggested a qualitatively different functional mechanism of facilitating agonist-induced signal transduction and contractility by acting as an adaptor protein for ERK1/2 and PKCα/ε in differentiated vascular smooth muscle cells. Little is known about the mechanistic function of the h3/acidic isoform except by analogy to h1CaP.
In the present study we examined the role of the h3/acidic isoform of CaP in ERK1/2 signaling, using REF52.2 fibroblasts as a model of nonmuscle cells with a distinctive actin cytoskeleton. Our results demonstrate that h3/acidic CaP constitutively associates with both ERK1/2 and PKCα in vivo and cotranslocates to the cell cortex and podosome-like structures together with these two signaling molecules after stimulation with the phorbol ester phorbol-12,13-dibutyrate (PDBu). The h3/acidic CaP translocation is blockable by a PKC inhibitor but does not require phosphorylation at Ser175 and/or Thr184 by PKC. By using a C-terminal deletion mutant of h3/acidic CaP, we also show that the acidic tail of the protein is not involved in the PDBu-mediated translocation process. Moreover, a small interfering RNA (siRNA)-mediated knockdown of h3/acidic CaP blocks not only PDBu-mediated ERK1/2 targeting, ERK1/2 activation, and phosphorylation of the ERK1/2 substrate l-caldesmon (low-molecular-weight form of CaD [l-CaD]) but also cell migration in a wound healing assay. Our results indicate that h3/acidic CaP plays an important role in REF52.2 fibroblast motility, probably due to a regulation of ERK1/2 mediated l-CaD phosphorylation.
MATERIALS AND METHODS
DNA Constructs
Rat h3/acidic CaP cDNA was excised from pET30b(+)-aCaP (plasmid was a gift from T. Tao, Boston Biomedical Research Institute, Watertown, MA) with restriction enzymes BamHI and HindIII and cloned into the pCMV-tag 2A mammalian expression vector (Clontech, Mountain View, CA). The plasmid, further denoted as pFLAG-aCaP-wt, codes for the FLAG-aCaP-wt fusion protein. To generate a pFLAG-aCaP-S175A construct, harboring the point mutation S175 to alanine, site-directed mutagenesis was performed with the oligonucleotide pair 5′-aaa tgt gcc gcc cag gcg gg-3′ and 5′-ccc gcc tgg gcg gca cat tt-3′, by using pFLAG-aCaP-wt as template. The pFLAG-aCaP-S175A/T184A mutant was generated using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) with the oligonucleotide pair 5′-gta tga cag cct atg ggg ctc gga ggc atc ttt atg-3′ and 5′-cat aaa gat gcc tcc gag ccc cat agg ctg tca tac-3′, by using pFLAG-aCaP-S175A as template. To generate rat aCaP-wt and aCaP-ΔCt deletion mutants, we amplified the fragments in a polymerase chain reaction (PCR) with the following oligonucleotide pairs: 5′-gaa aga tct atg acc cac ttc aac aag ggc-3′ and 5′-cgc gga tcc cta gta atc gat gcc-3′ for aCaP-wt; 5′-gaa aga tct atg acc cac ttc aac aag ggc-3′ and 5′-cgc gga tcc aca gta ctt ggg gtc-3′ for aCaP-ΔCt. PCR fragments were digested with BglII and BamHI and cloned into the pEGFP-C1 plasmid (Clontech). Generated plasmids are further denoted as pEGFP-aCaP-wt and pEGFP-aCaP-ΔCt (corresponding to amino acids [aa] 1–273). To generate bacterial expression vectors coding for either ferret h1CaP or rat h3/acidic CaP, we have chosen the pTYB2 vector (IMPACT-CN System; New England Biolabs, Ipswich, MA) which contains a C-terminal chitin binding domain (CBD). Ferret h1CaP fragment was amplified by PCR using oligonucleotides 5′-gga att cca tat gat gtc ctc tgc tca c-3′ and 5′-ctc gag ggc gga gtt ata gta gtt g-3′ and pET30b(+)-h1CaP as template. Rat h3/acidic CaP fragment was amplified with oligonucleotide pairs 5′-gga att cca tat gat gac cca ctt caa caa g-3′ and 5′-tcc ccc ggg gta atc gat gcc c-3′ by using pET30b(+)-aCaP as template. The h1CaP fragment was cloned via NdeI and XhoI restriction sites into the pTYB2 vector, and h3/acidic CaP via NdeI and SmaI. The validity of all constructs was confirmed by DNA sequencing.
Cell Culture and Transfection
The rat embryonic REF52.2 and the mouse NIH-3T3 fibroblast cell lines were kindly provided by Dr. K. H. Scheidtmann (University of Bonn, Bonn, Germany), and mouse BALB/c 3T3 fibroblast cells were kindly provided by Dr. Joyce Wong (Boston University, Boston, MA). All cell lines were maintained in DMEM high glucose (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal calf serum (FCS) (Invitrogen, Carlsbad, CA) and 1% Pen/Strep (Invitrogen). For siRNA transfection experiments, cells were seeded at 2 × 105/60-mm culture dish or at 1.7 × 104/coverslip and transiently transfected using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's protocol. A mix of four different siRNA sequences against rat h3/acidic calponin (SMARTpool Rat CNN3) or mouse h3/acidic calponin (SMARTpool Mouse CNN3) was purchased from Dharmacon RNA Technologies (Lafayette, CO). As control, a mix of four different nontargeting siRNA sequences was used (Dharmacon RNA Technologies; nontargeting siRNA pool #2). For DNA transfection, cells were seeded at 1.7 × 104/coverslip and transiently transfected by using the jetPEI transfection reagent (PolyPlus, New York, NY) according to the manufacturer's protocol. Calphostin (Calbiochem, San Diego, CA) was used at a final concentration of 0.5 or 1 μM for 30 min, after a prior serum starvation for 30 min and before PDBu stimulation. Cells could not be serum starved for longer times because cells being exposed to prolonged serum starvation displayed phenotypic anomalies indicative of cellular stress related to serum starvation. PDBu was purchased from Calbiochem and used at a final concentration of 1 μM for 30 min.
Immunofluorescence Analysis
For immunofluorescence analysis, cells were fixed with 3% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min at room temperature after treatment with 1% bovine serum albumin, 1% goat serum, and 0.1% Triton X-100 in PBS for at least 1 h and stained overnight at 4°C, depending on the experiment, with the rabbit polyclonal anti-h3/acidic CaP antibody (calponin 3 H-55; Santa Cruz Biotechnology, Santa Cruz, CA) at 1:1000 dilution, the mouse monoclonal anti-PKCα antibody (PKCα clone M4; Millipore, Billerica, MA) at 1:1000 dilution, the mouse monoclonal anti-phospho-ERK1/2 antibody (p44/42 MAPK (Thr202/Tyr204) E10; Cell Signaling Technology, Beverly, MA) at 1:1000 dilution, the mouse monoclonal anti-caldesmon antibody (Abcam, Cambridge, MA) at 1:250 dilution, the rabbit polyclonal anti-cortactin antibody (H222; Cell Signaling Technology) at 1:500 dilution, the rabbit polyclonal anti-matrix metalloproteinase-2 antibody (MMP2; Abcam) at 1:200 dilution, and the mouse monoclonal anti-FLAG M2 antibody (Sigma-Aldrich) at 1:2500 dilution. For costaining of h3/acidic CaP and ERK1/2, we used the rabbit polyclonal anti-MAP kinase (ERK1/2) CT antibody coupled to fluorescein isothiocyanate (Millipore) at 1:500 dilution and the rabbit polyclonal anti-h3/acidic CaP antibody (calponin 3 H-55; Santa Cruz Biotechnology) coupled to Alexa555 at a dilution of 1:500. Alexa555 dye coupling to anti-h3/acidic CaP was performed using the AlexaFluor555 labeling kit (Invitrogen) following manufacturer's protocol. As secondary antibodies, Alexa488-conjugated goat anti-mouse immunoglobulin (Ig)G (Invitrogen), Alexa568-conjugated goat anti-rabbit IgG (Invitrogen), or Alexa488-conjugated goat anti-rabbit IgG (Invitrogen) was used at 1:1000 dilution and incubated for 30 min. Filamentous actin was stained with Alexa488- or Alexa568-labeled phalloidin (Invitrogen) at 1:5000 dilution, nuclei were stained with 4,6-diamidino-2-phenylindole (Sigma-Aldrich) for 15 min and subsequent washing with PBS. Cells were examined with a TE 2000-E2 Perfect Focus epifluorescence microscope (Nikon Instruments, Melville, NY) and a 60×, numerical aperture 1.4 oil immersion objective. For deconvolution, three-dimensional images were acquired and recorded with a CoolSNAP HQ2 camera (Photometrics, Tucson, AZ). Out-of-focus fluorescent blur was removed by deconvolution using the NIS-Elements AR 3.0 software (Nikon Instruments) (Richardson-Lucy algorithm, constrained iterative-maximum likelihood estimation algorithm). Images were transferred with Adobe Photoshop CS4 software (Adobe Systems, Mountain View, CA).
SDS-Polyacrylamide Gel Electrophoresis (PAGE) and Western Blot Analysis
Cells were lysed in isotonic lysis buffer (10 mM NaPO4, pH 8.0, 140 mM NaCl, 3 mM MgCl2, 1 mM dithiothreitol, 0.5% Nonidet-P40, and 1× protease inhibitor mix [Roche Diagnostics, Indianapolis, IN]). Cell lysates were subjected to SDS-PAGE according to standard procedures and electrophoretically transferred onto polyvinylidene difluoride membranes (Millipore). Residual protein binding sites on the membrane were blocked with blocking buffer (Odyssey; LI-COR Biosciences, Lincoln, NE). The membranes were incubated either with the rabbit polyclonal anti-h3/acidic CaP antibody (calponin 3 H-55; Santa Cruz Biotechnology) at 1:2000 dilution, the mouse monoclonal anti-h1CaP antibody (calponin clone hCP; Sigma-Aldrich) at 1:100,000 dilution, the mouse monoclonal anti-h2CaP antibody (h2-calponin CP21; Hossain et al., 2005) at 1:1000 dilution, the rabbit polyclonal anti-ERK1/2 antibody (ERK1/2 CT; Millipore) at 1:500 dilution, the mouse monoclonal anti-phospho-ERK1/2 antibody (p44/42 MAPK [Thr202/Tyr204] E10; Cell Signaling Technology) at 1:2000 dilution, the mouse monoclonal anti-caldesmon antibody (Abcam) at 1:500 dilution, the rabbit polyclonal anti-phospho-caldesmon (S789) antibody (Millipore) at 1:500 dilution, the mouse monoclonal anti-α-tubulin antibody (Sigma-Aldrich) at 1:5000 dilution, the rabbit polyclonal anti-PAN actin antibody (Cytoskeleton, Denver, CO) at 1:2000 dilution or the mouse monoclonal anti-CBD antibody (New England Biolabs) at 1:2000 dilution. As secondary antibodies, IRDye680-conjugated goat anti-rabbit IgG (Odyssey; LI-COR Biosciences) and IRDye800CM-conjugated goat anti-mouse IgG (Odyssey; LI-COR Biosciences) were used at 1:2000 dilution and incubated for 30 min. Bound antibodies were detected with an Odyssey 1400 infrared imager (Odyssey; LI-COR Biosciences).
Protein Expression and Purification of CBD-tagged h3/Acidic and h1 Calponin
Ferret h1 and rat h3/acidic calponin proteins were expressed in Escherichia coli BL21 cells with a C-terminal CBD tag. The methods were according to instructions of the supplier. In brief, E. coli BL21 cell pellets were resuspended in lysis buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM EDTA, and 0.1% Triton X-100) and sonicated. After centrifugation, the supernatant containing the expressed proteins was used for purification with chitin beads (see In Vitro Pull-Down Assay).
In Vitro Pull-Down Assay
CBD-tagged h3/acidic or h1 calponin proteins, respectively. The CBD tag alone for controls were attached to affinity resins by passing E. coli BL21 lysates through chitin beads. After several washes with a buffer containing 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, and 1 mM EDTA, resins were stored at 4°C and used for pull-down assays. The pull-down assay was performed by incubating the beads with recombinant, active murine ERK2 protein (p42 MAP kinase; New England Biolabs) overnight at 4°C in 1× MAP kinase reaction buffer (New England Biolabs) containing 1× protease inhibitor mix (Roche Diagnostics). Resins were subsequently washed three times with phosphate lysis buffer. Proteins were eluted from the resin by boiling at 100°C for 5 min with SDS sample buffer. Eluted proteins were detected by Western blotting with specific antibodies.
Immunoprecipitation
REF52.2 cells were washed with ice-cold PBS and lysed in isotonic lysis buffer. The lysates were precleared with protein A agarose (Sigma-Aldrich) at 4°C for at least 1 h and then subjected to immunoprecipitation with the anti-MAP kinase/ERK1/2 agarose conjugate (Millipore) at 4°C overnight. As a negative control, cell lysates were incubated with normal rabbit IgG-agarose conjugate (Santa Cruz Biotechnology). The immunoprecipitates were washed three times with low-stringency lysis buffer (10 mM NaPO4, pH 8.0, 50 mM NaCl, 3 mM MgCl2, 1 mM dithiothreitol, 0.5% Nonidet-P40, and 1× protease inhibitor mix [Roche Diagnostics]); the samples were separated on 8.5% SDS-PAGE and further analyzed by Western blotting. For precipitating h3/acidic CaP, REF52.2 cell lysates were precleared with protein G-coupled Dynabeads (Invitrogen). To reduce background, the antibodies used in this assay were cross-linked to protein G-coupled Dynabeads by using the cross-linking reagent bis(sulfosuccinimidyl)suberate following manufacturer's instructions. The precleared cell lysates were incubated overnight at 4°C with the rabbit polyclonal anti-h3/acidic CaP antibody (Santa Cruz Biotechnology) cross-linked to protein G-coupled Dynabeads. As negative control, the rabbit polyclonal anti-green fluorescent protein (GFP) antibody (Clontech) cross-linked to protein G-coupled Dynabeads was used. Precipitated proteins were washed three times with isotonic lysis buffer and eluted by incubation at 80°C for 10 min in sample buffer. Samples were separated on 8.5% SDS-PAGE and further analyzed by Western blotting.
In Vitro Wound Healing Assay
REF52.2 cells (1 × 105) were seeded on 35-mm uncoated glass bottom culture dishes (MatTek, Ashland, MA). Sixteen hours later, cells were transfected with either siRNA against rat h3/acidic calponin or nontargeting siRNA as control. Seventy-two hours after transfection the cell culture medium was exchanged to DMEM containing 10% FCS, 1% Pen/Strep, and 20 mM HEPES buffer, pH 7.2, and the confluent cell layer was wounded using a 100-μl “yellow” pipette tip. Cultures were maintained in a stage top RC 30 microincubator (Warner Instruments, Hamden, CT) at 35°C on an IX 81 microscope (Olympus, Tokyo, Japan). Sterile tissue culture grade mineral oil (Sigma-Aldrich) covered the cell culture medium to prevent evaporation and to give an optically flat viewing area. Wound healing was monitored over 20 h with a 10× phase contrast objective, and images were collected every 15 min using a CoolSNAP HQ camera (Photometrics) controlled by IPLab software (BD Biosciences, San Jose, CA). Quantification of the wound healing process by measuring the wound area and manual tracking of single cells was performed with the ImageJ software (National Institutes of Health, Bethesda, MD).
Statistical Analysis
All values in the text are mean + SE. Differences between means were evaluated using a two-tailed Student's t test. Significant differences were taken at the p < 0.05 level.
RESULTS
Expression Level and Intracellular Localization of h3/Acidic CaP in Fibroblast Cell Lines and Tissue
The h3/acidic isoform of calponin is known to be expressed ubiquitously, even though it was previously speculated that its expression is highest in brain cells and its role was mainly studied in these cell types (Rozenblum and Gimona, 2008). Using a multitissue Western blot derived from ferret (Vetterkind and Morgan, 2009), we show here that h3/acidic CaP is indeed expressed in a variety of tissues, including brain, liver, kidney, lung, trachea, heart, aorta, and portal vein. There was, however, no detectable h3/acidic CaP protein in skeletal muscle (Figure 1A).
Figure 1.

Expression level and intracellular localization of h3/acidic CaP. (For brevity, the abbreviation aCaP is used for h3/acidic CaP in all figures.) (A) A ferret multiple tissue Western blot was stained with antibodies against PAN-actin, tubulin-α, and h3/acidic CaP (aCaP). (B–E) Whole cell lysates (50 μg) of rat REF52.2 (B), mouse NIH-3T3 (C), mouse BALB/c 3T3 (D), and rat A7r5 (E) cells were analyzed by SDS-PAGE and subsequent Western blotting with specific antibodies against h3/acidic CaP (aCaP), h1CaP, or h2CaP as indicated. (F) REF52.2 cells were stained with the h3/acidic CaP-specific antibody used in A–E (a) and with Al488-labeled phalloidin (b) to detect actin filaments. Images were obtained with deconvolution microscopy. The yellow signals in the merged image (d) indicate colocalization. Bar, 10 μm.
In the present study, we wanted to examine the role of h3/acidic CaP in nonmuscle cells but with a pronounced actin cytoskeleton, because the protein is tightly bound to actin filaments and probably exerts its function mainly through this cell compartment. Therefore, as a cell culture model we chose the fibroblast cell line REF52.2, which harbors a strong actin filament system. As shown in Figure 1B, immunostaining of rat REF52.2 cell homogenates with a specific antibody for h3/acidic CaP shows a single band at the molecular mass for h3/acidic CaP (∼36 kDa, lane 1) that does not cross react with the h2CaP isoform (∼32 kDa, lane 3) at a lower molecular mass. No detectable amount of smooth muscle specific h1CaP (∼32 kDa, lane 2) was seen. To rule out that REF52.2 fibroblasts are unusual in displaying a high expression level of h3/acidic CaP, we examined two more fibroblast cell lines: mouse NIH-3T3 cells exhibit the same CaP expression pattern as REF52.2 cells as can be seen in Figure 1C (lanes 1 and 3), whereas mouse BALB/c 3T3 cells express only the h3/acidic isoform of CaP as shown in Figure 1D, lane 1. In contrast, in rat smooth muscle A7r5 cells all three CaP isoforms are present and can be detected (Figure 1E, lanes 1–3). Using the same anti-h3/acidic CaP antibody as for the Western blot, cellular immunofluorescence shows that h3/acidic CaP is located at actin filaments in REF52.2 cells, as indicated by phalloidin costaining (Figure 1Fd, arrows). However, there is also some h3/acidic CaP protein located in the cytosol that does not colocalize with actin filaments (Figure 1F, a and d).
Interaction Studies with h3/acidic CaP, ERK1/2, and PKCα
To test whether h3/acidic CaP is involved in PKCα–ERK1/2 signaling pathways, we examined whether h3/acidic CaP, ERK1/2, and PKCα coexist in a complex. For this purpose, we performed coimmunoprecipitation assays to probe for protein–protein interactions. Whole REF52.2 cell lysates were incubated with anti-ERK1/2-agarose beads (Figure 2A, lane 3). Rabbit IgG directly coupled to agarose was used as a control (Figure 2A, lane 2). Although no ERK1/2, h3/acidic CaP or PKCα pull-down could be detected in the control sample (Figure 2A, lane 2), both h3/acidic CaP and PKCα were coprecipitated with ERK1/2 (Figure 2A, lane 3). A lysate input control (10 μg of total protein) is shown in lane 1. To further corroborate these data, we also performed coimmunoprecipitation studies with REF52.2 cell lysates by using the anti-h3/acidic CaP antibody for precipitation. As shown in Figure 2B, both ERK1/2 and PKCα are copurified with h3/acidic CaP (lane 3), whereas there is no ERK1/2, h3/acidic CaP, or PKCα protein in the negative control (lane 2). However, there is less apparent ERK2 coprecipitating with h3/acidic CaP compared with ERK1 (lane 3). This might be due to a preference of h3/acidic CaP to binding to ERK1. The input lysate control (lane 1; 10 μg of total protein), in contrast, indicates that there is less ERK2 protein than ERK1 protein in the lysate used for the coimmunoprecipitation assay. A different batch of ERK1/2 antibody was used in panel 2B than 2A, so differences in antibody affinities are possible. Hence, the difference of coprecipitated ERK1 and ERK2 together with h3/acidic CaP is more likely due to technical differences. In summary, the results from the coimmunoprecipitation studies show that h3/acidic CaP, PKCα, and ERK1/2 associate in vivo in unstimulated cells. Moreover, coprecipitated PKCα and h3/acidic CaP amounts did not increase upon PDBu treatment (data not shown), demonstrating that those three proteins constitutively form a complex.
Figure 2.
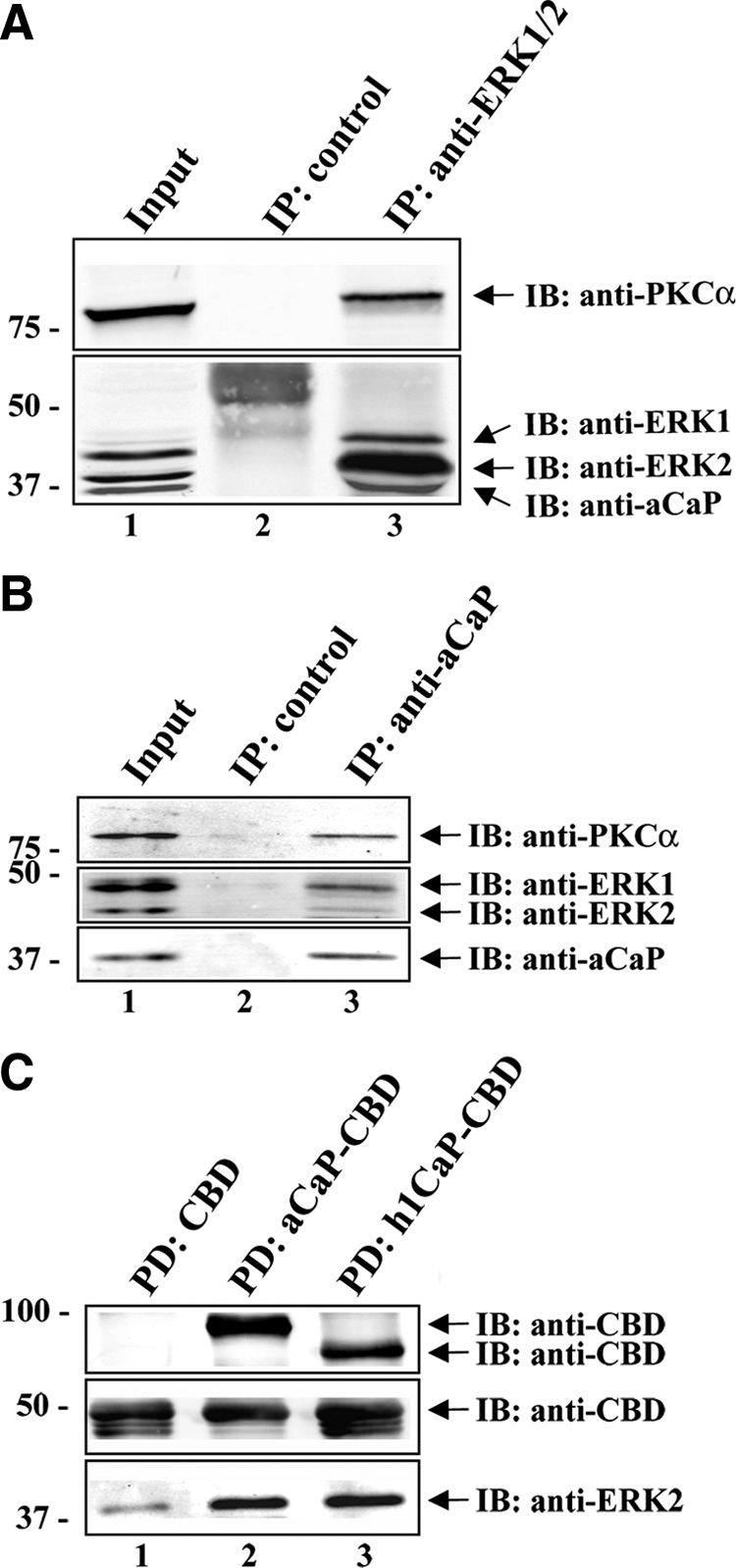
Interaction studies with h3/acidic CaP, ERK1/2, and PKCα. (A) REF52.2 whole cell lysates were subjected to coimmunoprecipitation by using the rabbit polyclonal anti-ERK1/2 antibody coupled to protein A agarose (lane 3). For negative control, lysates were incubated with rabbit IgG coupled to protein A agarose (lane 2). The immunoprecipitated proteins were separated on an 8.5% SDS-PAGE. A lysate input control (10 μg of total protein) was loaded in lane 1. Arrows indicate the positions of PKCα, ERK1, ERK2, and aCaP. A typical Western blot is shown here, and the experiment was repeated three times. (B) Lysates of REF52.2 cells were incubated with the specific h3/acidic CaP antibody (lane 3) cross-linked to protein G-coupled Dynabeads. Lysates incubated with the GFP-antibody cross-linked to protein G-coupled Dynabeads served as negative control (lane 2). Proteins were eluted from the beads and subsequently analyzed by Western blotting. Ten micrograms of total protein of the lysate was loaded as input (lane 1). Arrows indicate the positions of PKCα, ERK1, ERK2, and aCaP. A typical Western blot is shown here, and the experiment was repeated three times. (C) Recombinant h3/acidic CaP-CBD (aCaP-CBD; lane 2), h1CaP-CBD (lane 3), or the CBD tag alone (lane 1), bound to chitin beads were used in an in vitro pull-down assay to probe for direct interaction with recombinant ERK2 protein. Precipitated proteins were eluted from the chitin beads and analyzed by Western blotting. The membrane was immunostained with an anti-CBD antibody and the polyclonal anti-ERK1/2 antibody.
We next wanted to know whether h3/acidic CaP is able to bind ERK directly as it was shown for the basic isoform of calponin (Leinweber et al., 1999). To address this question, we performed an in vitro pull-down assay. Therefore, we used purified recombinant h1CaP as positive control and recombinant h3/acidic CaP, bound to chitin beads via a CBD tag. The CBD tag alone served as negative control. Recombinant active ERK2 protein was then incubated with the proteins and bound proteins were eluted from the beads and subsequently analyzed by Western blot using antibodies against the CBD tag and ERK2. For both h3/acidic (Figure 2C, lane 2) and basic CaP (Figure 2C, lane 3), we saw a strong interaction with ERK2, whereas there was only little ERK2 bound to the CBD tag alone (Figure 2C, lane 1). Note that the CBD tag visible in lanes 2 and 3 (Figure 2C, middle) derives from proteolysis of the recombinant h3/acidic CaP-CBD and h1CaP-CBD proteins. Hence, we conclude that h3/acidic CaP is able to bind ERK2 directly.
h3/Acidic CaP, ERK1/2, and PKCα Localize at the Cortex and in Podosome-like Structures upon PDBu Stimulation
We wanted to know whether h3/acidic CaP, ERK1/2, and PKCα translocate in PDBu-stimulated fibroblast cells, because activated PKC and activated ERK1/2 are reported to localize at the cell membrane, and h1CaP was shown to move to the cell cortex in stimulated differentiated vascular smooth muscle cells (Khalil and Morgan, 1993; Pouyssegur, 2000). The rat fibroblast cell line REF52.2 was stimulated with the PKC-activator PDBu and subsequently immunostained for either h3/acidic CaP, ERK1/2, or PKCα. Actin filaments were visualized using Alexa568-labeled phalloidin. As shown in Figure 3, all three proteins do translocate to the cell cortex and actin-rich aggregates (Figure 3, A–C, see arrows in merged images) in stimulated REF52.2 fibroblast cells. Because PDBu is known to induce podosomes in a variety of cell types, we were curious to see whether these actin-rich aggregates might represent podosomes. Therefore, we immunofluorescently labeled endogenous cortactin and MMP-2 proteins in stimulated REF52.2 cells. Indeed, the actin-rich aggregates forming upon PDBu treatment colocalize with cortactin (Figure 3D), a protein known to be required for the formation and function of podosomes. Moreover, the structures also colocalized with MMP-2 (Figure 3E), a protease known to be secreted at podosomes/invadopodia (Webb et al., 2006; Clark and Weaver, 2008; Crowley et al., 2009; Young et al., 2009). Because the nomenclature of podosomes is usually restricted to monocytes, we call these structures “podosome-like” in the REF52.2 fibroblast cell line used in this study. In summary, we show that h3/acidic CaP, ERK1/2, and PKCα translocate in PDBu-treated fibroblasts to the cell cortex and to podosome-like structures.
Figure 3.

Translocation of h3/acidic CaP, ERK1/2, and PKCα to podosome-like structures and the cortex in PDBu-treated rat fibroblast cells. REF52.2 fibroblasts were grown on glass coverslips and then stimulated with 1 μM PDBu for 30 min. After fixation with paraformaldehyde, cells were immunofluorescently labeled with specific antibodies against aCaP (A, a), ERK1/2 (B, a), PKCα (C, a), cortactin (D, a), and MMP-2 (E, a). Actin filaments were visualized with Al568-labeled phalloidin (b). The yellowish signals in the merged images (c) indicate colocalization. Bar, 10 μm.
PDBu-mediated ERK1/2, but Not PKCα, Translocation Requires h3/Acidic CaP in REF52.2 Fibroblasts
We next examined whether h3/acidic CaP, ERK1/2, and PKCα cotranslocate to the membrane as a complex by stimulating REF52.2 cells with 1 μM PDBu for 30 min. As a control, parallel cells were treated with vehicle alone (0.01% dimethyl sulfoxide [DMSO]). A fraction of h3/acidic CaP as well as PKCα showed a filamentous staining pattern in unstimulated cells with partial colocalization of the proteins (Figure 4A, a–d). PDBu treatment induced a cotranslocation of h3/acidic CaP and PKCα to the cell cortex and to podosome-like structures (Figure 4A, e–h, arrows). A similar cotranslocation upon stimulation to the cell cortex and podosome-like structures was observed for h3/acidic CaP and ERK1/2 proteins (Figure 5A, e–h, arrows). Because the antibodies used in immunofluorescence staining of h3/acidic CaP and ERK1/2 were both raised in the rabbit, they were directly labeled, resulting in lesser fluorescent intensity and some increase in background; however, it can be seen that fractions of both proteins show a filamentous staining pattern in unstimulated cells (Figure 5A, a–d) and that stimulation with PDBu induces a cotranslocation and colocalization of ERK1/2 and h3/acidic CaP to the cell cortex and podosome-like structures (Figure 5A, e–h, arrows).
Figure 4.

Knockdown of h3/acidic CaP does not affect PKCα targeting upon PDBu stimulation. (A) REF52.2 cells were transfected with siRNA against aCaP siRNA (i–p) or nontargeting control siRNA (a–h). Three days after transfection, cells were treated with 0.01% DMSO for control (a–d and i–l) or with 1 μM PDBu for 30 min (e–f and m–p) before staining for aCaP (a, e, i, and m) and PKCα (b, f, j, and n) with the rabbit polyclonal anti-h3/acidic CaP and the mouse monoclonal anti-PKCα antibodies. Images were obtained with deconvolution microscopy. The yellowish signal in the merged images indicates colocalization (d, h, l, and p). Bar, 10 μm. (B) REF52.2 cells transfected with siRNA against h3/acidic CaP or with nontargeting control siRNA were treated with 1 μM PDBu for 30 min. After an immunofluorescence staining for h3/acidic CaP and PKCα, cells showing a PKCα translocation to either the cell cortex or podosome-like structures were counted. The graph represents three independent experiments, where at least 100 cells were counted.
Figure 5.

Knockdown of h3/acidic CaP inhibits PDBu-mediated ERK1/2 targeting. (A) REF52.2 cells were transfected with siRNA against aCaP siRNA (i–p) or nontargeting control siRNA (a–h). Three days after transfection, cells were treated with DMSO for control (a–d and i–l) or with 1 μM PDBu for 30 min (e–f and m–p) before staining for aCaP (a, e, i, and m) and ERK1/2 (b, f, j, and n) with the rabbit polyclonal anti-h3/acidic CaP-Al555 and anti-ERK1/2-Alexa488 antibodies. Note that the background in images a, d, e, h, i, l, m, and p derives from uncoupled Alexa555 dye, which was resistant to extensive washing. Images were obtained with deconvolution microscopy. The yellowish signal in the merged images indicates colocalization (d, h, l, and p). Bar, 10 μm. (B) REF52.2 cells transfected with siRNA against h3/acidic CaP or with nontargeting control siRNA were treated with 1 μM PDBu for 30 min. After an immunofluorescence staining for h3/acidic CaP and ERK1/2, cells showing an ERK1/2 translocation to either the cell cortex or podosome-like structures were counted. The graph represents three independent experiments, where at least 100 cells were counted. Note that the difference is highly statistically significant in a two-tailed paired t test (p = 0.0001) marked by asterisks.
To address the question of whether h3/acidic CaP is necessary for PDBu-mediated PKCα or ERK1/2 translocation, we performed h3/acidic CaP knockdown experiments. Seventy-two hours after transfection with siRNA-targeting h3/acidic CaP or nontargeting siRNA as control, cells were stimulated with 1 μM PDBu or with vehicle alone (0.01% DMSO). Coimmunostaining for h3/acidic CaP and PKCα shows that a knockdown of h3/acidic CaP has no influence on PKCα translocation after PDBu treatment (Figure 4A, m–p, arrows). In contrast, PDBu-mediated movement of ERK1/2 to podosome-like structures and the cell cortex was inhibited in cells lacking h3/acidic CaP protein expression (Figure 5A, m–p). To further support these finding, we performed a statistical analysis, counting at least 100 cells per experiment and determined the percentage of cells showing PKCα or ERK1/2 translocation to podosome-like structures or the cell cortex after PDBu treatment. The results presented here are derived from three independent experiments. As can be seen in Figure 5B, a knockdown of h3/acidic CaP resulted in a highly statistically significant decrease of PDBu-mediated ERK1/2 translocation to podosome-like structures and the cell cortex. In contrast, the percentage of cells showing a PKCα translocation upon PDBu treatment did not change after h3/acidic CaP knockdown (Figure 4B). These data clearly demonstrate that h3/acidic CaP is necessary for ERK1/2 translocation to podosome-like structures and/or the cell cortex upon PDBu stimulation. Interestingly, the percentage of cells showing ERK1/2 localization in the nucleus without a stimulus (∼90% of control cells vs. 87% of h3/acidic CaP knockdown cells) or upon a stimulus (∼96% of control cells vs. 97% of h3/acidic CaP knockdown cells) is not affected by knockdown of h3/acidic CaP.
h3/Acidic CaP Translocation Is Independent of Phosphorylation at S175 and/or T184 but Can Be Blocked by a PKC Inhibitor
Because the translocation of PKCα, ERK1/2 and h3/acidic CaP to the cell cortex and podosome-like structures is inducible by the PKC stimulating agent PDBu, we wanted to know whether this process can be blocked by a PKC inhibitor. Calphostin is a cell-permeable reagent that specifically binds to the regulatory domain of PKC, thereby blocking diacylglycerol and phorbol ester binding and consequently PKC activity (Kobayashi et al., 1989; Bruns et al., 1991). For this experiment, REF52.2 cells growing on coverslips were serum starved for 30 min, followed by treatment with 0.5 or 1 μM calphostin for 30 min. Cells were then stimulated with 1 μM PDBu for 30 min. We probed for PKCα and h3/acidic CaP localization by immunofluorescence, and the percentage of cells showing a translocation of the two proteins to the cortex or podosome-like structures was counted. A significant inhibition of PDBu-mediated PKCα-h3/acidic CaP movement by pre-treatment with calphostin was seen, that was also dependent on the calphostin concentration (Figure 6A). Because it was previously shown that h1CaP binds to the regulatory domain of PKCα (Leinweber et al., 2000), we wanted to know whether calphostin is able to compete with h3/acidic CaP binding to PKCα. For this purpose, REF52.2 cells were treated with 1 μM calphostin. Cell lysates where then subjected to immunoprecipitation with the h3/acidic CaP-antibody and copurified PKCα was detected (Figure 6B, left). As shown in the graph (Figure 6B, right), there was no statistical significant change in the relative copurified PKCα protein amount with h3/acidic CaP when comparing DMSO-treated versus calphostin-treated cells. Hence, calphostin does not inhibit h3/acidic CaP association with PKCα.
Figure 6.

The C-terminal tail of h3/acidic CaP or h3/acidic CaP phosphorylation at Ser175 and/or Thr184 is not necessary for PDBu-mediated translocation, but can be blocked by a PKC inhibitor. (A) REF52.2 cells, grown on glass coverslips, were serum starved for 30 min, pretreated with either 0.5 or 1 μM calphostin or DMSO as control for 30 min, and then stimulated with 1 μM PDBu for 30 min. Endogenous aCaP and PKCα proteins were immunofluorescently labeled with the rabbit anti-h3/acidic CaP and the mouse anti-PKCα antibody. The percentage of cells showing a translocation of h3/acidic CaP/PKCα to the cell membrane/podosome-like structures was determined and graphed. Results are from three independent experiments. Note that the difference between DMSO pretreated control cells and both 0.5 μM calphostin (p = 0.0005) and 1 μM calphostin (p = 0.0001) pretreated cells is highly statistically significant. (B) Right, REF52.2 cells were treated with either DMSO as control (lane 2) or 1 μM calphostin (lane 3) for 30 min before preparing cell lysates. Lysates were incubated with the specific h3/acidic CaP antibody (lanes 2 and 3), and the immunocomplex was precipitated using protein G-coupled Dynabeads. Lysates incubated with the GFP-antibody served as negative control (lane 1). Proteins were eluted from the beads and subsequently analyzed by Western blotting. Arrows indicate the positions of PKCα and aCaP. A typical Western blot is shown here, and the experiment was repeated three times. Left, densitometry of all three Western blots showing the amount of PKCα coprecipitated together with h3/acidic CaP in the presence of calphostin (gray column) or DMSO as control (white column). The relative protein amount of PKCα was normalized to the relative amount of precipitated h3/acidic CaP. (C) REF52.2 cells were transfected with pFLAG-aCaP-wt, grown for 24 h, and then treated with DMSO as control (a–c) or 1 μM PDBu (d–f) for 30 min. The ectopically expressed FLAG-aCaP-wt protein was detected by immunofluorescence staining using the mouse monoclonal anti-FLAG antibody. Actin filaments were visualized with Alexa568-labeled phalloidin. Bar, 20 μm. (D) REF52.2 cells were transfected with pFLAG-aCaP-S175A/T184A and treated as described under A. Bar, 20 μm. (E) pFLAG-aCaP-wt or pFLAG-aCaP-S175A/T184A transfected cells, stimulated with 1 μM PDBu, were screened for translocation of the ectopically expressed proteins to the cell cortex/podosome-like structures. The percentage of cells showing a translocation is shown in the graph. The results are from three independent experiments, in each at least 100 cells were assessed. (F) REF52.2 cells were transfected with pEGFP-aCaP-wt, grown for 24 h and then treated with DMSO as control (a–c) or 1 μM PDBu (d–f) for 30 min. Actin filaments were visualized with Alexa568-labeled phalloidin. Bar, 20 μm. (G) REF52.2 cells were transfected with pEGFP-aCaP-ΔCt and treated as described under F. Bar, 20 μm. (H) pEGFP-aCaP-wt– or pEGFP-aCaP-ΔCt–transfected cells, stimulated with 1 μM PDBu, were screened for translocation of the ectopically expressed proteins to the cell cortex/podosome-like structures. The percentage of cells showing a translocation is shown in the graph. The results are from three independent experiments, in each at least 100 cells were assessed.
The basic isoform of CaP is a well known substrate for PKC, phosphorylating CaP on either Ser175 or Thr184 (Nakamura et al., 1993; Jin et al., 2000). Because both PKC phosphorylation sites are conserved in all three CaP isoforms, we speculate that PKC is also able to modify the h3/acidic isoform of CaP and that it might have the same effect as for h1CaP, namely weakening the binding affinity for actin (Winder and Walsh, 1990c). It is not known how CaP can leave the actin filaments to move to the cortex or podosome-like structures upon stimulation, but the phosphorylation by PKC might release CaP from the actin filaments, enabling translocation of the PKC-ERK1/2-CaP complex. To test this hypothesis, we generated a h3/acidic CaP-S175A/T184A mutant in which the putative PKC phosphorylation sites were mutated to alanine. REF52.2 cells were transfected with FLAG-tagged fusion proteins of h3/acidic CaP, either FLAG-aCaP-wt or FLAG-aCaP-S175A/T184A. Twenty-four hours after transfection, cells were stimulated with PDBu before immunostaining for the ectopically expressed FLAG-proteins and actin. As shown in Figure 6C, FLAG-aCaP-wt was mainly located at the actin filaments in unstimulated cells (Figure 6C, a–c), but translocates upon PDBu-treatment (Figure 6C, d–f, see arrows). The FLAG-aCaP-S175A/T184A protein was also located at filamentous structures, but a large amount was diffusely distributed in the cytoplasm in nontreated cells (Figure 6D, a–c). This was surprising to us because we predicted a stronger actin binding of this mutant due to the lack of the PKC phosphorylation sites. Moreover, after PDBu stimulation, FLAG-aCaP-S175A/T184A also moved to podosome-like structures (Figure 6D, d–f, see arrow). Thus, either phosphorylation of h3/acidic CaP by PKC is not necessary for PDBu-mediated translocation or PKC does not use the h3/acidic calponin isoform as a substrate, even though the consensus sites are highly conserved.
The Specific C-terminal Acidic Tail Is Not Involved in PDBu-mediated h3/Acidic CaP Translocation
To further test the hypothesis that the affinity of h3/acidic CaP to actin filaments plays a role in translocation of the protein to the cortex and podosome-like structures, we generated a C-terminal deletion mutant of h3/acidic CaP, lacking the specific highly acidic tail. Previous studies have shown that the h3/acidic CaP's acidic tail region is capable of down-regulating actin association of CaP (Burgstaller et al., 2002). Because the h3/acidic CaP protein is expected to dissociate from actin filaments to translocate to podosomes and the cell cortex, an h3/acidic CaP mutant binding stronger to actin should display less translocation upon cell stimulation. We therefore generated the GFP-aCaP-ΔCt construct which lacks aa 274–330 corresponding to its specific acidic tail. REF52.2 cells were transfected with either GFP-aCaP-wt or GFP-aCaP-ΔCt and stimulated using PDBu. As can be seen in Figure 6F, the GFP-aCaP-wt construct is located mainly at the actin filaments as indicated by costaining with phalloidin (Figure 6F, a–c). The GFP-aCaP-ΔCt fusion protein indeed seems to be attached more strongly to actin filaments because it displayed less diffuse background staining compared with GFP-aCaP-wt (Figure 6G, a–c). Interestingly, overexpression of GFP-aCaP-ΔCt in REF52.2 cells seems to result in an increase in actin bundling as can be seen in the phalloidin staining (Figure 6Gb). However, the h3/acidic CaP wild type (aCaP-wt) and the h3/acidic CaP C-terminal deletion construct (aCaP-ΔCt) translocate to about the same amount to podosome-like structures and the cell cortex upon PDBu treatment (see Figure 6, F and G, d–f, see arrows; and 6H). From these results we conclude that the affinity of h3/acidic CaP to actin filaments does not play an important role in h3/acidic CaP translocation.
Down-Regulation of h3/Acidic CaP Blocks ERK1/2 Activity in REF52.2 Cells and ERK2 Activity in NIH-3T3 Cells
To clarify the role of h3/acidic CaP in regulating ERK1/2 function, we knocked down endogenous h3/acidic CaP expression with siRNA in REF52.2 cells and assayed ERK1/2 activity by measuring ERK1/2 phosphorylation as well as phosphorylation of l-CaD at an ERK1/2 phosphorylation site (Figure 7). Cells were again treated with 1 μM PDBu for 30 min or 0.01% DMSO as vehicle control and whole cell extracts were probed by Western blotting for the appropriate proteins. Tubulin-α was used as a loading control (Figure 7C, bottom). Endogenous h3/acidic CaP expression was knocked down in cells transfected with siRNA against h3/acidic CaP (Figure 7C, top, lanes 3 and 4) to ∼85% compared with cells transfected with nontargeting control siRNA (Figure 7C, top, lanes 1 and 2; and 7Dd), whereas h2CaP expression was not affected by siRNA against h3/acidic CaP (Figure 7C, middle). As shown in Figure 7B (lane 2), ERK1 as well as ERK2 phosphorylation increases upon PDBu treatment of cells transfected with nontargeting control siRNA. Some basal ERK1 and ERK2 phosphorylation is also detectable in unstimulated cells (lane 1), and a longer serum starvation up to 24 h which resulted in cells displaying phenotypic anomalies indicative of cellular stress related to serum starvation, did not significantly reduce basal active levels of ERK1/2. In cells where h3/acidic CaP levels were knocked down due to transfection with siRNA against h3/acidic CaP (lanes 3 and 4), basal ERK1/2 phosphorylation was much lower (lane 3). After PDBu treatment, phospho-ERK1/2 levels increase but to a lesser amount (lane 4) in cells transfected with siRNA against h3/acidic CaP. Thus, it seems that both residual effects of serum exposure and PDBu represent stimuli that activate ERK1/2 and that both pathways require h3/acidic CaP for activation.
Figure 7.

h3/acidic CaP knockdown inhibits ERK1/2 activation in REF52.2 cells. Seventy-two hours after transfection with siRNA against rat h3/acidic CaP (+, lanes 3 and 4) or with nontargeting control-siRNA (−, lanes 1 and 2), REF52.2 cells were treated with 1 μM PDBu for 30 min (+, lanes 2 and 4) or DMSO as solvent control (−, lanes 1 and 3). Fifty micrograms of whole cell extracts was analyzed by SDS-PAGE and Western blotting with specific antibodies. Arrows indicate the positions of (A) l-caldesmon (CaD), phospho-l-caldesmon (pCaD); (B) ERK1, phospho-ERK1 (pERK1), ERK2, phospho-ERK2 (pERK2); and (C) h3/acidic calponin (aCaP), neutral calponin (h2CaP) and tubulin-α. (D) Densitometric measurement of Western blots, derived from three independent experiments, was performed and total protein levels were normalized to tubulin-α. For phosphorylation levels of ERK1/2 and l-CaD, the ratio of phosphorylated to unphosphorylated protein was calculated and graphed (a–c). The decrease of phosphorylated ERK1 (b; p = 0.0195) and ERK2 (a; p = 0.0478) in h3/acidic CaP knockdown cells stimulated with PDBu was statistically significant, as was the difference of l-CaD phosphorylation (c; p = 0.0012). For relative h3/acidic CaP levels, the amount of h3/acidic CaP in nontargeting control-siRNA transfected and DMSO treated cells was set as 1, and relative h3/acidic CaP levels of the other three samples was calculated (d).
To further corroborate the finding that h3/acidic CaP knockdown inhibits ERK1/2 activation, we monitored phosphorylation of the ERK1/2 substrate l-CaD at Ser527. Basal phospho-l-CaD levels were low in both cells transfected with siRNA against h3/acidic CaP or nontargeting control siRNA (Figure 7A, lanes 1 and 3); however, basal l-CaD phosphorylation was lower in h3/acidic CaP knockdown cells. On PDBu stimulation, there was an increase in l-CaD phosphorylation in cells transfected with nontargeting control siRNA (Figure 7A, lane 3) that was inhibited in cells transfected with siRNA against h3/acidic CaP (Figure 7A, lane 4). Thus, h3/acidic CaP is necessary for ERK1/2 activation and ERK1/2-mediated l-CaD phosphorylation. We also probed for an effect of h3/acidic CaP knockdown on PKCα activation as monitored by autophosphorylation upon PDBu treatment. To address this question, we stained the same Western blot membrane with a rabbit polyclonal anti-phospho-PKC PAN antibody (Cell Signaling Technology) that recognizes the autophosphorylation site at Ser660. However, there was no detectable difference in phospho-PKCα levels by either h3/acidic CaP knockdown or PDBu stimulation in this cell system (data not shown). To quantify the data, we performed densitometric measurements of Western blots derived from three independent experiments. Total protein levels were normalized to tubulin. For ERK1/2 and l-CaD phosphorylation levels, we graphed the ratio of phosphorylated to unphosphorylated proteins. Statistical analysis clearly shows a significant decrease of both ERK1 (Figure 7Db) and ERK2 (Figure 7Da) as well as l-CaD (Figure 7Dc) phosphorylation in h3/acidic CaP-knockdown cells upon stimulation.
To corroborate our findings of h3/acidic CaP regulating ERK 1/2 activity, we also assayed for ERK1/2, l-CaD, and PKCα phosphorylation in the mouse fibroblast cell line NIH-3T3 (Figure 8). Our results show that in NIH-3T3 cells, a knockdown of endogenous h3/acidic CaP lowers basal ERK2 activity significantly (Figure 8B, lane 2, and D). However, we saw no effects on phosphorylation of the ERK substrate l-CaD (Figure 8A). Just like in REF52.2 cells, we also saw no difference in phospho-PKCα levels in control cells compared with cells transfected with siRNA against h3/acidic CaP (data not shown). Apparently, even though h3/acidic CaP regulates ERK2 activity in these cells, the subsequent downstream signaling pathway differs.
Figure 8.

Knockdown of h3/acidic CaP decreases phospho-ERK2 levels in NIH-3T3 cells. NIH-3T3 cells were either transfected with nontargeting siRNA-control (lane 1) or siRNA against h3/acidic CaP (siRNA-aCaP; lane 2). Seventy-two hours after transfection, whole cell extracts were generated and examined by Western blotting. The membrane was stained with specific antibodies against (A) phospho-l-caldesmon (pCaD); (B) ERK1, phospho-ERK1 (pERK1), ERK2, phospho-ERK2 (pERK2); and (C) h3/acidic calponin (aCaP), neutral calponin (h2CaP) and tubulin-α as indicated. (D) Densitometric measurement of Western blots, derived from three independent experiments, was performed. Total protein levels were normalized to tubulin-α and the ratio of phosphorylated to unphosphorylated ERK2 protein was calculated and graphed. Note that the decrease of phosphorylated ERK2 (p = 0.00002) in h3/acidic CaP knockdown cells is highly statistically significant.
In Vitro Wound Healing Is Inhibited in h3/Acidic CaP Knockdown REF52.2 Cells
Because CaD phosphorylation has been shown to alter actin–myosin interactions (Chalovich et al., 1990), the influence of h3/acidic CaP on l-CaD phosphorylation in REF52.2 cells suggests an involvement of h3/acidic CaP in the regulation of cell motility. To test this prediction, we measured the effect of h3/acidic CaP knockdown in an in vitro wound healing assay. For this purpose, REF52.2 fibroblasts were either transfected with siRNA against h3/acidic CaP or with nontargeting control siRNA. Seventy-two hours after transfection, the cell layer was wounded by using a sterile pipette tip, followed by recording of images of the wound healing process at timed intervals on a microscope equipped for live cell imaging for 20 h. After the wound healing assay, cells were immunostained for endogenous h3/acidic CaP expression to evaluate knockdown efficiency (Figure 9B). Figure 9A shows a representative photo series from three independent experiments, demonstrating that cells transfected with siRNA against h3/acidic CaP close the wound more slowly compared to cells transfected with nontargeting control siRNA (also see Supplemental Movies). To quantify the wound healing procedure, the wound area was measured and calculated as a percentage of the wound area at 0 h. Measured data from three independent experiments are plotted in Figure 9C, demonstrating that the h3/acidic CaP knockdown cells (black columns) do migrate less into the wound compared with control cells (gray columns). To examine directionality of single cells in each group, we traced individual cells and plotted the migration tracks as a scatter plot (Figure 9D). This analysis shows that h3/acidic CaP knockdown cells (Figure 9D, right) move less directionally compared with control cells (Figure 9D, left). We also assessed the average speed of cells either transfected with siRNA against h3/acidic CaP or nontargeting control siRNA, revealing an average speed of ∼2.2 nm/s for the h3/acidic CaP knockdown cells compared with ∼2.8 nm/s for the control cells (Figure 9E). Hence, in summary, REF52.2 cells expressing lower levels of endogenous h3/acidic CaP, move significantly slower and less directionally and therefore exert abnormalities in migration and in vitro wound healing.
Figure 9.

h3/acidic CaP enhances wound healing in REF52.2 cells in an in vitro assay. (A) REF52.2 cells were seeded on glass-bottomed culture plates and either transfected with siRNA against rat h3/acidic CaP (siRNA-aCaP) or with nontargeting control-siRNA the next day. Seventy-two hours after transfection, the cell layer was wounded with a sterile pipette tip, and the healing procedure was observed for 20 h. Bar, 20 μm. (B) To verify h3/acidic CaP knockdown, cells were immunostained after the in vitro wound healing assay for endogenous aCaP expression by using the rabbit polyclonal anti-h3/acidic CaP antibody. Bar, 20 μm. (C) To quantify in vitro wound healing, the wound area was measured using ImageJ software and calculated as a percentage of the wound area at 0 h. Data represent three independent experiments. (D) Ten single cells of each group (left, control cells; right, h3/acidic CaP knockdown cells) were manually traced using ImageJ software and migration tracks are shown as scatter plots. (E) The average speed of 30 individual cells in total, coming from three independent experiments, for either h3/acidic CaP knockdown or control cell groups was calculated and plotted in a graph bar. Note that the difference is statistically significant (p = 0.017).
DISCUSSION
The serine/threonine-kinase ERK1/2 is implicated in many cellular signaling pathways. One of the main findings of the present study is that the widely expressed h3/acidic CaP protein plays an important role in activation and PDBu-mediated translocation of ERK1/2 in the REF52.2 fibroblast cell line. h3/acidic CaP, ERK1/2, and PKCα colocalize and coprecipitate, even in the absence of a stimulus. Moreover h3/acidic CaP cotranslocates with both ERK1/2 and PKCα to the cell cortex and podosome-like structures in PDBu-stimulated REF52.2 cells. As for the podosome-like structures, we can only speculate about their role in the fibroblast cell line. Due to the fact that endogenous MMP-2 is located at these loci it is conceivable that they play a role in matrix degradation, which in turn is an important step in cell migration in a natural environment/tissue. Moreover, the podosome-like structures seem to be involved in PKC signaling pathways, because the kinase translocates to these loci after a stimulus.
A cause-and-effect relationship between h3/acidic CaP and ERK signaling is shown by the fact that down-regulation of h3/acidic CaP prevents both basal and PDBu-mediated ERK1/2 activation. These results indicate an important role for h3/acidic CaP in ERK1/2 signaling in REF52.2 fibroblasts. However, we cannot conclude from our data whether h3/acidic CaP affects ERK1/2 activity in general or regulates only specific subfractions of ERK1/2. Interestingly, we saw no difference in nuclear localization of ERK1/2 when comparing control cells to h3/acidic CaP knockdown cells. These results point to an apparent effect of h3/acidic CaP to regulate only the activity of cytosolic ERK1/2 and not nuclear ERK1/2 in REF52.2 cells; however, further experiments are necessary to investigate this hypothesis. In contrast to REF52.2 cells, NIH-3T3 fibroblasts display only a decrease in ERK2 activity upon h3/acidic CaP knockdown, whereas the ERK1 activity is unaffected.
Our data also demonstrate that h3/acidic CaP controls phosphorylation of l-CaD in REF52.2 cells, a substrate of ERK1/2. CaD is an actin-binding protein and has been shown capable of regulating actomyosin interactions and hence contractile events in muscle and nonmuscle cells (Matsumura and Yamashiro, 1993). Nonsmooth muscle cells express l-CaD, which lacks a spacer region but shares all other functional domains with the high-molecular-weight CaD isoform expressed in smooth muscle cells (Humphrey et al., 1992). Interaction of l-CaD with actin is controlled by a phosphorylation at Ser497 and 527, which leads to a conformational change that increases the access of myosin to actin (Kordowska et al., 2006). Although cdc2 seems to be responsible for l-CaD phosphorylation in dividing cells, phosphorylation by ERK1/2 is known to be important for actin remodeling processes in migrating cells (Yamboliev and Gerthoffer, 2001; Goncharova et al., 2002).
Previous studies have shown that h1CaP can support PKCα autophosphorylation in vitro (Leinweber et al., 2000); thus, we thought an influence of h3/acidic CaP on PKCα activation might be possible in vivo. The failure to detect any effect of h3/acidic CaP on PKCα targeting may result from the fact that PKCα is constitutively autophosphorylated in REF52.2 fibroblasts and no increase occurs on addition of PDBu. Moreover, previous findings demonstrated that the direct interaction between the basic isoform of CaP and PKCα takes place at the regulatory domain of the kinase (Leinweber et al., 2000). Hence, it was tempting to speculate that calphostin might not only inhibit the kinase activity but also competes away CaP binding. However, our knockdown data clearly show that h3/acidic CaP is not necessary for phorbol ester-stimulated PKCα translocation. Moreover, a coimmunoprecipitation of h3/acidic CaP and PKCα in calphostin-treated cells demonstrated that there is no difference in interaction of the two proteins. Therefore, blocking of PDBu binding rather than blocking of h3/acidic CaP binding to the regulatory domain of PKCα seems to be the reason for calphostin-inhibited PKCα movement following stimulation.
To clarify the mechanism as to how h3/acidic CaP translocates to the cell cortex in stimulated cells, we tested the hypothesis that phosphorylation by PKC affects h3/acidic CaP's actin binding affinity, as was shown previously for h1CaP (Winder and Walsh, 1990c). However, by using an h3/acidic CaP construct that harbors inactive putative PKC phosphorylation sites (“aCaP-S175A/T184A”), we demonstrated that PKC phosphorylation of h3/acidic CaP at neither Ser175 nor Thr184 is necessary for the translocation of h3/acidic CaP to the membrane upon phorbol ester stimulation in REF52.2 cells. We also ruled out the possibility that the specific acidic tail of h3/acidic CaP is involved in the translocation process because a deletion mutant (“aCaP-ΔCt”) displays no differences in intracellular localization in stimulated cells compared with the h3/acidic CaP wild-type protein. However, we noticed that overexpression of the h3/acidic CaP C-terminal deletion mutant induces strong actin bundling in transfected REF52.2 cells that is not seen with the h3/acidic CaP wild-type protein. From previous studies it is known that deletion of the C-terminal tail of either h2CaP or h3/acidic CaP results in stronger F-actin binding (Burgstaller et al., 2002). In this context, it is interesting that overexpressed h1CaP is capable of stabilizing actin filaments in cultured cells (Danninger and Gimona, 2000) and that it is known to enhance the bundling of actin filaments in vitro (Tang et al., 1997), whereas the h2CaP isoform whose specific C terminus impairs its affinity to actin is not able to stabilize actin filaments (Danninger and Gimona, 2000). Hence, the observed thicker actin filaments in transfected REF52.2 cells might result from h3/acidic CaP's F-actin binding/stabilizing ability which is increased when its acidic C terminus is deleted.
The effect of h3/acidic CaP to promote REF52.2 fibroblast motility is partially in contrast to previous findings for the other CaP isoforms. Danninger and Gimona (2000) showed a reduced cell motility of h1CaP-GFP overexpressing NIH-3T3 mouse fibroblasts. h2CaP also seems to slow down cell migration, because macrophages derived from h2CaP knockout mice displayed a faster migration rate in an in vitro wound healing assay (Huang et al., 2008). In contrast, it was shown that a knockdown of h2CaP in endothelial cells slowed down cell motility in an in vitro wound healing assay (Tang et al., 2006). It is known that h3/acidic CaP has minimal effect on actomyosin ATPase activity compared with h1CaP and h2CaP, so it is likely that the inhibition of motility seen with the basic and possibly also the neutral isoforms is due to greater inhibition of actomyosin ATPase activity (Fujii et al., 2002). The differences in the manner in which the h3/acidic isoform of CaP controls cell motility compared with the neutral and the basic isoforms, is also likely to be related to sequence diversity. The difference between the isoforms is mainly located in the C terminus, which varies in length and pI and apparently contributes to the actin binding capacity. It has been shown that h3/acidic CaP and h2CaP have a less tight association with actin compared with h1CaP due to differences in their C-terminal tails (Burgstaller et al., 2002). Furthermore, it has been shown that h3/acidic CaP is involved in dendritic spine morphology, probably via regulation of actin cytoskeleton reorganization and dynamics (Rami et al., 2006). These differences in the actions of h3/acidic CaP on the cytoskeleton are thus likely to lead to specific novel functions in the cell types where it is expressed.
Together, our findings on h3/acidic CaP's regulation of ERK1/2 signaling and targeting have functional relevance for REF52.2 fibroblast motility and are consistent with the model illustrated in Figure 10C. As shown in this model, h3/acidic CaP, ERK1/2 and PKCα form a complex at actin filaments in unstimulated REF52.2 cells. On stimulation, 1) PKCα becomes activated and moves together with h3/acidic CaP and ERK1/2 to the cell cortex. At the cell cortex, ERK1/2 becomes phosphorylated in a signaling complex including Raf and MEK (Cacace et al., 1996; Cai et al., 1997; Gangopadhyay et al., 2009). The subfraction of ERK1/2 that is activated at the cortex (2) moves back to the actin filaments (see Figure 10B, a–d, arrow) where it comes in contact with its substrate CaD (CaD is located at actin filaments in this cell line; Figure 10A). 3) Phosphorylation of CaD results in increased actin-myosin interactions and hence 4) increased contractility, facilitating cell motility. siRNA-mediated knockdown of h3/acidic CaP results in the loss of the connection between PKCα and ERK1/2. Therefore, the PKCα molecule translocates without ERK1/2 to the cell cortex, and the cytosolic/actin bound fraction of ERK1/2 is not activated (Figure 10B, e–h). This leads to a reduced CaD phosphorylation and therefore to a reduced cell motility. A specific and selective downstream role of CaD is, however, speculative at this time because we did not monitor for changes in phosphorylation of other possible ERK1/2 substrates such as myosin light chain kinase (MLCK), which are also known to affect cell contractility and motility (Huang et al., 2004).
Figure 10.

Model of h3/acidic CaP function in the regulation of REF52.2 cell motility. (A) REF52.2 cells, cultured on glass coverslips, were fixed with paraformaldehyde and stained for CaD with the mouse monoclonal anti-caldesmon antibody and for actin with Alexa568-labeled phalloidin. Images were obtained with deconvolution microscopy. The yellowish color in c indicates colocalization. The arrow points out an example of colocalization. Bar, 20 μm. (B) REF52.2 cells, either transfected with nontargeting control-siRNA (top) or siRNA against h3/acidic CaP (siRNA-aCaP; bottom), were fixed 72 h after transfection and labeled for phospho-ERK1/2 (pERK1/2) using the mouse monoclonal phospho-ERK1/2–specific antibody (a) and for aCaP (b). Images were obtained with deconvolution microscopy and colocalization of phospho-ERK1/2 and h3/acidic CaP on filaments is indicated by the arrow in the merged image in c. Bar, 20 μm. (C) Model: PKCα, h3/acidic CaP (aCaP) and ERK1/2 are located in a complex at the actin filaments in untreated cells. 1. A stimulus such as PDBu leads to translocation of the PKCα–h3/acidic CaP–ERK1/2 complex to the cell cortex. Due to full activation of PKCα at the cell membrane, the kinase is now able to activate Raf, which in turn activates MEK, and MEK activates ERK1/2. 2. Activated ERK1/2 moves back to actin filaments where it binds and phosphorylates its substrate, CaD. 3. This leads to a conformational change in CaD, allowing interaction between actin and myosin filaments and thereby 4. Increasing contraction coupled to cell motility. From our experiments we can, however, not rule out the possibility that activated ERK1/2 phosphorylates other substrates such as MLCK (as indicated in the model with a question mark), which are also involved in regulating cell motility/contractility.
Thus, in summary, the h3/acidic isoform of CaP, in a cause-and-effect manner, facilitates ERK1/2 activation and enhances cell motility in nonmuscle cells, resulting in increased in vitro wound healing activity in REF52.2 fibroblasts.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. K. H. Scheidtmann for the REF52.2 and NIH-3T3 cells and Dr. J. Wong for the BALB/c 3T3 cells. This study was supported by National Institutes of Health grants HL-80003, HL-31704, and HL-86655 (to K.G.M.) and HL-86720 (to J.P.J.).
Abbreviations used:
- aCaP
h3/acidic calponin
- CaD
caldesmon
- CaP
calponin
- ERK
extracellular signal-regulated kinase
- h1CaP
basic calponin
- h2CaP
neutral calponin
- PDBu
phorbol-12,13-dibutyrate
- PKC
protein kinase C.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-06-0451) on February 24, 2010.
REFERENCES
- Allen B. G., Walsh M. P. The biochemical basis of the regulation of smooth-muscle contraction. Trends Biochem. Sci. 1994;19:362–368. doi: 10.1016/0968-0004(94)90112-0. [DOI] [PubMed] [Google Scholar]
- Applegate D., Feng W., Green R. S., Taubman M. B. Cloning and expression of a novel acidic calponin isoform from rat aortic vascular smooth muscle. J. Biol. Chem. 1994;269:10683–10690. [PubMed] [Google Scholar]
- Bruns R. F., Miller F. D., Merriman R. L., Howbert J. J., Heath W. F., Kobayashi E., Takahashi I., Tamaoki T., Nakano H. Inhibition of protein kinase C by calphostin C is light-dependent. Biochem. Biophys. Res. Commun. 1991;176:288–293. doi: 10.1016/0006-291x(91)90922-t. [DOI] [PubMed] [Google Scholar]
- Burgstaller G., Kranewitter W. J., Gimona M. The molecular basis for the autoregulation of calponin by isoform-specific C-terminal tail sequences. J. Cell Sci. 2002;115:2021–2029. doi: 10.1242/jcs.115.10.2021. [DOI] [PubMed] [Google Scholar]
- Cacace A. M., Ueffing M., Philipp A., Han E. K., Kolch W., Weinstein I. B. PKC epsilon functions as an oncogene by enhancing activation of the Raf kinase. Oncogene. 1996;13:2517–2526. [PubMed] [Google Scholar]
- Cai H., Smola U., Wixler V., Eisenmann-Tappe I., Diaz-Meco M. T., Moscat J., Rapp U., Cooper G. M. Role of diacylglycerol-regulated protein kinase C isotypes in growth factor activation of the Raf-1 protein kinase. Mol. Cell Biol. 1997;17:732–741. doi: 10.1128/mcb.17.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter C. L. Actin cytoskeleton and cell signaling. Crit. Care Med. 2000;28:N94–N99. doi: 10.1097/00003246-200004001-00011. [DOI] [PubMed] [Google Scholar]
- Chalovich J. M., Hemric M. E., Velaz L. Regulation of ATP hydrolysis by caldesmon. A novel change in the interaction of myosin with actin. Ann. NY Acad. Sci. 1990;599:85–99. doi: 10.1111/j.1749-6632.1990.tb42367.x. [DOI] [PubMed] [Google Scholar]
- Chu C. L., Reenstra W. R., Orlow D. L., Svoboda K. K. Erk and PI-3 kinase are necessary for collagen binding and actin reorganization in corneal epithelia. Invest. Ophthalmol. Vis. Sci. 2000;41:3374–3382. [PMC free article] [PubMed] [Google Scholar]
- Clark E. S., Weaver A. M. A new role for cortactin in invadopodia: regulation of protease secretion. Eur. J. Cell Biol. 2008;87:581–590. doi: 10.1016/j.ejcb.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley J. L., Smith T. C., Fang Z., Takizawa N., Luna E. J. Supervillin reorganizes the actin cytoskeleton and increases invadopodial efficiency. Mol. Biol. Cell. 2009;20:948–962. doi: 10.1091/mbc.E08-08-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danninger C., Gimona M. Live dynamics of GFP-calponin: isoform-specific modulation of the actin cytoskeleton and autoregulation by C-terminal sequences. J. Cell Sci. 113. 2000;21:3725–3736. doi: 10.1242/jcs.113.21.3725. [DOI] [PubMed] [Google Scholar]
- English J., Pearson G., Wilsbacher J., Swantek J., Karandikar M., Xu S., Cobb M. H. New insights into the control of MAP kinase pathways. Exp. Cell Res. 1999;253:255–270. doi: 10.1006/excr.1999.4687. [DOI] [PubMed] [Google Scholar]
- Fujii T., Yabe S., Nakamura K., Koizumi Y. Functional analysis of rat acidic calponin. Biol. Pharm. Bull. 2002;25:573–579. doi: 10.1248/bpb.25.573. [DOI] [PubMed] [Google Scholar]
- Gangopadhyay S. S., Kengni E., Appel S., Gallant C., Kim H., Leavis P., Degnore J., Morgan K. G. SMAV is an ERK scaffolding protein. J. Biol. Chem. 2009;284:17607–17615. doi: 10.1074/jbc.M109.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharova E. A., Vorotnikov A. V., Gracheva E. O., Wang C. L., Panettieri R. A., Jr, Stepanova V. V., Tkachuk V. A. Activation of p38 MAP-kinase and caldesmon phosphorylation are essential for urokinase-induced human smooth muscle cell migration. Biol. Chem. 2002;383:115–126. doi: 10.1515/BC.2002.012. [DOI] [PubMed] [Google Scholar]
- Hai C. M., Gu Z. Caldesmon phosphorylation in actin cytoskeletal remodeling. Eur. J. Cell Biol. 2006;85:305–309. doi: 10.1016/j.ejcb.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Hossain M. M., Crish J. F., Eckert R. L., Lin J. J., Jin J. P. h2-Calponin is regulated by mechanical tension and modifies the function of actin cytoskeleton. J. Biol. Chem. 2005;280:42442–42453. doi: 10.1074/jbc.M509952200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Jacobson K., Schaller M. D. MAP kinases and cell migration. J. Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- Huang Q. Q., Hossain M. M., Wu K., Parai K., Pope R. M., Jin J. P. Role of H2-calponin in regulating macrophage motility and phagocytosis. J. Biol. Chem. 2008;283:25887–25899. doi: 10.1074/jbc.M801163200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey M. B., Herrera-Sosa H., Gonzalez G., Lee R., Bryan J. Cloning of cDNAs encoding human caldesmons. Gene. 1992;112:197–204. doi: 10.1016/0378-1119(92)90376-z. [DOI] [PubMed] [Google Scholar]
- Je H. D., Gangopadhyay S. S., Ashworth T. D., Morgan K. G. Calponin is required for agonist-induced signal transduction—evidence from an antisense approach in ferret smooth muscle. J. Physiol. 2001;537:567–577. doi: 10.1111/j.1469-7793.2001.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J. P., Walsh M. P., Sutherland C., Chen W. A role for serine-175 in modulating the molecular conformation of calponin. Biochem. J. 2000;350:579–588. [PMC free article] [PubMed] [Google Scholar]
- Khalil R. A., Morgan K. G. PKC-mediated redistribution of mitogen-activated protein kinase during smooth muscle cell activation. Am. J. Physiol. 1993;265:C406–C411. doi: 10.1152/ajpcell.1993.265.2.C406. [DOI] [PubMed] [Google Scholar]
- Khokhlatchev A. V., Canagarajah B., Wilsbacher J., Robinson M., Atkinson M., Goldsmith E., Cobb M. H. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell. 1998;93:605–615. doi: 10.1016/s0092-8674(00)81189-7. [DOI] [PubMed] [Google Scholar]
- Kobayashi E., Nakano H., Morimoto M., Tamaoki T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1989;159:548–553. doi: 10.1016/0006-291x(89)90028-4. [DOI] [PubMed] [Google Scholar]
- Kordowska J., Huang R., C.-Wang L. A. Phosphorylation of caldesmon during smooth muscle contraction and cell migration or proliferation. J. Biomed. Sci. 2006;13:159–172. doi: 10.1007/s11373-005-9060-8. [DOI] [PubMed] [Google Scholar]
- Leinweber B., Parissenti A. M., Gallant C., Gangopadhyay S. S., Kirwan-Rhude A., Leavis P. C., Morgan K. G. Regulation of protein kinase C by the cytoskeletal protein calponin. J. Biol. Chem. 2000;275:40329–40336. doi: 10.1074/jbc.M008257200. [DOI] [PubMed] [Google Scholar]
- Leinweber B. D., Leavis P. C., Grabarek Z., Wang C. L., Morgan K. G. Extracellular regulated kinase (ERK) interaction with actin and the calponin homology (CH) domain of actin-binding proteins. Biochem. J. 1999;344:117–123. [PMC free article] [PubMed] [Google Scholar]
- Lewis T. S., Shapiro P. S., Ahn N. G. Signal transduction through MAP kinase cascades. Adv. Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- Matsumura F., Yamashiro S. Caldesmon. Curr. Opin. Cell Biol. 1993;5:70–76. doi: 10.1016/s0955-0674(05)80010-9. [DOI] [PubMed] [Google Scholar]
- Menice C. B., Hulvershorn J., Adam L. P., Wang C. A., Morgan K. G. Calponin and mitogen-activated protein kinase signaling in differentiated vascular smooth muscle. J. Biol. Chem. 1997;272:25157–25161. doi: 10.1074/jbc.272.40.25157. [DOI] [PubMed] [Google Scholar]
- Mezgueldi M., Mendre C., Calas B., Kassab R., Fattoum A. Characterization of the regulatory domain of gizzard calponin. Interactions of the 145–163 region with F-actin, calcium-binding proteins, and tropomyosin. J. Biol. Chem. 1995;270:8867–8876. doi: 10.1074/jbc.270.15.8867. [DOI] [PubMed] [Google Scholar]
- Mino T., Yuasa U., Nakamura F., Naka M., Tanaka T. Two distinct actin-binding sites of smooth muscle calponin. Eur. J. Biochem. 1998;251:262–268. doi: 10.1046/j.1432-1327.1998.2510262.x. [DOI] [PubMed] [Google Scholar]
- Nakamura F., Mino T., Yamamoto J., Naka M., Tanaka T. Identification of the regulatory site in smooth muscle calponin that is phosphorylated by protein kinase C. J. Biol. Chem. 1993;268:6194–6201. [PubMed] [Google Scholar]
- Newton A. C. Regulation of protein kinase C. Curr. Opin. Cell Biol. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- Pouyssegur J. Signal transduction. An arresting start for MAPK. Science. 2000;290:1515–1518. doi: 10.1126/science.290.5496.1515. [DOI] [PubMed] [Google Scholar]
- Rami G., Caillard O., Medina I., Pellegrino C., Fattoum A., Ben-Ari Y., Ferhat L. Change in the shape and density of dendritic spines caused by overexpression of acidic calponin in cultured hippocampal neurons. Hippocampus. 2006;16:183–197. doi: 10.1002/hipo.20145. [DOI] [PubMed] [Google Scholar]
- Rozenblum G. T., Gimona M. Calponins: adaptable modular regulators of the actin cytoskeleton. Int. J. Biochem. Cell Biol. 2008;40:1990–1995. doi: 10.1016/j.biocel.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Shirinsky V. P., Biryukov K. G., Hettasch J. M., Sellers J. R. Inhibition of the relative movement of actin and myosin by caldesmon and calponin. J. Biol. Chem. 1992;267:15886–15892. [PubMed] [Google Scholar]
- Strasser P., Gimona M., Moessler H., Herzog M., Small J. V. Mammalian calponin. Identification and expression of genetic variants. FEBS Lett. 1993;330:13–18. doi: 10.1016/0014-5793(93)80909-e. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Abe M., Hiwada K., Kokubu T. A novel troponin T-like protein (calponin) in vascular smooth muscle: interaction with tropomyosin paracrystals. J. Hypertens. Suppl. 1988;6:S40–S43. doi: 10.1097/00004872-198812040-00008. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Hiwada K., Kokubu T. Isolation and characterization of a 34,000-dalton calmodulin- and F-actin-binding protein from chicken gizzard smooth muscle. Biochem. Biophys. Res. Commun. 1986;141:20–26. doi: 10.1016/s0006-291x(86)80328-x. [DOI] [PubMed] [Google Scholar]
- Tang J., et al. A critical role for calponin 2 in vascular development. J. Biol. Chem. 2006;281:6664–6672. doi: 10.1074/jbc.M506991200. [DOI] [PubMed] [Google Scholar]
- Tang J. X., Szymanski P. T., Janmey P. A., Tao T. Electrostatic effects of smooth muscle calponin on actin assembly. Eur. J. Biochem. 1997;247:432–440. doi: 10.1111/j.1432-1033.1997.00432.x. [DOI] [PubMed] [Google Scholar]
- Toker A. Signaling through protein kinase C. Front, Biosci. 1998;3:D1134–D1147. doi: 10.2741/a350. [DOI] [PubMed] [Google Scholar]
- Vetterkind S., Morgan K. G. The pro-apoptotic protein Par-4 facilitates vascular contractility by cytoskeletal targeting of ZIPK. J. Cell. Mol. Med. 2009;13:887–895. doi: 10.1111/j.1582-4934.2008.00374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B. A., Eves R., Mak A. S. Cortactin regulates podosome formation: roles of the protein interaction domains. Exp. Cell Res. 2006;312:760–769. doi: 10.1016/j.yexcr.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Winder S., Walsh M. Inhibition of the actomyosin MgATPase by chicken gizzard calponin. Prog. Clin. Biol. Res. 1990a;327:141–148. [PubMed] [Google Scholar]
- Winder S. J., Walsh M. P. Smooth muscle calponin. Inhibition of actomyosin MgATPase and regulation by phosphorylation. J. Biol. Chem. 1990b;265:10148–10155. [PubMed] [Google Scholar]
- Winder S. J., Walsh M. P. Structural and functional characterization of calponin fragments. Biochem. Int. 1990c;22:335–341. [PubMed] [Google Scholar]
- Yamboliev I. A., Gerthoffer W. T. Modulatory role of ERK MAPK-caldesmon pathway in PDGF-stimulated migration of cultured pulmonary artery SMCs. Am. J. Physiol. Cell Physiol. 2001;280:C1680–C1688. doi: 10.1152/ajpcell.2001.280.6.C1680. [DOI] [PubMed] [Google Scholar]
- Young J. S., Guttman J. A., Vaid K. S., Shahinian H., Vogl A. W. Cortactin (CTTN), N-WASP (WASL), and clathrin (CLTC) are present at podosome-like tubulobulbar complexes in the rat testis. Biol. Reprod. 2009;80:153–161. doi: 10.1095/biolreprod.108.070615. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.