

Abstract
Purine riboswitches are RNA regulatory elements that control purine metabolism in response to intracellular concentrations of the purine ligands. Conformational changes of the guanine riboswitch aptamer domain induced by guanine binding lead to transcriptional regulation of genes involved in guanine biosynthesis. The guanine riboswitch aptamer domain has three RNA helices designated P1, P2, and P3. An overall model for the Mg2+- and guanine-dependent relative orientations and dynamics of P1, P2, and P3 has not been reported, and the conformational role of guanine under physiologically relevant conditions has not been fully elucidated. In this study, an ensemble and single-molecule fluorescence resonance energy transfer (FRET) study was performed on three orthogonally labeled variants of the xpt guanine riboswitch aptamer domain. The combined FRET data support a model in which the unfolded state of the aptamer domain has a highly dynamic P2 helix that switches rapidly between two orientations relative to nondynamic P1 and P3. At ≪1 mM Mg2+ (in the presence of a saturating level of guanine) or ≥1 mM Mg2+ (in the absence of guanine), the riboswitch starts to adopt a folded conformation in which loop−loop interactions lock P2 and P3 into place. At >5 mM Mg2+, further compaction occurs in which P1 more closely approaches P3. Our data help to explain the biological role of guanine as stabilizing the globally folded aptamer domain conformation at physiologically relevant Mg2+ concentrations (≤1 mM), whereas in the absence of guanine, much higher Mg2+ concentrations are required to induce this folding event.
The ability of many bacterial mRNAs to self-regulate their own transcription is exemplified by the guanine binding riboswitch of the xpt-pbuX operon in Bacillus subtilis(1). As with all known bacterial riboswitches (2−4), the guanine riboswitch aptamer domain is located in the 5′-untranslated region (5′-UTR) of the regulated mRNA. Binding of the metabolite guanine by the aptamer domain promotes formation of a terminator stem in the downstream expression platform domain, leading to transcriptional attenuation (1,5). Upon binding of their cognate ligands, other riboswitches may either repress translation or upregulate gene expression (2); upregulation of either transcription (1,6) or translation (7) is observed for adenine riboswitches. Structural rearrangement of a riboswitch expression platform is presumably caused by ligand-induced conformational changes in the aptamer domain, although in nearly all cases the detailed mechanism of this signal transduction remains to be elucidated (8).
Purine riboswitches are each composed of a three-way junction formed by three RNA helices designated P1, P2, and P3 [paired regions 1, 2, and 3 (Figure 1)] (9). Helices P2 and P3 are capped with loops L2 and L3, respectively. The remaining helix, P1, includes both the 5′- and 3′-ends of the aptamer domain and is directly connected to the downstream expression platform. The nonconserved P2 and P3 helices interact through L2−L3 base pairing, while the conserved P1 is crucial for regulating the riboswitch on and off state. Analyses of the secondary and tertiary structures of purine aptamer domains have revealed several distinct RNA−ligand interactions that are correlated with riboswitch activity. The connecting elements that join the P1, P2, and P3 helices create a solvent-inaccessible active site within the folded riboswitch. X-ray crystal structures of the guanine-responsive aptamer domain in complex with guanine or its analogue hypoxanthine reveal that base stacking and base triple interactions stabilize both the ligand binding pocket and the P1 helix, while residues from each connecting element form hydrogen bonds with the ligand (10,11). The Watson−Crick face of the guanine is base paired with a specific cytosine of the riboswitch. The adenine riboswitches have essentially the same structure except that the riboswitch cytosine is replaced with a uracil; this base pairing contact is the sole determinant of purine specificity (11−15). Extensive mutagenesis efforts as well as the X-ray crystal structures have identified the key RNA nucleotides involved in purine binding (16). Mutational analysis of the guanine riboswitch demonstrated that the L2−L3 interaction is required for the binding of lower-affinity ligands such as hypoxanthine (10). However, recognition by the purine aptamer domain of a higher-affinity analogue such as 2,6-diaminopurine can occur (albeit with decreased affinity) even when L2−L3 interactions are weakened or removed entirely (17,18).
Figure 1.
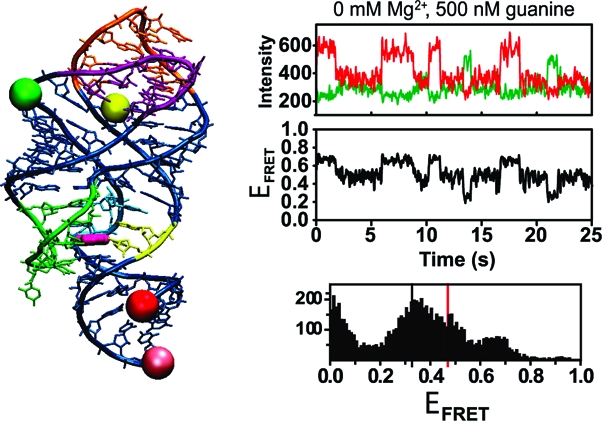
X-ray crystal structure of the guanine-bound xpt guanine riboswitch aptamer domain (10), marked with dye positions for the variants used in this FRET study. Guanine is colored dark pink. The green (Cy3) and red (Cy5) spheres mark dye locations on the variant designed for probing dynamics between the P1 and P2 helices (P1−P2 variant). Similarly, yellow (Cy3) and light pink (Cy5) spheres mark dye locations on the P1−P3 variant, and green (Cy3) and yellow (Cy5) spheres mark dye locations on the P2−P3 variant. Nucleotide color scheme (same as in ref (10)): J1/3 (joining region connecting P1 and P3), yellow; J2/3, green; J1/2, blue; L2 (loop capping P2), purple; L3, orange.
In some but not all cases, purine riboswitch activity is controlled by the kinetics of transcription and ligand association rather than thermodynamic binding affinity (7,17,19), which was also suggested for an FMN riboswitch (20). An optical tweezers-based single-molecule study of an in situ transcribed adenine riboswitch demonstrated that the aptamer domain folds at a rate comparable to the standard RNA synthesis rate (21,22). Collectively, these studies support the suggestion that ligand-induced tertiary interactions within the aptamer domain are sufficiently rapid to control mRNA transcription on a biologically relevant time scale. Understanding the conformational dynamics of the riboswitch aptamer domain will therefore provide insight into the biological functioning of the riboswitch.
Fluorescence resonance energy transfer (FRET)1 spectroscopy is commonly used to investigate biomolecular folding events, including conformational changes in riboswitches and other RNA molecules at the ensemble (23−25) and single-molecule levels (17,26−34). A single-molecule FRET investigation suggested that the folding of the pbuE adenine riboswitch aptamer domain involves an intermediate state, which was attributed to stacking of the P1 and P3 helices before P2 could fully interact with P3 (17). Interaction between P2 and P3 was observed without adenine, suggesting that a purine riboswitch aptamer domain may fluctuate from a relatively low-affinity conformation to a structure that is competent for purine binding even in the absence of the purine ligand itself. Additionally, an ensemble time-resolved fluorescence study observed multiple distinct adenine-bound aptamer domain conformations (35).
In this study, we comprehensively investigated the structure and dynamics of the xpt guanine riboswitch aptamer domain using both ensemble and single-molecule FRET spectroscopy. Our efforts significantly extend the previous single-molecule FRET study of the adenine riboswitch aptamer domain, which was performed on only one construct that probed the L2−L3 interaction between the dye-labeled P2 and P3 helices (17). In our work, we synthesized three G riboswitch aptamer domain variants that have orthogonal combinations of dye positions to investigate the structure and dynamics of each of the three helices (P1, P2, and P3) relative to one another. Our findings support a multistep folding model for the xpt G riboswitch aptamer domain. Our findings also provide further explanation for the role of guanine in modulating the riboswitch aptamer domain structure at physiologically relevant Mg2+ concentrations.
Experimental Procedures
Synthesis of xpt G Riboswitch Aptamer Domain Variants
All RNA substrates containing 2′-amino modifications were synthesized at the Yale W. M. Keck Center (New Haven, CT) or the University of Illinois at Urbana−Champaign W. M. Keck Center. RNA substrates were deprotected according to the supplier’s protocol, desalted on Sephadex G-25 NAP columns (GE Healthcare), and purified via 20% denaturing PAGE. Unmodified RNA substrates were transcribed from template DNA using T7 RNA polymerase and purified by 6 to 20% denaturing PAGE. The three G riboswitch aptamer domain variants (denoted P1−P2, P1−P3, and P2−P3 according to which two helices bear the FRET dye labels) were synthesized as described below. Additional details for aptamer domain synthesis are reported elsewhere (36).
P1−P2 Variant
The 5′-portion of the P1−P2 aptamer construct, with the 25 nt sequence 5′-GCACUCAUAUAAUCGCGUGGAUAUG-3′ (U30 in bold functionalized with 2′-amine), was conjugated with Cy3 NHS ester in sodium phosphate buffer (pH 8.0) with 0.4 mM EDTA and 18% DMSO for 24 h at 4 °C. The 3′-portion of the P1−P2 aptamer construct, with the 64 nt sequence 5′-GCACGCAAGUUUCUACCGGGCACCGUAAAUGUCCGACUAUGGGUGAGCCUCGCUGCCGUCGCCA-3′, was prepared by in vitro transcription using a PCR-generated DNA template and T7 RNA polymerase. The underlined portion of the 64-mer is an 18 nt extension of the 3′-end for annealing to a biotinylated and Cy5-functionalized 18-mer DNA (5′-biotin-TGGCGACGGCAGCGAGGC-Cy5-3′), which was synthesized at Integrated DNA Technologies (Coralville, IA). The two RNA substrates were joined by incubation with a cDNA splint and T4 RNA ligase in 50 mM Tris (pH 7.5), 10 mM DTT, 10 mM MgCl2, and 1 mM ATP for 2 h at 37 °C. T4 RNA ligase rather than T4 DNA ligase was found to be more effective for this particular ligation reaction.
P1−P3 Variant
The 5′-portion of P1−P3, with the 73 nt sequence 5′-GGAUGGCGACGGCAGCGAGGCGCACUCAUAUAAUCGCGUGGAUAUGGCACGCAAGUUUCUACCGGGCACCGUA-3′, was prepared by in vitro transcription using a PCR-generated DNA template and T7 RNA polymerase. The underlined portion represents the 18 nt extension described for P1−P2; the corresponding 18-mer DNA was derivatized with biotin at its 3′-end and Cy5 at its 5′-end. The 3′-portion of P1−P3, with the 19 nt sequence 5′-AAUGUCCGACUAUGGGUGA-3′ (U67 in bold functionalized with 2′-amine), was conjugated with Cy3 NHS ester as described for P1−P2. The two RNA substrates were joined by incubation with a DNA splint and T4 RNA ligase as described for P1−P2.
P2−P3 Variant
This riboswitch variant was assembled from three RNA fragments. The 5′-portion of P2−P3 included the same 25 nt modified with Cy3 at U30 as described for P1−P2. This RNA was joined to the central portion of P2−P3 with the 27 nt sequence 5′-GCACGCAAGUUUCUACCGGGCACCGUA-3′ using a cDNA template and T4 DNA ligase. The 3′-portion of P2−P3 was the 19 nt sequence described for P1−P3, with Cy5 rather than Cy3 at U67. A 21 nt hybridization sequence, 5′-GGAUGGCGACGGCAGCGAGGC-3′, was prepared for attachment at the 5′-end of P2−P3; the corresponding 18-mer DNA was derivatized with biotin at its 3′-end and had no Cy5 label. The 3′-portion and hybridization sequence RNAs were simultaneously attached to the remainder of P2−P3 using two cDNA splints and T4 RNA ligase.
In-Line Probing (37)
Each aptamer domain variant, as well as an unmodified RNA of the same sequence (without the 2′-amino group or dye labels), was 5′-32P-radiolabeled and incubated at 25 °C for 40 h in 50 mM Tris (pH 8.5), 20 mM MgCl2, and 100 mM KCl in the absence of ligand or with 1 μM guanine or adenine, the latter as a negative control. Samples (0.25 pmol) were then subjected to alkaline hydrolysis by incubation for 20 min at 90 °C in 50 mM sodium bicarbonate (pH 9.2 at 25 °C) and 0.1 mM EDTA. In parallel, RNase T1 digestion of each sample was performed in 50 mM Tris (pH 7.5) and 1.0 mM EDTA with 1.25 units of RNase T1 (Ambion) for 2 min at 25 °C. Cleavage products were separated via 10% denaturing PAGE and analyzed using a PhosphorImager. Each probing lane was quantified by normalizing the corresponding line graph of pixel intensity versus lane position. The normalization was performed relative to a control band within the same lane whose intensity was considered to be independent of guanine. Normalized line graphs in the absence and presence of guanine were superimposed to provide a visual measure of the effect of guanine on the extent of in-line probing cleavage at each nucleotide position (Figure S1 of the Supporting Information).
Ensemble FRET Measurements
Ensemble FRET titrations were performed with a ThermoSpectronic AB2 fluorescence spectrometer. The excitation wavelength was 550 nm for Cy3 and 649 nm for Cy5. Constructs were first annealed to biotinylated DNA (included here to sequester the RNA extension used for surface immobilization in the single-molecule FRET experiments) by being heated at 95 °C in 10 mM HEPES (pH 7.5) and 50 mM NaCl for 3 min and then slowly cooled to room temperature (>3 h). When the Cy3 and Cy5 dyes were attached to different oligonucleotide strands, the strand concentrations were 14 and 7 nM, respectively. For titration experiments that included 500 nM guanine, the guanine was added after annealing and immediately prior to temperature equilibration. Aliquots of 10 μM to 900 mM MgCl2 in the annealing buffer were mixed with the sample by manual pipetting; the sample was equilibrated at 25 °C in a recirculating water bath for 5 min before each measurement. Ensemble FRET efficiency (E) values were obtained through (ratio)A analysis (38) and are the average from two experiments. The FRET data were fit with the two-component titration equation described previously (39).
Single-Molecule FRET Measurements
Single-molecule FRET data were obtained using a wide-field prism-type total internal reflection microscope at room temperature (40). Sample chambers were prepared from quartz slides and glass coverslips and were incubated with 1 mg/mL BSA-biotin in 10 mM Tris (pH 8.0) and 50 mM NaCl (T50 buffer) for 10 min before flow-through with T50 buffer and incubation with 0.2 mg/mL neutravidin in T50 buffer. After the excess neutravidin had been rinsed from the chamber, 100 pM aptamer annealed to the biotinylated DNA strand (as described for the ensemble measurements) in T50 buffer was passed into the chamber and incubated for 5 min. Imaging buffer [10 mM HEPES (pH 7.5) and 50 mM NaCl] and an oxygen scavenging system were passed through the chamber, removing non-surface-bound aptamers. The oxygen scavenging system included 0.5% glucose, 165 units/mL glucose oxidase, 217 units/mL catalase, and 1 mM Trolox to suppress Cy5 blinking (41). Dye excitation was performed with a 532 nm laser (CrystaLaser). Fluorescence emission was collected with a water immersion objective (1.2 NA, 60×, Olympus) and imaged with a charge-coupled device (CCD) camera (iXon, Andor Technology) using 30 ms time integration.
Analyses of single-molecule trajectories were performed using custom programs written in MATLAB (42). The FRET efficiency (E) values were determined as the acceptor intensity divided by the total donor and acceptor intensities after correction for leakage between donor and acceptor channels. For construction of each single-molecule FRET histogram, E values were averaged from the first 300 ms of each of >104 single-molecule time traces for each combination of aptamer variant and buffer condition. For determination of each dwell time for the P2−P3 variant, an average of ∼140 molecules in each buffer condition were manually selected such that each molecule contained both donor and acceptor, as determined via observation of photobleaching events. The average times spent in high-FRET and low-FRET states were determined for each molecule individually by recording and averaging dwell times above and below a FRET threshold (E ∼ 0.45 for P1−P2, and E ∼ 0.6 for P2−P3), and scatter plots were made using these averages. For the determination of average rate constants from high-FRET to low-FRET transitions, the averages of the log values for all high-FRET and low-FRET average dwell times represented in the scatter plots were taken, and the exponentials of these averages yielded the reported high-FRET and low-FRET log-averaged rate constants, respectively.
Results
Three orthogonally labeled xpt guanine riboswitch aptamer domain variants were synthesized for ensemble and single-molecule FRET analyses at different Mg2+ and guanine concentrations. Shown in Figures 2−4 are results for the P1−P2, P1−P3, and P2−P3 variants, respectively, each of which is named according to the two helices that were labeled with a suitable combination of FRET dyes Cy3 (donor) and Cy5 (acceptor). In all cases, the P1 helix was extended by an 18 nt segment of single-stranded DNA to allow surface immobilization via a biotinylated cDNA strand. Each aptamer variant remained free in solution for the ensemble FRET measurements, whereas the RNA was bound to a neutravidin-coated glass coverslip for the single-molecule FRET measurements. Choices of dye labeling sites were guided by the xpt G riboswitch X-ray crystal structure (10,11). The P1 helix was labeled with Cy5 away from the guanine binding site by attaching the dye to the biotinylated DNA strand. Nucleotides U30 and U67 within P2 and P3, respectively, were chosen for dye labeling on the basis of the large distance change anticipated for the forming and breaking of the L2−L3 loop−loop interaction, while minimizing potential interference with ligand binding and loop−loop interactions. In-line probing (37) of each aptamer variant with and without the dyes confirmed that all three modified aptamers are capable of binding guanine and that each aptamer structure is minimally perturbed by the modifications (Figure S1 of the Supporting Information).
Figure 2.

Ensemble and single-molecule FRET analyses of the P1−P2 aptamer domain variant. (A) Design of P1−P2 for smFRET analysis. For ensemble FRET, the neutravidin surface was omitted. (B) Ensemble FRET titration of P1−P2 with Mg2+ at 0 or 500 nM guanine. (C) smFRET histograms for P1−P2. The vertical guidelines are at E values of 0.32 and 0.47. (D) Representative single-molecule smFRET time traces for P1−P2.
Figure 4.

Ensemble and single-molecule FRET analyses of the P2−P3 aptamer domain variant. (A) Design of P2−P3 for smFRET analysis. For ensemble FRET, the neutravidin surface was omitted. (B) Ensemble FRET titration of P1−P2 with Mg2+ at 0 or 500 nM guanine. Each curve was fitted to a standard two-component titration curve with a Hill coefficient of 1 for each component (39). (C) smFRET histograms for P2−P3. (D) Representative single-molecule smFRET time traces for P2−P3. Time traces at other Mg2+ and guanine concentrations (up to 1 mM Mg2+) were qualitatively similar, although the quantitative dwell time values varied slightly (see panel E). (E) Plots of rate constants kfold and kunfold as determined from single-molecule dwell times. Error bars represent the standard error from averaging rates of all molecules under the indicated buffer condition. (F) The average duration of the high-FRET (folded) and low-FRET (unfolded) dwell times of 154 P2−P3 molecules in 100 μM Mg2+ reveals broad heterogeneity. Inclusion of guanine did not considerably change the heterogeneity. Scatter plots at other Mg2+ concentrations are shown in Figure S5 of the Supporting Information.
Conformational changes in the aptamer domain variants were monitored via both ensemble and single-molecule FRET. Ensemble FRET data were acquired at 25 °C. Single-molecule FRET data were obtained at room temperature using a total internal reflection fluorescence microscope (40). The effects of Mg2+ and guanine concentrations were examined for each of the three G riboswitch aptamer domain variants. The results enabled construction of a global model of the relative orientations and dynamics of the P1, P2, and P3 helices and allowed assessment of the conformational role of guanine under physiologically relevant conditions.
Ensemble FRET Measurements
Ensemble FRET measurements were taken for all three aptamer domain variants. These experiments were performed in 10 mM HEPES (pH 7.5) and 50 mM NaCl at 25 °C by titration of Mg2+ from 0.1 μM to 100 mM in the presence of either 0 or 500 nM guanine, where the binding constant for guanine is ≤5 nM (1). FRET efficiency (E) values were determined by the (ratio)A method (38). For the P1−P2 variant, E was nearly unchanged up to 5 mM Mg2+ and was approximately independent of the guanine concentration [0 or 500 nM (Figure 2B)], indicating no overall distance change between the P1 and P2 helices upon tertiary folding of the riboswitch. A slight increase in E was observed at the highest tested Mg2+ concentrations (5−100 mM), which are considered nonphysiological. For the P1−P3 variant, E initially decreased as the Mg2+ concentration was increased to 1 mM and then increased as the Mg2+ concentration was raised even higher, indicating that P1 and P3 move farther apart and then closer as Mg2+ is added (Figure 3B). These changes in E depended on the guanine concentration. In the absence of guanine, the Mg2+ midpoint for the decrease in E was ∼0.4 mM, whereas at a saturating guanine concentration of 500 nM, the Mg2+ midpoint was ∼50 μM. Finally, for the P2−P3 variant, E increased substantially as the Mg2+ concentration was increased through the entire tested range, with a slightly lower Mg2+ midpoint at 500 nM guanine relative to 0 nM guanine (Figure 4B). Therefore, P2 and P3 move closer together at higher Mg2+ concentrations, as expected upon stable formation of the L2−L3 loop−loop interaction.
Figure 3.

Ensemble and single-molecule FRET analyses of the P1−P3 aptamer domain variant. (A) Design of P1−P3 for smFRET analysis. For ensemble FRET, the neutravidin surface was omitted. (B) Ensemble FRET titration of P1−P3 with Mg2+ at 0 or 500 nM guanine. Each curve was fitted to a standard two-component titration curve with Hill coefficients of 2 and 1 for the low- and high-Mg2+ components, respectively (39). (C) smFRET histograms for P1−P3. The vertical guideline is at an E of 0.4. (D) Representative single-molecule smFRET time traces for P1−P3.
Single-Molecule FRET Measurements
The single-molecule FRET (smFRET) measurements were performed in 10 mM HEPES (pH 7.5) and 50 mM NaCl at room temperature in the presence of either 0 or 500 nM guanine to obtain more detailed information about the conformations and dynamics within the G riboswitch aptamer domain. For each dual-labeled aptamer variant, at least 104 molecules in each incubation condition were monitored for up to 1 min to enable construction of FRET (E) histograms, and an appropriate subset of these molecules was chosen for more detailed dwell time determinations. The smFRET results for each of the three variants are presented below.
P1−P2 Variant
smFRET efficiency histograms were constructed for P1−P2 at various Mg2+ concentrations at either 0 or 500 nM guanine (Figure 2C). At 0 nM guanine and 0 mM Mg2+, two FRET states were evident with E values of ∼0.3 and ∼0.65, in addition to a population with an E of <0.1 that likely represents molecules without a functional Cy5 acceptor, i.e., Cy3 donor-only molecules. As the Mg2+ concentration was increased, the histogram collapsed to a single FRET state with an E of ∼0.4; this collapse occurred rather sharply between 100 μM and 1 mM Mg2+. With further increases in Mg2+ concentration, E increased slightly to ∼0.5.
Data were also obtained for P1−P2 in the presence of a saturating concentration of guanine. At 500 nM guanine and 0 mM Mg2+, in addition to the two FRET states with E values of ∼0.3 and 0.65, a third state with an E of ∼0.45 was clearly evident (Figure 2C). As observed in the absence of guanine, increasing the Mg2+ concentration led solely to the third FRET state, for which the E of ∼0.45 increased to ∼0.55 at very high Mg2+ concentrations. These values are slightly yet reproducibly higher than the E of ∼0.4−0.5 observed at similar Mg2+ concentrations in the absence of guanine, suggesting subtle guanine-dependent structural differences such as greater compaction of the aptamer. Including up to 50 μM adenine instead of guanine did not produce the third FRET state (Figure S2 of the Supporting Information).
The single-molecule time traces for the P1−P2 variant were examined individually (Figure 2D). In the absence of guanine, at 0 mM Mg2+, individual molecules toggled back and forth on the subsecond to second time scale between the two non-zero E states. A substantial fraction (∼40%) of the traces revealed >1 s−1 transitions (Figure S3 of the Supporting Information). In contrast, at 1 mM Mg2+, homogeneous behavior was observed. Importantly, when 500 nM guanine was included, the single-molecule time traces showed that all three FRET states can be visited by a single molecule, even at 0 mM Mg2+. Again, homogeneous behavior was seen at higher Mg2+ concentrations.
P1−P3 Variant
In contrast to the relatively complex behavior observed for P1−P2, for P1−P3 the single-molecule FRET behavior was simple: a single FRET peak was observed (Figure 3C). This peak shifted its position in a manner consistent with the ensemble FRET data, in that the E value decreased and then increased as the Mg2+ concentration was increased. Single-molecule time traces showed homogeneous FRET values, with little dynamics under all tested conditions (Figure 3D).
P2−P3 Variant
At either 0 or 500 nM guanine and 0 mM Mg2+, two distinct populations of P2−P3 with E values of ∼0.4 and 0.9 were observed in the single-molecule histograms (Figure 4C). As the Mg2+ concentration was increased above 1 mM, these two FRET peaks coalesced to a single population with an E of ∼0.8 (which is slightly lower than the high E value of 0.9 observed at lower Mg2+ concentrations). These findings are similar to observations for the adenine riboswitch aptamer domain, for which a high-FRET P2−P3 state predominated above 2 mM Mg2+(17). The inclusion of 500 nM guanine had a small but reproducible effect on the P2−P3 variant. In the ensemble FRET measurements, 500 nM guanine shifted the Mg2+ titration curve to lower concentrations by a factor of ∼3 (Figure 4B). The single-molecule data (Figure 4C) showed a similarly modest effect. At 100 μM Mg2+, the two FRET states with E values of ∼0.4 and 0.9 were observed regardless of the presence of guanine, although including 500 nM guanine favored the high-FRET state with an E of ∼0.8. The ratio of high-E (0.9 or 0.8) to low-E (0.4) populations was ∼0.5 with 0 nM guanine and ∼0.8 with 500 nM guanine, as determined by integrating the number of molecules in each FRET peak.
Single-molecule time traces for P2−P3 were examined in more detail. These traces revealed rapid switching (with an average frequency of ∼1 s−1) between the low-E and high-E states (Figure 4D). Despite modest differences in dynamic behavior between P1−P2 and P2−P3 (compare Figures 2D and 4D), the average dwell times for the high-E and low-E states of both P1−P2 and P2−P3 were comparable, as were the variations in the average high-E and low-E dwell times of many individual molecules (Figures S3 and S5A,B of the Supporting Information).
The dwell times in the high-E and low-E states of P2−P3 were determined at Mg2+ concentrations from 1 μM to 1 mM (Figure 4E); at higher Mg2+ concentrations, only the high-E state was observed. The kfold for the transition from the low-E state to the high-E state was calculated as the reciprocal of the average low-E dwell time. Similarly, kunfold for the transition from high E to low E was determined as the reciprocal of the average high-E dwell time. The time required for the actual transition between the states was shorter than the experimental time resolution (30 ms), consistent with a riboswitch known to operate at the transcriptional level (1,5,10). In the absence of guanine, both kfold and kunfold increased with Mg2+ concentration up to 100 μM, with kunfold slightly greater than kfold. The addition of 500 nM guanine had little effect at very low Mg2+ concentrations, but guanine stabilized the high-E state slightly at 100 μM Mg2+. The high-E state was considerably stabilized at 1 mM Mg2+ in a manner independent of the presence of guanine, with kfold and kunfold significantly increasing and decreasing, respectively, relative to their values at lower Mg2+ concentrations. At 100 μM Mg2+, guanine stabilized the P2−P3 folded high-E state over the unfolded low-E state by ∼0.5 kcal/mol (ΔΔG°), as calculated from the equilibrium constants kfold and kunfold (Figure S4 of the Supporting Information). Under all conditions, ∼100-fold variations in high-E and low-E dwell times revealed heterogeneity similar to that observed for the P2−P3 folding dynamics of the adenine riboswitch (Figure S5A of the Supporting Information) (17). Scatter plots of the average high-FRET and low-FRET dwell times for 154 molecules in 100 μM Mg2+ indicate that introduction of guanine only slightly affects the distribution of dwell times (Figure 4F). There is a modest increase in the fraction of molecules that have long dwell times in the folded state (2 of 154 molecules with dwell times above 10 s without guanine; 15 of 154 molecules above 10 s with guanine). This effect is also observed in the average dwell times (Figure 4E), where kunfold is affected more than kfold by the presence of guanine at 100 μM Mg2+.
Discussion
A Model for Conformational Changes of the xpt G Riboswitch Aptamer Domain
RNA conformational changes induced by ligand binding to riboswitches have biologically important consequences (2−4). In this study, we have comprehensively examined how Mg2+ and guanine influence the relative orientations and dynamics of the three RNA helices that compose the xpt guanine riboswitch aptamer domain. On the basis of our combined ensemble and single-molecule FRET data, we have constructed a model of the Mg2+- and guanine-dependent conformational changes that are experienced by the G riboswitch aptamer domain (Figure 5).
Figure 5.

Proposed folding model for the G riboswitch aptamer domain.
Effect of Mg2+ on the Aptamer Domain Structure and Dynamics
The ensemble data (Figures 2B, 3B, and 4B) require that at least two different conformations be sequentially induced as the Mg2+ concentration is increased. The constant E value for the P1−P2 variant at lower Mg2+ concentrations (Figure 2B) suggests a model in which there is little relative change in the equilibrium relationship of P1 and P2 as the Mg2+ concentration is varied; only at very high and nonphysiological Mg2+ concentrations (>5 mM) do P1 and P2 begin to move closer together. In contrast, the biphasic nature of the changes in E for the P1−P3 variant (Figure 3B) suggests that P1 and P3 initially move apart and then move closer together as the Mg2+ concentration is increased. Finally, the substantial increase in E for the P2−P3 variant (Figure 4B) is consistent with formation of L2−L3 loop−loop interactions at higher Mg2+ concentrations, as expected from many studies on purine riboswitches (2,3,9). The overall model in Figure 5 is consistent with all of these ensemble FRET data, both in the absence and in the presence of guanine.
The single-molecule FRET data reveal an underlying dynamic complexity that is not apparent solely from the ensemble measurements, which provide only an averaged description of the RNA structure. The smFRET data for the P1−P3 variant (Figure 3C,D) indicate that P1 and P3 form a nondynamic platform, upon which P2 changes its orientation. The smFRET data for the P1−P2 variant (Figure 2C,D) show that two conformations with different relationships between P1 and P2 contribute to the constant ensemble E at lower Mg2+ concentrations. As the Mg2+ concentration is increased, a single state with intermediate E is populated, indicating the formation of a structure in which P1 and P2 no longer dynamically change their relative orientation. Similarly, the smFRET data for the P2−P3 variant (Figure 4C,D) reveal two contributing conformations that have different orientations between P2 and P3 at lower Mg2+ concentrations, and a single population with a high E at higher Mg2+ concentrations.
As shown in Figure 5, the simplest model consistent with all of these data depicts the P2 helix as switching dynamically between two conformations, for which the distance between P2 and P1 and the distance between P2 and P3 both change. The data indicate that P2 switches between two specific favored orientations, rather than uniformly exploring all potential orientations; the structural details of these two favored orientations are not defined by our data. As the Mg2+ concentration is increased, in addition to establishment of L2−L3 loop−loop interactions that hold together P2 and P3, the P1 and P3 helices also change their relative positions, presumably adopting the coaxially stacked orientation observed in the X-ray crystal structure (10,11). The further compaction observed at high (>5 mM) Mg2+ concentrations is likely not biologically relevant.
The high-E FRET value for the P2−P3 variant at low Mg2+ concentrations (<100 μM) is ∼0.9, whereas the single E value for P2−P3 at high Mg2+ concentrations (≥1 mM) is ∼0.8 (Figure 4C). This slight but clear decrease in E indicates that binding of Mg2+ causes a structural adjustment to the aptamer domain, resulting in a small relative movement of P2 and P3. This movement could be caused by motion of either P2 or P3 (or both), although the precise origin of the small change in E at high Mg2+ concentrations cannot be determined from these data.
Effect of Guanine on the Aptamer Domain Structure and Dynamics
The absence or presence of guanine influences both the structure and dynamics of the G riboswitch aptamer domain. This influence is manifested in several ways in both the ensemble and smFRET data. As revealed by the smFRET data for the P1−P2 variant (Figure 2C), inclusion of 500 nM guanine leads to the appearance of the intermediate-E population even in the absence of Mg2+. Therefore, guanine induces a particular conformation of the P2 helix relative to the P1/P3 helical scaffold, and this conformation predominates in the folded aptamer domain. At Mg2+ concentrations of ≥1 mM, guanine causes a slight increase in the E of the stable folded state (Figure 2C, vertical red lines), indicating the possibility of a more compact folded aptamer in the presence of ligand (21). Similarly, for the P1−P3 variant, the ensemble FRET data (Figure 3B) show a decrease in the Mg2+ requirement for the observed structural change. For the P2−P3 variant, the ensemble FRET data (Figure 4B) indicate a 3-fold decrease in the Mg2+ requirement for folding at 500 nM guanine. The P2−P3 smFRET data (Figure 4C) show a shift to the high-E population with guanine, and from kfold and kunfold values, the higher-E state is stabilized by ∼0.5 kcal/mol in the presence of guanine at 100 μM Mg2+. These observations all support the model in Figure 5. Either Mg2+ or guanine can stabilize the folded riboswitch structure, and the P2 helix dynamics are suppressed in the folded conformation.
Conformational Role of Guanine in Folding of the G Riboswitch Aptamer Domain
In vivo, the free intracellular Mg2+ concentration is generally millimolar or lower (43). The P1−P2 smFRET data (Figure 2C) at 100 μM Mg2+ provide clear evidence of the physiologically relevant role of guanine in G riboswitch aptamer domain folding. Although higher Mg2+ concentrations alone (i.e., without guanine) are capable of inducing folding as reflected in the population of the intermediate-E state, at 100 μM Mg2+ this state is substantially populated only when guanine is present. Therefore, the presence of guanine allows adoption of the folded aptamer domain conformation at physiologically relevant Mg2+ concentrations.
Relationship of the New Findings to Previous Work on Purine Riboswitch Aptamer Domain Folding
Previous work has revealed much about the conformations of purine riboswitches in addition to their static X-ray crystal structures (10,11). A careful kinetic study of the G riboswitch aptamer domain led Gilbert et al. to propose a conformationally dynamic unliganded state that is stabilized by initial Watson−Crick interactions between the aptamer and the guanine ligand, followed by formation of the three-helix junction that defines the overall architecture of the ligand-bound riboswitch (12). Other biochemical (1,18) and NMR spectroscopic (13) studies agree that the ligand binding pocket becomes organized only after ligand binding. Nevertheless, the aptamer domain has global organization even in the absence of guanine (12,13), and the guanine ligand can bind in the absence of Mg2+(44). Similarly for the adenine riboswitch, the loop−loop interactions that globally organize the structure can form in the absence of adenine, although the ligand is required to complete the overall folding process (17,21,22).
Our data provide a new perspective on these previous findings. By monitoring all three distance relationships (P1−P2, P1−P3, and P2−P3) via both ensemble and single-molecule FRET, we have developed the comprehensive model in Figure 5 for the overall Mg2+- and guanine-dependent conformational changes in the G riboswitch. Our results are fully consistent with all previous work, while showing that the dynamics of the unfolded aptamer domain primarily involve P2 helix motion around the relatively static platform formed by the P1 and P3 helices. In the adenine riboswitch aptamer domain, P2 is the first cotranscriptionally formed helix and the most stable of the aptamer’s structural elements (21,22). We also quantitatively established the Mg2+ and guanine concentrations that are together needed to coalesce multiple interconverting conformational states into a single folded structure. Importantly, our model provides a straightforward explanation for the role of the guanine ligand in modulating the G riboswitch structure under physiologically relevant conditions.
Because the guanine and adenine riboswitches share considerable sequence and structural homology, observations on purine riboswitches are readily compared. The previous study by Lemay et al. provided significant information about the pbuE adenine riboswitch via analysis of the P2−P3 vector alone (17). Our folding model in Figure 5, devised on the basis of measuring all three FRET vectors among the P1, P2, and P3 helices, is more comprehensive, enabling identification of at least three P2 conformational states and revealing an increase in the P1−P3 helical distance upon folding. Our other observations are consistent with most but not all of their findings. In particular, while they observed a fairly substantial decrease in the heterogeneity of the unfolded and folded dwell times upon inclusion of adenine (their Figure 5E), we found at most a slight guanine-dependent change in heterogeneity (Figure 4F). This difference may relate to the examination of adenine versus guanine riboswitches, although ultimately more data are needed to be sure.
Conclusions
By the combined application of ensemble and single-molecule FRET to three orthogonally labeled variants of the xpt G riboswitch aptamer domain, we have developed an overall folding model for this biological RNA that regulates gene expression. In the unfolded state, the model depicts the P1 and P3 helices as a nondynamic platform upon which the P2 helix dynamically switches between two orientations. With an increase in either the Mg2+ or guanine concentration, the folded state is formed. In this folded state, L2−L3 loop−loop interactions hold together P2 and P3, while P1 and P3 change relative orientations to adopt the coaxially stacked conformation that is observed in the X-ray crystal structure. These structural changes require a rather high (>1 mM) Mg2+ concentration in the absence of guanine, whereas a saturating guanine concentration enables adoption of the folded conformation when the Mg2+ concentration is ≤1 mM. Therefore, our findings clarify the role of guanine in G riboswitch aptamer domain structure at physiologically relevant Mg2+ concentrations.
Acknowledgments
We thank Dana Baum for assistance with sample management.
Supporting Information Available
Additional experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health Grants R01 GM065966 (to S.K.S.) and R01 GM065367 (to T.H.) and National Science Foundation Grant 0822613 (to T.H.).
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: FRET, fluorescence resonance energy transfer; smFRET, single-molecule FRET; nt, nucleotide.
Supplementary Material
References
- Mandal M.; Boese B.; Barrick J. E.; Winkler W. C.; Breaker R. R. (2003) Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell 113, 577–586. [DOI] [PubMed] [Google Scholar]
- Roth A.; Breaker R. R. (2009) The Structural and Functional Diversity of Metabolite-Binding Riboswitches. Annu. Rev. Biochem. 78, 305–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montange R. K.; Batey R. T. (2008) Riboswitches: Emerging themes in RNA structure and function. Annu. Rev. Biophys. 37, 117–133. [DOI] [PubMed] [Google Scholar]
- Blouin S.; Mulhbacher J.; Penedo J. C.; Lafontaine D. A. (2009) Riboswitches: Ancient and Promising Genetic Regulators. ChemBioChem 10, 400–416. [DOI] [PubMed] [Google Scholar]
- Johansen L. E.; Nygaard P.; Lassen C.; Agersø Y.; Saxild H. H. (2003) Definition of a second Bacillus subtilis pur regulon comprising the pur and xpt-pbuX operons plus pbuG, nupG (yxjA), and pbuE (ydhL). J. Bacteriol. 185, 5200–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M.; Breaker R. R. (2004) Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol. 11, 29–35. [DOI] [PubMed] [Google Scholar]
- Rieder R.; Lang K.; Graber D.; Micura R. (2007) Ligand-Induced Folding of the Adenosine Deaminase A-Riboswitch and Implications on Riboswitch Translational Control. ChemBioChem 8, 896–902. [DOI] [PubMed] [Google Scholar]
- Garst A. D.; Batey R. T. (2009) A switch in time: Detailing the life of a riboswitch. Biochim. Biophys. Acta 1789, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. N.; Breaker R. R. (2008) Purine sensing by riboswitches. Biol. Cell 100, 1–11. [DOI] [PubMed] [Google Scholar]
- Batey R. T.; Gilbert S. D.; Montange R. K. (2004) Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature 432, 411–415. [DOI] [PubMed] [Google Scholar]
- Serganov A.; Yuan Y. R.; Pikovskaya O.; Polonskaia A.; Malinina L.; Phan A. T.; Höbartner C.; Micura R.; Breaker R. R.; Patel D. J. (2004) Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol. 11, 1729–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert S. D.; Stoddard C. D.; Wise S. J.; Batey R. T. (2006) Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain. J. Mol. Biol. 359, 754–768. [DOI] [PubMed] [Google Scholar]
- Noeske J.; Richter C.; Grundl M. A.; Nasiri H. R.; Schwalbe H.; Wöhnert J. (2005) An intermolecular base triple as the basis of ligand specificity and affinity in the guanine- and adenine-sensing riboswitch RNAs. Proc. Natl. Acad. Sci. U.S.A. 102, 1372–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulhbacher J.; Lafontaine D. A. (2007) Ligand recognition determinants of guanine riboswitches. Nucleic Acids Res. 35, 5568–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert S. D.; Reyes F. E.; Edwards A. L.; Batey R. T. (2009) Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure 17, 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert S. D.; Love C. E.; Edwards A. L.; Batey R. T. (2007) Mutational analysis of the purine riboswitch aptamer domain. Biochemistry 46, 13297–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay J. F.; Penedo J. C.; Tremblay R.; Lilley D. M. J.; Lafontaine D. A. (2006) Folding of the adenine riboswitch. Chem. Biol. 13, 857–868. [DOI] [PubMed] [Google Scholar]
- Stoddard C. D.; Gilbert S. D.; Batey R. T. (2008) Ligand-dependent folding of the three-way junction in the purine riboswitch. RNA 14, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickiser J. K.; Cheah M. T.; Breaker R. R.; Crothers D. M. (2005) The kinetics of ligand binding by an adenine-sensing riboswitch. Biochemistry 44, 13404–13414. [DOI] [PubMed] [Google Scholar]
- Wickiser J. K.; Winkler W. C.; Breaker R. R.; Crothers D. M. (2005) The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol. Cell 18, 49–60. [DOI] [PubMed] [Google Scholar]
- Greenleaf W. J.; Frieda K. L.; Foster D. A.; Woodside M. T.; Block S. M. (2008) Direct observation of hierarchical folding in single riboswitch aptamers. Science 319, 630–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman S. K. (2008) A Forced March across an RNA Folding Landscape. Chem. Biol. 15, 211–213. [DOI] [PubMed] [Google Scholar]
- Lilley D. M. J.; Wilson T. J. (2000) Fluorescence resonance energy transfer as a structural tool for nucleic acids. Curr. Opin. Chem. Biol. 4, 507–517. [DOI] [PubMed] [Google Scholar]
- Klostermeier D.; Millar D. P. (2001) Time-resolved fluorescence resonance energy transfer: A versatile tool for the analysis of nucleic acids. Biopolymers 61, 159–179. [DOI] [PubMed] [Google Scholar]
- Lilley D. M. (2004) The Varkud satellite ribozyme. RNA 10, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T.; Zhuang X.; Kim H. D.; Orr J. W.; Williamson J. R.; Chu S. (1999) Ligand-induced conformational changes observed in single RNA molecules. Proc. Natl. Acad. Sci. U.S.A. 96, 9077–9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X.; Bartley L. E.; Babcock H. P.; Russell R.; Ha T.; Herschlag D.; Chu S. (2000) A single-molecule study of RNA catalysis and folding. Science 288, 2048–2051. [DOI] [PubMed] [Google Scholar]
- Zhuang X.; Kim H.; Pereira M. J.; Babcock H. P.; Walter N. G.; Chu S. (2002) Correlating structural dynamics and function in single ribozyme molecules. Science 296, 1473–1476. [DOI] [PubMed] [Google Scholar]
- Tan E.; Wilson T. J.; Nahas M. K.; Clegg R. M.; Lilley D. M.; Ha T. (2003) A four-way junction accelerates hairpin ribozyme folding via a discrete intermediate. Proc. Natl. Acad. Sci. U.S.A. 100, 9308–9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z.; Srividya N.; Sosnick T. R.; Pan T.; Scherer N. F. (2004) Single-molecule studies highlight conformational heterogeneity in the early folding steps of a large ribozyme. Proc. Natl. Acad. Sci. U.S.A. 101, 534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodak J. H.; Downey C. D.; Fiore J. L.; Pardi A.; Nesbitt D. J. (2005) Docking kinetics and equilibrium of a GAAA tetraloop-receptor motif probed by single-molecule FRET. Proc. Natl. Acad. Sci. U.S.A. 102, 10505–10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobitski A. Y.; Nierth A.; Helm M.; Jaschke A.; Nienhaus G. U. (2007) Mg2+-dependent folding of a Diels-Alderase ribozyme probed by single-molecule FRET analysis. Nucleic Acids Res. 35, 2047–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira M. J.; Nikolova E. N.; Hiley S. L.; Jaikaran D.; Collins R. A.; Walter N. G. (2008) Single VS ribozyme molecules reveal dynamic and hierarchical folding toward catalysis. J. Mol. Biol. 382, 496–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner M.; Karunatilaka K. S.; Sigel R. K.; Rueda D. (2008) Single-molecule studies of group II intron ribozymes. Proc. Natl. Acad. Sci. U.S.A. 105, 13853–13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandari S.; Prychyna O.; Leung J.; Avdic D.; O'Neill M. A. (2007) Ligand-directed dynamics of adenine riboswitch conformers. J. Am. Chem. Soc. 129, 11308–11309. [DOI] [PubMed] [Google Scholar]
- Scanlan M. S. (2007) Fluorescence Investigations of RNA Folding: Group I Intron P4−P6 Domain and xpt G-Riboswitch Aptamer Domain. Ph.D. Thesis, University of Illinois at Urbana−Champaign, Urbana, IL.
- Regulski E. E.; Breaker R. R. (2008) In-line probing analysis of riboswitches. Methods Mol. Biol. 419, 53–67. [DOI] [PubMed] [Google Scholar]
- Clegg R. M. (1992) Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211, 353–388. [DOI] [PubMed] [Google Scholar]
- Silverman S. K.; Cech T. R. (1999) RNA Tertiary Folding Monitored by Fluorescence of Covalently Attached Pyrene. Biochemistry 38, 14224–14237. [DOI] [PubMed] [Google Scholar]
- Roy R.; Hohng S.; Ha T. (2008) A practical guide to single-molecule FRET. Nat. Methods 5, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasnik I.; McKinney S. A.; Ha T. (2006) Nonblinking and long-lasting single-molecule fluorescence imaging. Nat. Methods 3, 891–893. [DOI] [PubMed] [Google Scholar]
- McKinney S. A.; Declais A. C.; Lilley D. M.; Ha T. (2003) Structural dynamics of individual Holliday junctions. Nat. Struct. Biol. 10, 93–97. [DOI] [PubMed] [Google Scholar]
- Romani A.; Scarpa A. (1992) Regulation of cell magnesium. Arch. Biochem. Biophys. 298, 1–12. [DOI] [PubMed] [Google Scholar]
- Noeske J.; Buck J.; Fürtig B.; Nasiri H. R.; Schwalbe H.; Wöhnert J. (2007) Interplay of 'induced fit' and preorganization in the ligand induced folding of the aptamer domain of the guanine binding riboswitch. Nucleic Acids Res. 35, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.