

Abstract
Age-related macular degeneration (AMD) is the most prevalent form of irreversible blindness worldwide in the elderly population. The pathology of dry AMD consists of degeneration of photoreceptors and the RPE, lipofuscin (A2E) accumulation, and drusen formation. Mice have been widely used for generating models that simulate human AMD features for investigating the pathogenesis, treatment and prevention of the disease. Although the mouse has no macula, focal atrophy of photorecptors and RPE, lipofuscin accumulation, and increased A2E can develop in aged mouse eyes. However, drusen are rarely seen in mice because of their simpler Bruch’s membrane and different process of lipofuscin extrusion compared with humans. Thus, analyzing basal deposits at the ultrastructural level and understanding the ultrastructural pathologic differences between various mouse AMD models are critical to comprehending the significance of research findings and response to possible therapeutic options for dry AMD.
Based on the multifactorial pathogenesis of AMD, murine dry AMD models can be classified into three groups. First, genetically engineered mice that target genes related to juvenile macular dystrophies are the most common models, and they include abcr−/− (Stargardt disease), transgenic ELOVL4 (Stargardt-3 dominant inheritary disease), Efemp1R345W/R345W (Doyne honeycomb retinal dystrophy), and Timp3S156C/S156C (Sorsby fundus dystrophy) mice. Other murine models target genes relevant to AMD, including inflammatory genes such as Cfh−/−, Ccl2−/−, Ccr2−/−, Cx3cr1−/−, and Ccl2−/−/cx3cr1−/−, oxidative stress associated genes such as Sod1−/− and Sod2 knockdown, metabolic pathway genes such as neprilysin −/− (amyloid β), transgenic mcd/mcd (cathepsin D), Cp−/−/Heph−/Y (ferroxidase ceruloplasmin/hepaestin, iron metabolism), and transgenic ApoE4 on high fat and high cholesterol diet (lipid metabolism). Second, mice have also been immunologically manipulated by immunization with carboxyethylpyrrole (CEP), an oxidative fragment of DHA found in drusen, and found to present with dry AMD features. Third, natural mouse strains such as arrd2/arrd2 (Mdm gene mutation) and the senescence accelerated mice (SAM) spontaneously develop features of dry AMD like photoreceptor atrophy and thickening of Bruch’s membrane.
All the aforementioned models develop retinal lesions with various features that simulate dry AMD lesions: focal photoreceptor degeneration, abnormal RPE with increased lipofuscin, basal infolding, decreased melanosomes and degeneration. However, Bruch’s membrane changes are less common. Most mice develop retinal lesions at an older age (6–24 months, depending on the models), while the Ccl2−/−/cx3cr1−/− mice develop lesions by 4–6 weeks. Although murine models present various degrees of retinal and/or RPE degeneration, classical drusen is extremely rare. Using electron microscopy, small drusenoid deposits are found between RPE and Bruch’s membrane in a few models including Efemp1 R345W/R345W, Ccl2−/−/cx3cr1−/−, neprilysin −/−, transgenic mcd/mcd, and ApoE4 transgenic mice on a high fat diet. High A2E levels are measured in the retinas of abcr−/−, transgenic ELOVL4, and Ccl2−/−/cx3cr1−/− mice. In summary, murine models provide useful tools for studying AMD pathogenesis and evaluating novel therapies for this disease. This review compares the major dry AMD murine models and discusses retinal pathology at the ultrastructural level.
Keywords: Age-related macular degeneration, AMD, dry, mouse model, retina, ultrastructure, pathology
1. Introduction
Age-related macular degeneration (AMD) is the leading cause of irreversible visual impairment and blindness in the elderly population (World Health Organization 2009; Friedman et al., 2004). The development of AMD is a slow progressive process that occurs with aging and mainly affects people over age 60 (Gehrs et al., 2006). People with AMD experience a loss of high-resolution vision and perceive distortions and loss of images in the center of their visual field, making driving and reading very difficult. Many environmental risk factors, especially smoking, have been associated with an increased risk of developing AMD. The genetics of macular degenerative diseases, or maculopathies, is complicated, and genes associated with AMD are being identified. Mendelian inherited macular degeneration (MD) that often develops in the early ages, can be either autosomal dominant or recessive. Complex MD can result from chloroquine and clofazimine drug toxicities (Wolfensberger, 1998). AMD still has an unknown etiology, though oxidative stress to the retinal pigment epithelium (RPE) cells along with immune dysregulation are leading theories of AMD pathogenesis (Ding et al., 2009).
1.1. Epidemiology of AMD
AMD accounts for more than 50% of blindness cases in Caucasian Americans, the group most affected by early and late AMD in the United States (Congdon et al., 2004). The prevalence of the disease increases with age, and by age 75 early AMD affects 30% of the population. An estimated 1.75 million Americans were affected by AMD in 2000, and the number is expected to rise to 2.95 million in 2020 (Friedman et al., 2004). Worldwide, 30–50 million people are affected by AMD, and limited population studies in Asia demonstrate a prevalence of AMD that is lower than the United States (Wong et al., 2006; Gehrs et al., 2006).
1.2. Pathology of AMD
Aging induces retinal changes in RPE cell size and shape and results in the accumulation of lysosomal residual bodies called lipofuscin granules. With age, photoreceptor cells (rods much more than cones, peripheral more than central retina) and ganglion cells, decrease in number and density; RPE cells also show parallel changes and progressive cell loss (Gao and Hollyfield, 1992; Panda-Jonas et al., 1995). Recently, it has been noted that abnormalities in the distal cone axon occurs with aging and increases susceptibility to AMD (Shelley et al., 2009). There is an age-related increase in thickness in Bruch’s membrane and the internal limiting membrane (Sarks, 1976; Ding et al., 2009). Cholesterol and calcium have been found to accumulate in Bruch’s membrane throughout adulthood (Sarks, 1976; Curcio et al., 2001; Pauleikhoff et al., 1990). These age-associated changes set the stage for AMD development in individuals with a genetic predisposition and exposure to environmental risk factors.
AMD was first described as “symmetric central chorioretinal disease occurring in senile persons” (Hutchinson and Tay, 1875). The signs of degenerative retinal change in AMD have been described in histopathologic studies of AMD eyes (Green, 1993; Sarks, 1976). The classic pathology of early AMD is multiple small or intermediate drusen (drusen size of ≥ 63 microns but < 125 microns) in the macular area (Coleman et al., 2008). The classic pathology of advanced AMD is described as having two forms: the ‘geographic atrophic (dry)’ and ‘neovascular/exudative (wet)’ forms. The ‘dry’ form of AMD, non-neovascular AMD, including intermediate AMD with many intermediate or large drusen (drusen size of ≥ 125 microns) and RPE alterations and advanced AMD with geographic atrophy, consists of many perturbations in the RPE, Bruch’s membrane, photoreceptors, and choriocapillaris, leading to photoreceptor degeneration and atrophy. New blood vessel formation from the choroid, known as choroidal neovascularization (CNV), is the hallmark of ‘wet’ AMD. This form, often accompanied by hemorrhages, serous exudates, and edema in the neuroretina, is called exudative or neovascular AMD.
1.2.1. Early stage AMD
In the early stages of AMD, pathologic findings are found mostly in the RPE and Bruch’s membrane. The RPE progressively accumulates lipofuscin, leaving partially digested photoreceptors in the cytoplasm. The number and density of RPE cells in the macula also decrease. Early in the disease process, basal deposits, a type of waste between the RPE and Bruch’s membrane, can be identified by electron microscopy. There are two types of basal deposits: basal laminar deposits (BlamD) and basal linear deposits (BlinD). BlamD are a granular electron dense material with wide-spaced collagen between the plasma membrane and basal lamina of the RPE (Green, 1993). BlinD are lipid-rich electron dense substances with granules and vesicles external to the RPE basement membrane in the inner collagenous zone. BlinD are a more specific marker of AMD progression to late disease, and BlamD are a more reliable indicator of RPE atrophy and photoreceptor degeneration (van der Schaft et al., 1992; Curcio and Millican, 1999; Sarks, 1976). Thick BlamD closely correlate with the presence of AMD (Spraul and Grossniklaus, 1997).
Other features of early AMD include thickening and loss of the normal architecture of Bruch’s membrane and drusen formation. Drusen are subretinal extracellular deposits composed of glycoproteins and lipids in the inner collagenous zone of Bruch’s membrane (Sarks, 1976). Drusen can be described as soft or hard. Large soft drusen are thought to arise from BlinD, which are 24-fold more common in eyes with AMD than age-matched controls (Curcio and Millican, 1999). Hard, or nodular, drusen are smooth surfaced, dome-shaped structures between the RPE and Bruch’s membrane that are clinically common. At the ultrastructural level, drusen are a fine, granular, amorphous material the same electrodensity of the RPE basement membrane with vesicles, tubular structures, curly membranes and abnormal collagen (Sarks, 1976).
Studies analyzing the composition of drusen have shown that drusen contain acute phase proteins (C-reactive protein, vitronectin, α-antichymotrypsin, amyloid P component, and fibrinogen), complement pathway components (C3, C5 and C5b-9 complex) and inhibitors (clusterin), apolipoproteins B and E, mucopolysaccarides, lipids, mannose, and sialic acid (Farkas et al., 1971; Mullins et al., 2000). Lower levels of the endoplasmic reticulum chaperone protein ERp29 are found in neurodegenerative diseases, in the AMD maculae, and the aging retina. Low ERp29 levels impacts chaperone function and lead to the accumulation of misfolded proteins which may aggregate and interfere with intracellular movement, allowing for the aggregation of incompletely folded compounds such as lipofuscin and drusen to cause RPE damage (Li et al., 2004; Ethen et al., 2006). Loss of mitochondria is also reported in AMD retina (Ding et. al, 2009). Lipofuscin accumulation in RPE cells, basal deposits, drusen, and thickening of Bruch’s membrane are all seen in early AMD and may continue to be present in late stages of the disease. Additionally, patients with drusen and CNV have higher circulating levels of anti-retinal antibodies compared to controls (Patel et al., 2005), and this profile does not significantly change with progression from early AMD to advanced stage AMD (Cherepanoff et al., 2006).
1.2.2. Advanced stage AMD
In the advanced stage of dry AMD called geographic atrophy, the RPE has well-demarcated atrophy and the neurosensory retina is severely affected (Coleman et al., 2008). Areas of geographic atrophy may have a single layer of macrophages between the RPE basement membrane and the inner collagenous layer of Bruch’s membrane with residual pigmented material. The retina adjacent to the areas of geographic atrophy (junctional zones) is hyperpigmented and has hypertrophied RPE cells with occasional multinucleated giant cells (Dastgheib and Green, 1994). RPE atrophy is often accompanied by degeneration and loss of the outer layers of the retina (photoreceptors, outer nuclear layer, and external limiting membrane). Cone degeneration and red-green cone hypertrophy are also found. Photoreceptor cell loss is most prominent in the parafovea and can progress to total photoreceptor loss and disciform degeneration (Curcio et al., 1996; Sarks, 1976; Shelley et al., 2009). Additionally, sclerosis of the choriocapillaris is found without breaks in Bruch’s membrane (Sarks, 1976).
When Bruch’s membrane becomes calcified and focal breaks occur, new blood vessels from the choroid form in the space under the RPE, and this CNV is the hallmark of exudative/neovascular, or ‘wet’ AMD (Spraul and Grossniklaus, 1997). IgG antibodies against retinal antigens are present in a complex and specific pattern in wet AMD eyes, which differs from dry AMD eyes and healthy controls (Joachim et al., 2007). Neovascular AMD often causes sudden loss of vision and presents with serous or hemorrhagic detachment of either the retinal pigment epithelium or sensory retina. Subretinal fibrous tissue, or minimal subretinal fibrosis, and widespread RPE atrophy are also often noted (Coleman et al., 2008). Hemorrhages, exudates, and neovascular fibrous tissue may replace normal retinal architecture and result in photoreceptor atrophy (Green, 1999; Kim et al., 2002). The presence of an inflammatory cell component at the site of breaks in Bruch’s membrane strongly suggests that the immune system is intricately involved in disease progression (van der Schaft et al., 1992; Spraul and Grossniklaus, 1997; Anderson et al., 2002; Chan et al., 2008; Patel and Chan, 2008).
In this review, we focus on murine models for dry AMD, including intermediate stage and geographic atrophy. We will emphasize the histopathology, particularly the retinal ultrastructure, and molecular pathologic findings in the presented mouse models.
2. The use of murine models for AMD
Since 1924 when the first inherited form of retinal degeneration was identified, the mouse has been the main model organism for the study of diseases involving retinal degeneration (Keeler, 1924; Hafezi et al., 2000). Mouse strains with spontaneously arising retinal degeneration (the Rd mice) have been described, and genes causing retinal degeneration have been identified, characterized, and manipulated at the molecular level (Hafezi et al., 2000). Mouse models for retinitis pigmentosa (RP), cone-rod dystrophies (CORD), and other eye diseases have led to a wealth of understanding about the molecular pathogenesis of their responsible diseases and have provided model systems in which therapeutic options have been tested (Chang et al., 2005). AMD, unlike retinal degenerative diseases that affect peripheral vision and the peripheral retina, preferentially affects central vision and the macula. It is difficult to model this disease in the mouse because the mouse lacks a macula. However, certain clinical, pathological, physiological and biochemical features of AMD, such as focal deep retinal lesions, progressive A2E accumulation, abnormal ERGs, and RPE and photoreceptor degeneration, can be demonstrated in retinal lesions in mice (Rakoczy et al., 2006)
AMD is difficult to study because of its late onset, complex genetics, and the influence of environmental factors. Therefore, it can be very useful to apply models that demonstrate pathologic findings of the disease with an earlier presentation and Mendelian inheritance (Zack et al., 1999). These models help reveal the biochemical and genetic perturbations that occur in the disease state and allow scientists and clinicians to test therapeutic options and get answers to questions that would be more difficult and expensive to ask in clinical trials. A success story is the analysis of a rare juvenile form of glaucoma that led to the discovery of a gene that was found to be responsible for some types of primary open angle glaucoma (Stone et al., 1997). Groups that have studied Stargardt disease and Best disease, early forms of macular degeneration with Mendelian inheritance patterns, have analyzed the role of the perturbed genes and proteins at the molecular level, and these models will be discussed in more detail (Weng et al., 1999). Modeling macular degeneration in mice helps us readily learn aspects of AMD that may be difficult to tease out in animals with a macula. Aged primates may develop AMD, but they only exhibit symptoms in their second decade of life. The slow time course of the disease, exorbitant cost, and the ethical and technical issues in making transgenic primates make this model system less useful (Marmorstein and Marmorstein, 2007). The power to genetically modify and manipulate the structure of the mouse retina will elucidate the biological importance of the different cellular components of the retina and immune system and can bring us closer to understanding the etiology of AMD.
2.1. Differences between the human and mouse retina
The main difference between the human and mouse retina is that the mouse has no maculae. The photoreceptor cell populations also differ. Because mice are derived from a nocturnal species, the mouse retina is composed of mainly rod photoreceptor cells (97%), and cones comprise the remaining 3% of photoreceptors (Carter-Dawson, 1979). The mouse also has only two types of cones: S-cones (blue light) and M-cones (green light), whereas humans have three types of cones: S-cones, M-cones, and L-cones (red light) (Szel et al., 1992). The mouse retina has a distribution of 3.1% horizontal cells, 41% bipolar cells, 16% Müller cells, and 39% amacrine cells. Primates have 9% horizontal cells, 40% bipolar cells, 22% Müller cells, and 28% amacrine cells (Jeon et al., 1998). The average cone density in the mouse retina is the same as the primate retina at 3–4 mm eccentricity. Evolutionarily, cones evolved first, and it is possible that a fixed number of cones set the framework for retinal organization (Wikler and Rakic, 1990). Therefore, the mouse retina is most structurally similar to the human peripheral retina, making it a useful model for retinal degenerative diseases (Figure 1).
Figure 1. A comparison of the mouse and human retina and AMD-like pathology.

a) A normal human fundus, b) light microscope histologic picture of the normal human maculae, c) the fundus of a patient with dry AMD with drusen and scarring in the maculae, d) light microscope histologic picture of a patient with dry AMD, e) A normal mouse fundus, f) light microscope histologic picture of the normal mouse retina, g) fundus picture of a 3 month-old Ccl2−/−/Cx3cr1−/− mouse with drusenoid deposits and a retinal scar, h) light microscope histologic picture of a 3 month-old Ccl2−/−/Cx3cr1−/− mouse with RPE hyperpigmentation and hypertrophy and photoreceptor outer segment disorganization and atrophy
In the aged human eye, residual body phagosomes often pass through the cytoplasm toward Bruch’s membrane, and lysosomal bodies can be seen in the basal portion of the RPE cytoplasm or near the basement membrane (Hogan and Alvarado, 1967; Mishima and Hasebe, 1978). Drusen formation in Bruch’s membrane has been hypothesized to come from RPE phagosomes via the basal pathway of secretion into Bruch’s membrane (Mishima and Hasebe, 1978). However, in mice, the accumulation of lysosomal bodies occurs apically in the RPE cytoplasm, causing dilation of RPE microvilli over time, and lysosomal bodies are extruded into the subretinal spaces to preserve RPE function (Mishima and Kondo, 1981). That is, mice do not have a tendency to accumulate lysosomes basally or secrete residual bodies into Bruch’s membrane like humans (Mishima and Kondo, 1981). For this reason, drusen and residual bodies in Bruch’s membrane are rarely seen in aged mice or transgenic mouse models.
Bruch’s membrane has a simple structure in mice, consisting of the RPE basement membrane, intermediate connective tissue collagen fibers, and the choriocapillaris basement membrane. The total thickness is about 0.4 μm, increasing to 1.1 μm in 2 year-old mice. Age-related changes in mice are less remarkable than in humans and include thickening, hyalinization, and patchy basophilia. The number of collagen fibrils and amorphous material increase little with age (Mishima and Hasebe, 1978). Extended and enlarged basal cytoplasmic infoldings are found in mice over 12 months of age, and large amounts of filamentous fibrous material with amorphous deposits can be seen between basal infoldings in the mouse RPE. However, striations of banded material and electron-dense clumps in the basal infoldings of human RPE is not seen (Mishima and Hasebe, 1978). Aged mice have parallel undulating filaments in the RPE, large accumulations of lysosomal dense bodies (similar to what is seen in humans with only a difference in cytoplasmic location), and enlarged basal infoldings (Mishima and Hasebe, 1978). A great deal can be learned about the pathogenesis of macular degeneration from studying models of retinal degeneration and transgenic mouse models that produce lesions similar to those found in patients with AMD. The power to genetically modify and manipulate the structure of the mouse retina will elucidate the importance of cone versus rod biology and the etiology of AMD.
2.2. Pathophysiologic findings of murine AMD models
Despite the fact that the mouse does not have a macula, AMD mouse models are used to recapitulate the pathology and genetics of AMD. In mouse models, the pathologic hallmarks of AMD remain relatively the same: lipofuscin and basal deposit accumulation, soft drusen development, RPE and photoreceptor atrophy, and CNV. Many models show one or more of the features of AMD, and few show late-stage ‘dry’ AMD progression to ‘wet’ AMD (Rakoczy et al., 2006). Aged mice have been shown to have an accumulation of lysosomal dense bodies in the apical portion of the cytoplasm, vacuolization of the cytoplasm, and slightly extended basal infolding. There is also more lipofuscin and its byproduct A2E in the aged mouse retina (Mishima and Kondo, 1981). The production of lipofuscin and A2E is increased in the abcr−/−, ELOVL4-mutant, Efemp1R345W/R345W, Ccr2−/−, sod1−/−, and Neprilysin−/− mouse models (Imamura et al., 2006; Iwata et al., 2001; Vasireddy et al., 2009; Weng et al., 1999; Marmorstein et al., 2007; Ambati et al., 2003). Mouse models are examined for the presence of drusen, which has been shown to confer risk for AMD. The first sign of drusen is the development of BlinD, which has been seen in AMD patients but is rarely encountered in mice (e.g. Neprilysin−/− and mcd/mcd mice). There are basal deposits in the Timp3S156C/S156C, mcd/mcd, ApoE4 TR, APO B100, APO*E3-Leiden, Ccl2−/−, Ccr2−/−, Ccl2−/−/Cx3cr1−/−, Efemp1R345W/R345W, Sod2 knockdown, neprilysin−/−, mcd/mcd, CEP-immunized, and SAMP8 among other mouse models (Iwata et al., 2001; Kliffen et al., 2000; Heckenlively et al., 1995; Fogarasi et al., 2008; Zhang et al., 2002; Espinosa-Heidmann et al., 2004; Chan et al., 2008; Weber et al., 2002; Rakoczy et al., 2002; Marmorstein et al., 2007; Sandbach et al., 2001; Justilien et al., 2007; Hollyfield et al., 2008; Majji et al., 2000). Photoreceptor loss or derangement is seen in the abcr−/−, ELOVL4-mutant, cfh−/−, Ccl2−/−, Ccr2−/−, Ccl2−/−/Cx3cr1−/−, Sod2 knockdown, mcd/mcd, Cp−/−/Heph−/Y, ApoE4 TR, arrd2/arrd2, Mfrprd6, Nr2e3rd7, and cpfl3 among other mouse models (Vasireddy et al., 2006; Weng et al., 1999; Ambati et al., 2003; Heckenlively et al., 1995; Coffey et al., 2007; Chan et al., 2008; Justilien et al., 2007; Malek et al., 2005; Chang et al., 2008; Kameya et al., 2002; Akhmedov et al., 2000; Chang et al., 2006). The Ccr2−/−, Ccl2−/−/Cx3cr1−/−, ApoE(−), and APO*E3-Leiden mouse models, demonstrate the presence of dry and wet AMD features with RPE degeneration, photoreceptor loss, and CNV (Chan et al., 2008; Ambati et al., 2003; Dithmar et al., 2000; Kliffen et al., 2000). Animal models of CNV are reviewed by Grossniklaus and colleagues (in this issue). The detailed pathological findings of murine dry AMD models will be discussed in detail in this review.
3 Murine models of dry AMD
3.1. Genetically engineered mice
The majority of dry AMD murine models have been genetically engineered to disrupt genes that are postulated to have a role in AMD pathogenesis (Rakoczy et al., 2006). A summary of the mouse models, their relevant genetic modifications, and human disease associations is included (Table 1). Mouse models that were created by genetic engineering will be described with their relevance to dry AMD.
Table 1.
Genetics of dry AMD models. This table presents a summary of the mouse and human genetics of the transgenic, immunologically modified, and naturally occurring dry AMD murine models and related diseases.
| No. | Mouse model | Human Disease association | Human Gene |
Chromosome Location |
OMIM # | Genetic Modification |
Mouse gene(s) |
Encoding protein(s) |
Reference(s) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | abcr−/− | Stargardt disease | ABCA4 | 1p21-p13 | *601691 | knock-out | Abca4 | RmP | Weng et al., 1999; Mata et al., 2000 |
| 2 | ELOVL4-mutant | Stargardt-3 dominant inheritary disease | ELOVL4 | 6q14 | *605512 | transgenic | Elovl4 | Elov4 | Karan et al., 2005 |
| 3 | Efemp1R345W/R345W | Doyne honeycomb retinal dystrophy | EFEMP1 | 2p16 | *601548 | transgenic | Efemp1 | fibulin-3 | Marmorstein et al., 2007; Fu et al., 2007 |
| 4 | Timp3S156C/S156C | Sorsby fundus dystrophy | TIMP3 | 22q12.1-q13.2 | *188826 | knock-in | Timp3 | TIMP3 | Weber et al., 2002 |
| 5 | cfh−/− | AMD | CFH | 1q32 | +134370 | knock-out | Cfh | CFH | Coffey et al., 2007 |
| 6 | ccl2−/− | coronary artery disease, tuberculosis | CCL2 (MCP1) | 17q11.2-q12 | +158105 | knock-out | Mcp1 | MCP1 | Ambati et al., 2003 |
| 7 | ccr2−/− | atherosclerosis, rheumatoid arthritis | CCR2 | 3p21 | *601267 | knock-out | Ccr2 | MCP1 receptor | Ambati et al., 2003 |
| 8 | cx3cr1−/− | AMD, coronary artery disease, HIV | CX3CR1 | 3pter-p21 | *601470 | knock-out | Cx3cr1 | Fractalkine receptor CX3CR1 | Combadiere et al., 2007 |
| 9 | ccl2−/−/cx3cr1−/− | coronary artery disease | CCL2, CX3CR1 | 17q11.2-q12, 3pter-p21 | +158105, *601470 | double knock-out | Mcp1, Cx3cr1 | MCP1, CX3CR1 | Tuo et al., 2007 |
| 10 | sod1−/− | amyotrophic lateral sclerosis | SOD1 | 21q22.1 | *147450 | knock-out | Sod1 | SOD1 | Imamura et al., 2006 |
| 11 | sod2−/− | Leber’s hereditary optic neuropathy, idopathic cardiomyopathy | SOD2 | 6q25.3 | *147460 | knock-down | Sod2 | SOD2 | Sandbach et al., 2001; Justilien et al., 2007 |
| 12 | neprilysin −/− | Alzheimer’s disease | NEPRILYSIN | 3q21-q27 | *120520 | knock-out | Neprilysin | Amyloid β | Iwata et al., 2001 |
| 13 | mcd/mcd mice | neuronal ceroid lipofuscinosis | CTSD | 11p15.5 | *116840 | transgenic | mcd | Cathepsin D | Rakoczy et al., 2002 |
| 14 | Cp−/−/Heph−/Y | aceruloplasminemia | CP | 3q23-q24 | *117700 | knock-out | Cp, sla | ferroxidase ceruloplasmin/hepaestin | Hahn et al., 2004 |
| 15 | ApoE−/− | hyperlipoproteinemia | APOE | 19q13.2 | +107741 | knock-out | ApoE | ApoE | Dithmar et al., 2000 |
| 16 | APO*E3-Leiden | hyperlipoproteinemia | APOE | 19q13.2 | +107741.0015 | transgenic | - | apoE3 | Kliffen et al 2002 |
| 17 | ApoE4 TR | Type III/V hyperlipoproteinemia, Alzhimer’s disease | APOE | 19q13.2 | +107741.0016 | transgenic | - | apoE4 | Malek et al., 2005 |
| 18 | APO B100 | hypobetalipoproteinemia | APOB | 2p24 | +107730 | transgenic | - | apoB100 | Espinosa-Heidmann et al. 2004; Callow et al. 1994 |
| 19 | arrd2/arrd2 | progressive rod-cone degeneration, AMD | MDM1 | 12q14.3 | *164785 | none | Mdm1 | Mdm1 | Chang et al., 2008 |
| 20 | Mfrprd6 | retinitis pigmentosa | MFRP | 11q23 | *606227 | none | Mfrp | MFRP | Kameya et al., 2002 |
| 21 | Nr2e3rd7 | Enhanced S-cone syndrome (ESCS) | NR2E3 | 15q23 | *604485 | none | Nr2e3 | NR2E3 | Ahkmedov et al., 2000 |
| 22 | cpfl3 | achromotopsia | GNAT2 | 1p13 | +139340 | none | Gnat2 | GNAT2 | Chang et al., 2006 |
| 23 | SAMP | aging | unknown | unknown | unknown | unknown | Mpmv, Pmv | unknown | Takada et al., 1994 |
| 24 | SAMR | aging | unknown | unknown | unknown | unknown | Pmv-35 | unknown | Majji et al., 2000; Takada et al., 1994 |
3.1.1. Genes that lead to macular or retinal degeneration
The most well established murine models of dry AMD target genes previously identified to have a key role in the pathogenesis of AMD with predictable Mendelian inheritance. These diseases include Stargardt disease, Stargardt-3 dominant disease, and Malattia leventinese or Doyne’s honeycomb retinal dystrophy. These models are especially popular because the mutated genes are known to cause juvenile macular dystrophy with the phenotype of RPE and photoreceptor atrophy in the macula.
3.1.1.1. abcr−/− (Stargardt disease) murine model
Mutations in the human ABCR gene for Rim protein (RmP), a glycoprotein in outer segment disc rims in the ATP-binding cassete (ABC) transporter family, result in the phenotype known as Stargardt’s disease (STGD), a recessive form of macular degeneration that presents in childhood with progressive central visual loss and RPE atrophy overlying the macula (Stargardt, 1909; Allikmets et al., 1997). Heterozygous mutations in ABCR have been shown to be responsible for AMD, and abcr+/− mice have been shown to have delayed dark adaptation, a clinical feature of AMD and STGD (Mata et al., 2001). Ultrastructural examination of the RPE in abcr−/− mice at 44 weeks of age reveals an apical accumulation of electron-dense bodies, likely melanosome or melanosome-phagosome fusion particles (Feeney-Burns and Eldred, 1983), and thickening and disorganization of the basal RPE underlying Bruch’s membrane that is more prominent in the abcr−/− mouse compared to the abcr+/− mouse (Mata et al., 2001). The baseline number of lipofuscin granules is increased in the abcr−/− mouse compared to WT mice, and accumulation of lipofuscin granules also occurs at a faster rate with age. As expected, A2E levels were elevated in both the abcr+/− and abcr−/− mouse models compared to WT mice (Mata et al., 2001; Weng et al., 1999). However, no significant RPE degenerative changes were noted. At 44 weeks, the photoreceptor layers were relatively unremarkable (Weng et al., 1999). Additionally, abcr−/− mice have thickening of Bruch’s membrane but no evidence of drusen (Weng et al., 1999).
3.1.1.2 ELOVL4-mutant (Stargardt-3 dominant inheritary disease) murine model
Stargardt-like macular degeneration (STGD3) is an autosomal dominant disease with progressive central vision and color vision loss that begins in the second decade of life and has been associated with mutations in the elongation of very long chain fatty acids-4 (ELOVL4) gene (Stone et al., 1994). Three different mutations in the ELOVL4 gene have been identified: a 5-bp deletion mutation, two 1-bp mutations, and a nonsense mutation (Zhang et al., 2001). ELOVL4 is normally heavily expressed in the endoplasmic reticulum of rod and cone photoreceptor cells; however, mutant ELOV4 acts as a dominant negative protein (Vasireddy et al., 2005) and loses its ER retention signal, causing deranged intercellular trafficking and accumulation of mutant ELOV4 in the Golgi apparatus (Ambasudhan et al., 2004). Complete loss of the Elovl4 gene causes embryonic lethality, and haploinsufficiency results in an essentially normal retinal phenotype (Raz-Prag et al., 2006). Transgenic mice expressing a mutant form of human ELOVL4 (TG E_mut+/−) develop RPE changes, including vacuolization with accumulation of debris and undigested outer segments in the subretinal space and pigment granule deposits as early as two months of age. The number of lipofuscin granules and A2E levels were significantly higher in TG E_mut+/− mice at 7 months and 4 months, respectively, compared to controls (Vasireddy et al., 2009; Karan et al., 2005). The Elovl4 5-bp deletion knock-in mouse model demonstrated progressive photoreceptor degeneration, predominately of cones, from 6–18 months of age (Vasireddy et al., 2006).
3.1.1.3 Efemp1R345W/R345W (Doyne honeycomb retinal dystrophy) murine model
Malattia leventinese (ML), or Doyne’s honeycomb retinal dystrophy (DHRD), is an autosomal dominant, fully penetrant, inherited maculopathy that presents in early adulthood with drusen in a honeycomb form in the macula and optic nerve head (Collins, 1913). The burden of drusen increases, leading to geographic atrophy, CNV, and central vision loss by age 40–50 (Tree, 1937). Doyne’s retinal dystrophy has been shown to be caused by a R345W mutation in the EGF-containing fibrillin-like ECM protein 1 (EFEMP1) gene, which encodes fibulin-3, a protein of unknown function (Stone et al., 1999). EFEMP1 is a member of the fibulin family of extracellular matrix (ECM) proteins that are expressed in epithelial basement membranes and play a role in the assembly of elastin fibers (Timpl et al., 2003). Genetic studies have implicated other fibulins in the pathogenesis of AMD (Stone et al., 2004). Efemp1−/− mice were shown to have features of premature aging but not macular degeneration (McLaughlin et al., 2007). The R345W mutation was first identified in individuals with familial DHRD/ML but was not identified in a large cohort of control or AMD subjects (Stone et al., 1999).
An Efemp1−R345W point mutation knock-in mouse was made and found to have many pathologic features of dry AMD (Fu et al., 2007). Efemp1R345W/R345W mice have vacuoles in RPE cells that appear at 6 months and increase in number; by 12 months of age, the normal organization of the basolateral infolds of the RPE is lost(Marmorstein et al., 2007; Fu et al., 2007). Activated C3 is detected in RPE cells and Bruch’s membrane (Fu et al., 2007). The Efemp1R345W/R345W mice have Bruch’s membrane abnormalities: deposits of wide-spaced collagen are noted at two months, and significant amorphous electron dense debris of membrane coated vesicles are diffusely in the collagenous and elastic layers of Bruch’s membrane at 12 months of age (Marmorstein et al., 2007; Fu et al., 2007). Basal deposits appear in Efemp1R345W/R345W and Efemp1+/R345W mice at 4 months of age; at 8 months of age, they are larger in size, and at 12 months of age, the deposits merge into continuous patches preferentially in the central retina (Marmorstein et al., 2007). Basal deposits contain Timp3, an Efemp1 interacting protein, and mutant fibulin-3. The neural retina in the Efemp-1 R345W/R345W mutant mouse was normal from 6 to 18 months of age (Marmorstein et al., 2007; Fu et al., 2007).
3.1.1.4 Timp3S156C/S156C (Sorsby fundus dystrophy) murine model
Sorsby fundus dystrophy (SFD) is a rare, late-onset, hereditary degenerative disease of the retina and choroid that causes rapid central vision loss and progressive peripheral vision loss, ultimately leading to blindness (Sorsby et al., 1949). Histopathologic studies of SFD patients have shown that these patients have sub-RPE deposits of extracellular matrix material (collagen, elastin and glycoaminoglycans) and Bruch’s membrane thickening, leading to CNV and RPE atrophy, reminiscent of AMD pathology (Chong et al., 2000). SFD patients were found to have point mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) gene, and TIMPS, the inhibitors of the metalloproteinases (MMPs), have a role in the composition of the ECM (Weber et al., 1994; Matrisian, 1992).
A mouse model was constructed with a mutant Timp3 gene (Weber et al., 2002). There was no significant difference in the thickness or structure of Bruch’s membrane when Timp3+/S156C, Timp3S156C/S156C, and wild-type mice were compared at 8 months of age, but Bruch’s membrane thickness increased in Timp3S156C/S156C mice as granular debris in the inner layers of the ECM accumulated by 30 months of age (Weber et al., 2002). The RPE cells in the Timp3+/S156C mice had local disorientation of the apical processes and reduced thickness and complexity of basal microvilli. The RPE cells in Timp3S156C/S156C aged mice had a disrupted basal layer with loss of the palisade-like orientation of microvilli. The RPE processes no longer faced the outer segment of the photoreceptors; rather, the processes were horizontally aligned near the RPE cell bodies and formed a vesiculated barrier between the outer segment and RPE (Weber et al., 2002). The Timp3+/S156C and Timp3S156C/S156C mice had no derangements of the neural retina from 8 months to 30 months old.
3.1.2. Genes relevant to AMD
While it is known that genetics and environmental factors both contribute to AMD development and progression, the roles of genetic factors in AMD have been described in detail in recent years (Swaroop et al., 2007). Twin studies have demonstrated that genetic factors contribute to 46–71% of the overall variation in severity of macular degeneration. When one or both twins have AMD, concordance is 55% among monozygotic twins and 25% among dizygotic twins for AMD (Seddon et al., 2005). Single nucleotide polymorphisms in several genes, especially the Y402H variant of complement factor H (CFH) (Haines et al., 2005; Klein and Weleber, 1998; Klein et al., 2005; Edwards et al., 2005; Hageman et al., 2005), have been associated with AMD pathogenesis (Swaroop et al., 2007; Ding et al., 2009). Missense mutations in the gene encoding fibulin-5 have also been associated with AMD (Stone et al., 2004). Several studies have demonstrated an increased risk for AMD in patients with polymorphisms in C2, BF, CX3CR1, ARMS2/HTRA1, and APOE (Gold et al., 2006; Maller et al., 2006; Klaver et al., 1998; Francis, 2008).
3.1.2.1. Inflammatory genes
Chronic para-inflammation, a tissue adaptive response to noxious stress, contributes to the initiation and progression of obesity, type 2 diabetes mellitus, atherosclerosis, and age-related neurodegenerative diseases including AMD (Xu et al., 2008; Xu et al., 2009). The pathology of AMD lesions demonstrates chronic persistent inflammatory damage, including macrophage infiltration, microglial accumulation, and inflammatory components in drusen, such as complement factors, pro-inflammatory cytokines and chemokines (van der Schaft et al., 1992; Spraul and Grossniklaus, 1997; Anderson et al., 2002; Chan et al., 2008; Patel and Chan, 2008). Mutations in many of the genes that have been attributed to AMD result in a pro-inflammatory state in the eye, which are signified by increased C-reactive protein and decreased inhibitory complement factors (Boekhoorn et al., 2007; Seddon et al., 2004). Because of the central role of inflammation in AMD, mouse models that target specific aspects of the inflammatory process have been created to understand their significance in AMD and will be discussed in detail.
3.1.2.1.1. Cfh−/− murine model
CFH negatively regulates the complement system by inhibiting the alternative pathway either by promoting Factor I-mediated inactivation of C3b or by displacing Factor Bb from the C3bBb complex (Alsenz et al., 1985). CFH dysfunction may lead to excessive inflammation and tissue damage and contribute to the pathogenesis of AMD (Johnson et al., 2006). Homozygous deficiency of CFH in humans and mice results in complement factor alternative pathway dysregulation, the inflammatory renal disease membranoproliferative glomerulonephritis type II (MPGN2), and an AMD-like phenotype (Raines et al., 1989; Leys et al., 1991).
To further understand the role of CFH in the eye with relation to AMD, a cfh−/− mouse model was made and found to have poor visual function at two years of age (Coffey et al., 2007). The cfh−/− mice had C3 accumulation in the retina, secondary C3 deficiency, and uncontrolled C3 activation, which may increase phagocytic uptake and cause neural damage to the retina (Mullins et al., 2000; Coffey et al., 2007). Ultrastructurally, cfh−/− mice have significant thinning of Bruch’s membrane (by 29%) and consistently disorganized, misaligned photoreceptor outer segments. There is a decrease in the amount of sub-RPE electron-dense material when compared to age-matched wild-type mice (Coffey et al., 2007). CFH itself is a main component of drusen, and the loss of this protein may reduce the volume of sub-RPE desposits. The outer segment photoreceptors in cfh−/− mice are disorganized and misaligned with an inappropriate interface with RPE cells. Melanosomes, lipofuscin granules, and melanosome-lipofuscin granules were dispersed throughout the RPE in cfh−/− mice, compared to primarily apical localization of organelles observed in controls (Coffey et al., 2007). Though there is a touted association between CFH and AMD, the cfh−/− mice do not have many features of AMD; rather, thinning of Bruch’s membrane and decreased drusen were found. However, disorganized photoreceptor outer segments and organelle localization in the RPE can be found in AMD eyes.
3.1.2.1.2 Ccl2−/− and Ccr2−/− murine models
To analyze the role of macrophage dysfunction in AMD, mice genetically altered to affect macrophage recruitment were created (Ambati et al., 2003). The monocyte chemoattractant protein-1 (MCP-1, or Ccl-2) binds to the C-C chemokine receptor-2 (Ccr-2) and mediates adhesion of inflammatory cells to vessels, controlling their extravasation to tissues (Kuziel et al., 1997). MCP-1/CCL2 is essential for monocyte recruitment and influences cytokine expression related to helper T-cell responses (Lu et al., 1998). Both Ccl2−/− and Ccr2−/− murine models were found to have features of AMD (Ambati et al., 2003).
After 9 months of age, subretinal deposits with features of drusen were observed in all Ccl2−/− mice, and the number of deposits increased with age. Bruch’s membrane was markedly thickened with internal fragmentation and disruption of the collagen and elastin layers in 10 to 12 month old Ccl2−/− mice compared to age-matched wild-type mice (Ambati et al., 2003). At 9 months of age, RPE cells became swollen, and vacuolated and high electron density intracellular dense bodies, likely melanosomes or melanolipofuscin fusion granules, accumulated. Additionally, the number of lipofuscin granules and the A2E signal increased with age in senescent Ccl2−/− mice (Ambati et al., 2003).
As Ccl2−/− mice aged, they exhibited many of the findings of advanced AMD, including geographic atrophy, progressive outer retinal degeneration, and CNV (Ambati et al., 2003). At 14 months of age, photoreceptors were healthy, but attenuated and pyknotic photoreceptors developed by 16 months of age, while wild type mice still had normal photoreceptors (Ambati et al., 2003). The RPE cells in Ccl2−/− mice had marked vacuolization with a degenerative nucleus and few pigment granules at 16 months of age. There were abundant melanocytes filled in the choroid but no choriocapillaries in the 16-month old Ccl2−/− mouse. At 20 months of age, Ccl2−/− mice began to overexpress VEGF in the RPE, had dilated choriocapillaries, and began to have fragmentation in the outer layer of Bruch’s membrane (Ambati et al., 2003). In this model, deposition of IgG, C5, vitronectin, CD46, serum amyloid P, and advanced glycosylation endproducts were present in Ccl2−/− mice in a distribution similar to what has been previously reported in humans AMD eyes (Ambati et al., 2003; Mullins et al., 2000).
The Ccr2−/− mice developed a phenotype very similar to Ccl2−/− mice. After 9 months of age, subretinal deposits similar to drusen were noted in the Ccr2−/− mice. Bruch’s membrane was noticeably thickened with collagen and elastin layer disruption at 10 months of age. Lipofuscin granules accumulated at 9 months and increased in number with age. Vacuolization of the RPE cells was also noted at 9 months (Ambati et al., 2003). Ccr2−/− mice also displayed signs of late AMD with geographic atrophy, outer retinal degeneration, and CNV. Fundoscopic evidence of geographic atrophy was visible in Ccr2−/− mice at 18 months of age. Changes in the neural retina, especially cell loss and atrophy of the outer nuclear layer of photoreceptors, became evident around 16 months of age in the Ccr2−/− mice and were similar to findings in the Ccl2−/− mouse (Ambati et al., 2003). The choriocapillaris became severely attenuated, and the RPE became hypopigmented and devoid of basal infoldings and most organelles by 16 months of age. At 24 months of age, the Ccr2−/− mouse had CNV with endothelial cell and fibrocyte invasion of the sub-RPE space through a Bruch’s membrane defect. Complement deposition and activation was in a pattern similar to the Ccl2−/− mice (Ambati et al., 2003). The similar phenotype and features of both early ‘dry’ and late ‘wet’ AMD in the Ccl2−/− and Ccr2−/− mouse models highlight the role of macrophage recruitment in the pathogenesis of AMD.
3.1.2.1.3 Cx3cr1−/− murine model
Studies of the CCR2 and CCL2 receptors in transgenic mice have been shown to alter macrophage recruitment and influence AMD progression to CNV (Tsutsumi C, 2003). The role of retinal microglial cells in AMD is being investigated. Retinal migroglial cells express the CX3C cheomkine receptor 1 (CX3CR1). Homozygosity of the CX3CR1 M280 allele and V249I allele, associated with impaired microglial cell migration, has been shown to increase the risk of AMD (Combadiere et al., 2007; Tuo et al., 2004). The perimacular area in AMD eyes has lower expression of CX3CR1 than the perimacular area of control subjects, possibly implicating decreases in CX3CR1 in age-related changes in the macula (Tuo et al., 2004; Chan et al., 2005). Activated microglia have been shown to directly injure photoreceptor cells (Roque et al., 1999) and increase autofluorescence due to lipofuscin deposition (Xu et al., 2008).
At 12 months of age, ultrastructural analysis of senescent Cx3cr1−/− mice revealed subretinal cells that contained intracellular lipid deposits and outer segment remnants between the photoreceptor outer segment and RPE. At 18 months of age, the engorged subretinal microglial cells appeared drusen-like on fundoscopy (Combadiere et al., 2007). At this age, the Cx3cr1−/− mice had significant (40%) thinning of the outer retina, mainly the outer nuclear layer, which progressed to marked degeneration of the photoreceptor layer by 4 months of age when compared to wild-type mice. After laser injury, Cx3cr1−/− mice showed signs of significant CNV compared to wild-type mice (Combadiere et al., 2007). These studies suggest a role of microglial cells in aging and AMD pathogenesis (Xu et al., 2009; McGeer et al., 2005).
3.1.2.1.4 Ccl2−/−/Cx3cr1−/− murine model
The Ccl2−/−/Cx3cr1−/− mouse model has demonstrated early onset of the AMD phenotype with high penetrance (Tuo et al., 2007). Ccl2−/−/Cx3cr1−/− mice began exhibiting drusen-like lesions on fundoscopy as early as 4–6 weeks of age. These lesions progressed to large, confluent areas of yellow deposits in the deep retina and subretinal space by 4–6 months of age and flattened atrophic areas by 6 months of age (Chan et al., 2008). Other AMD features include an abnormal and thickened Bruch’s membrane with irregular deposits. Drusen, while present, were smaller (5–15 μm) than classic human drusen seen in AMD patients. Abnormalities of RPE cells included RPE hypopigmentation, depigmentation, vacuolization, loss of melanosomes, an increase in lipofuscin (Figure 2), and elevated A2E, which led to RPE degeneration. More basal infoldings were observed in Ccl2−/−/Cx3cr1−/− mice when compared to age-matched wild-type controls (Figure 2). Photoreceptor outer segment disorganization and photoreceptor atrophy were also observed in Ccl2−/−/Cx3cr1−/− mice (Figure 2). Compared to wild-type mice, Ccl2−/−/Cx3cr1−/− mice have significantly fewer photoreceptor terminals and synapses (Zhang et al., unpublished). Spontaneous CNV was present in about 15% of Ccl2−/−/Cx3cr1−/− mice (Tuo et al., 2007).
Figure 2.
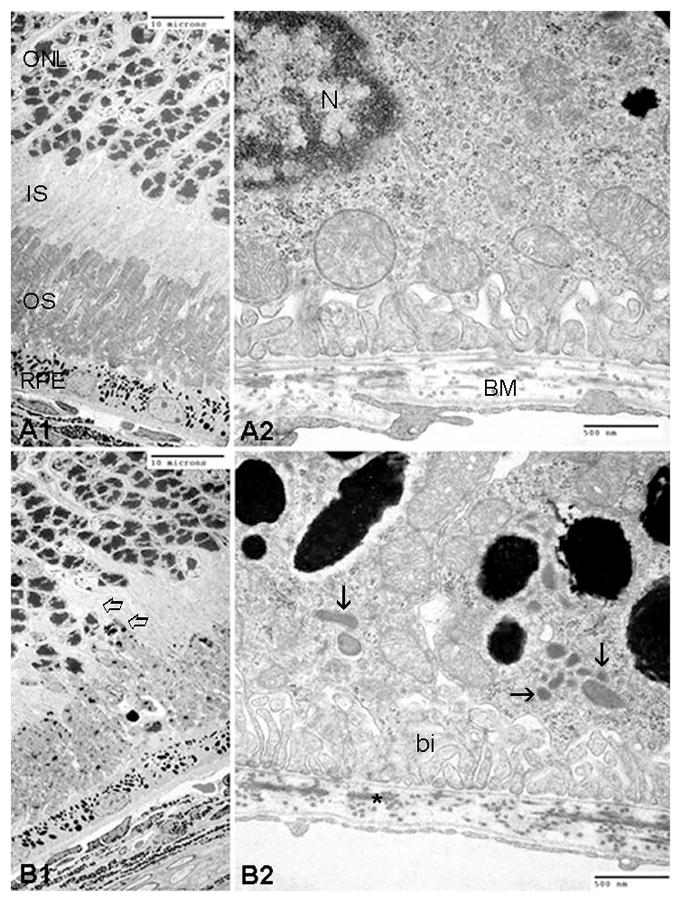
Transmission electron micrographs illustrated outer retinal degeneration in a 21-days old Ccl2−/−/Cx3cr1−/− mouse. Comparing with WT mice (A1–2), Ccl2−/−/Cx3cr1−/− mice revealed disorganization and migration of photoreceptors (open arrow in B1), rich lipofuscin (arrows in B2) and more basal infoldings (bi in B2) in RPE, as well as higher electron-dense of elastic layer in BM (asterisk in B2). Scale bars, 10 μm (A1, B1); 500 nm (A2, B2). (ONL, outer nuclear layer or photoreceptor nuclei; IS, inner segments of photoreceptors; OS, outer segments of photoreceptors; RPE, retinal pigment epithelial cells; BM, Bruch’s membrane)
Characterization of the immunologic mileu in Ccl2−/−/Cx3cr1−/− mice revealed several abnormalities. Ccl2−/−/Cx3cr1−/− mice had increased C3 and CD46. C3d deposition was increased in Bruch’s membrane, drusen-like lesions, RPE, photoreceptors, and choroidal capillaries (Ross et al., 2008), similar to previous reports in humans with AMD (Hageman et al., 2005). Lower levels of the chaperone protein ERp29 are found in Ccl2−/−/Cx3cr1−/− mice compared to wild-type controls (Tuo et al., 2007). Macrophage infiltration, microglial accumulation, and anti-retinal antibody levels were increased in Ccl2−/−/Cx3cr1−/− mice compared to wild-type controls (Ross et al., 2008). The Ccl2−/−/Cx3cr1−/− mouse model demonstrates many AMD features: small drusenoid deposits, RPE and Bruch’s membrane degenerations, photoreceptor atrophy, CNV, and simulation of the immunologic and pathologic environments found in AMD patients (Herzlich et al., 2008; Ross et al., 2008; Ding et al., 2009). It is one of the few models to have high penetrance and early onset- all of which facilitate experimentation (Chan et al., 2008).
3.1.2.2. Oxidative stress associated genes
The retina has the highest oxygen consumption of any tissue in the body (Sickel, 1972). Oxidative stress, or cellular and molecular damage caused by reactive oxygen species (ROS), has been implicated in aging and age-related eye diseases (Finkel and Holbrook, 2000). There is increasing evidence that cumulative oxidative damage is involved in AMD pathogenesis. Smoking, a common mechanism for generating oxidative stress, is a well-documented AMD risk factor, and a recent meta-analysis showed that current smoking nearly triples AMD incidence (Neuner et al., 2009). ROS include unstable species, such as superoxide anions and hydroxyl radicals, and stable freely diffusible substances like hydrogen peroxide. In the neutrophil, NADPH oxidases as part of a cytosolic enzyme system, contribute to oxidative stress. Most intracellular ROS production, however, is derived from the mitochondria in the electron transport chain.
Endogenous protection against oxidative stress is encountered and regulated through enzymatic and nonenzymatic antioxidant denfense by catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPx) (Finkel and Holbrook, 2000). One of the main retinal antioxidant systems is the superoxide dismutase (SOD) family of three isoenzymes that catalyze the dismutation of the superoxide radical: Cu, Zn-SOD (SOD1) is cytosolic; Mn-SOD (SOD2) is in the mitochondrial matrix, and extracellular SOD (SOD3) is interstitial and secreted (Valentine et al., 2005). In the retina, SOD1 has the highest activity (Behndig et al., 1998). To investigate the role of oxidative stress in AMD pathogenesis, transgenic mice targeting the SOD oxidative stress-recovery pathways have been made and examined.
3.1.2.2.1. Sod1−/− murine model
SOD1 levels are very high in the retina, and SOD1 is known to protect against oxidative damage, a possible trigger for AMD. Imamura et al. showed that Sod1−/− mice have accelerated age-related pathologic changes in the retina reminiscent of AMD, including drusen, thickening of Bruch’s membrane, and CNV (Imamura et al., 2006).
Drusen were visible on fundoscopy of Sod1−/− mice after 7 months of age, and lesions increased in number with age. At 10 months of age, 86% of mice had drusen, compared to very few drusen in age-matched wild-type mice (Imamura et al., 2006). These sub-RPE dome-shaped deposits have many characteristics of drusen in human AMD eyes, including positive staining for vitronectin, CD46, activated complement components, TIMP3, and Igs (Crabb et al., 2002; Imamura et al., 2006). Based on human studies that show light exposure is a risk factor for AMD (Wenzel et al., 2005; Cruickshanks et al., 1993), Sod1−/− mice were exposed to fluorescent light, and more light exposure was found to induce more drusen (Imamura et al., 2006).
At 12 months of age, RPE vacuolization and degenerative changes were noted in Sod1−/− mice. Ultrastructural analysis demonstrated disrupted junctional integrity of the RPE in Sod1−/− mice, signified by altered expression of junctional adherence proteins N-cadherin and β-catenin and their translocation from the cell membrane to the cytoplasm (Imamura et al., 2006). A biomarker of oxidative DNA damage was present at higher levels in Sod1−/− mice when compared to controls. Bruch’s membrane was markedly thickened, with a thickness approximately 6-fold greater in Sod1−/− mice compared to age-matched wild-type mice. Photoreceptor loss was shown in 17% percent of Sod1−/− mice. Approximately 10% of mice older than 10 months had CNV (Imamura et al., 2006). The similarities of the lesions in this mouse model to the human AMD lesions suggest that the Sod1−/− mouse is a suitable murine model for AMD.
3.1.2.2.2. Sod2 knockdown murine model
The antioxidant enzyme manganese superoxide dismutase (MnSOD), encoded by SOD2, is in the mitochondrial matrix and converts superoxide anions produced in aerobic respiration to hydrogen peroxide. To study the role of this enzyme, Sod2tm1Cje−/− mice were made, but they all died of dilated cardiomyopathy by 10 days of age (Li et al., 1995). The Sod2tm1Cje−/− mice were treated with the Sod mimetic mangenese 5,10,15,20-tetrakis (4-benzoic acid) porphyrin (MnTBAP), and the animal survival time nearly doubled. These animals died of spongiform encephalopathy with normal brain mitochondria (Melov et al., 1998). Ocular pathology of the MnTBAP-treated Sod2tm1Cje−/− mice revealed that the 10 day-old Sod2tm1Cje−/− mice had thinning only of the central photoreceptor layer (Sandbach et al., 2001). By 20 days of age, most retinal layers were all significantly reduced and contained markedly edematous mitochondria in the Sod2tm1Cje−/− mice compared to control animals. Photoreceptor damage at three weeks of age is early when compared to the Sod1 and Gpx-1 antioxidant knockdown models, which only show retinal damage at 1 year of age (Gosbell et al., 2006; Imamura et al., 2006). There were no differences in the RPE basement membrane or basal infoldings, and lipofuscin granules were rare in Sod2tm1Cje−/− mice and controls (Sandbach et al., 2001).
To further study the role of Sod2 deficiency in older mice, a Sod2-deficient mouse was made using a subretinal injection of an AAV-ribozyme-mediated knockdown of Sod2 mRNA in the RPE of wild-type mice (Justilien et al., 2007). The Sod2 knockdown mice had RPE and Bruch’s membrane changes and accumulated A2E and lipofuscin granules in the RPE (Justilien et al., 2007). One month after injection, the RPE exhibited hypopigmentation with a normal neural retina. Between 2 and 4 months after injection, the RPE cells underwent vacuolization and atrophy with shortening and disorganization of the outer and inner segment of the photoreceptors and thinning of the outer nuclear layer (Justilien et al., 2007). After 4 months, there was a 40% increase in Bruch’s membrane thickness, specifically in the inner and outer collagenous zones, in Sod2 knockdown mice compared to age-matched wild-type mice. Basal laminar deposits (BlamD) are present between the basal laminar infoldings and the plasma and basement membrane. ERG a-wave and b-wave amplitudes were noted to decrease by 33% and 41%, respectively, after 4 months. A2E levels were greater than twofold higher in Sod2 knockdown mice 4.5 months after injection, compared to controls (Justilien et al., 2007). The Sod2 knockdown murine model emphasizes the role of oxidative stress in AMD pathogenesis, as the phenotype includes RPE hypopigmentation and increased lipofuscin, Bruch’s membrane thickening, basal laminar deposits, and photoreceptor disorganization.
3.1.2.3. Metabolic pathway associated genes
When lipid and protein-rich sub-RPE and Bruch’s membrane deposits accumulate in AMD patients, hydraulic conductivity and the transport of fluids and hydrophilic substances through Bruch’s membrane becomes impaired (Moore et al., 1995). Derangements in cholesterol metabolism may affect hydraulic conductivity across Bruch’s membrane, cause the accumulation of its toxic metabolites such as 7-ketocholesterol, and contribute to AMD pathogenesis (Javitt and Javitt, 2009). Photoreceptors depend on choriocapillary circulation and the RPE for nutrients and clearance of metabolic waste products; impairment of this process is related to the pathogenesis of late AMD (Moore et al., 1995; Sarks, 1976). Lysosomal degradation of the photoreceptor outer segments is mediated in part by protease Cathepsin D, and the inactivation of this protease has been shown to cause metabolic derangements in RPE cells and induce AMD-like lesions in mcd/mcd transgenic mice (Zhang et al., 2002). Increased deposition of Amyloid β, as seen in Alzhimer’s disease, has been shown to cause AMD-like retinal pathology in the neprilysin−/− murine model (Yoshida et al., 2005). Derangements in iron metabolism leading to iron overload have been shown to cause retinal degeneration and the accumulation of inclusion bodies in the RPE (He et al., 2007). Mouse models with increased serum cholesterol include ApoE and ApoB-100 transgenic mice, and these mice have been shown to have AMD-like lesions (Cousins et al., 2002; Klaver et al., 1998). The retinal pathology of the aforementioned models with metabolic dysfunction will be discussed in detail.
3.1.2.3.1. Neprilysin−/− murine model
Amyloid β (Aβ) is a physiologic peptide, the steady state of which is maintained by a balance between synthesis and degradation (Saido, 1998). Immediately after production, Aβ is normally degraded by peptidases such as neprilysin (Iwata et al., 2001). Aging is associated with decreased levels of neprilysin and increased Aβ peptide deposition and aggregation (Iwata et al., 2002). Aβ oligomers and aggregates in the brain form senile plaques, which are pathologic, immunogenic, and implicated in Alzheimer’s disease (Saido, 1998; Wisniewski et al., 1997). Aβ oligomers result in microglia recruitment and neuronal apoptosis (Uchihara et al., 1997). Recent evidence has shown that drusen contains Aβ, a specific finding in drusen from AMD eyes (Johnson et al., 2002; Anderson et al., 2004; Dentchev et al., 2003).
In vitro data has shown that the oligomeric form of Aβ: OAβ(1–42) reduces mitochondrial redox potential and increases the production of reactive oxygen species without inducing apoptosis in RPE cell cultures, a finding that is partially reversible with antioxidant pre-treatment (Bruban et al., 2009). Subretinal injection of OAβ(1–42) in wild-type mice induces RPE abnormalities and photoreceptor loss; however, the retinal ultrastructure was not depicted in this model (Bruban et al., 2009). The retinal pathology of senescent transgenic mice with disruption of the neprilysin gene was also examined (Yoshida et al., 2005). In 27 month-old Neprilysin−/− mice, the RPE developed vacuolization, loss of the tight and adherence junctions, prominent basal infoldings, and subretinal accumulation of amorphous deposits and photoreceptor outer segments, implying RPE dysfunction. Neprilysin−/− mice have extensive BlamD and some BlinD. Bruch’s membrane appears normal, as does the photoreceptor layer. The Neprilysin−/− mice have enhanced VEGF expression and diminished PEDF expression with no evidence of CNV or leakage on fluorescein angiography (Yoshida et al., 2005).
3.1.2.3.2. mcd/mcd murine model
Phagocytosis of photoreceptor outer segments is a fundamental vision-preserving physiologic function of the RPE (Young and Bok, 1969). An early sign of RPE dysfunction in AMD eyes is indigestion of photoreceptor outer segments, with visible remnants in the RPE cytoplasm. Accumulation of this material results from lysosomal abnormalities in the RPE (Hayasaka, 1983) or Bruch’s membrane changes inhibiting exocytosis (Starita et al., 1996). Cathepsin D (CatD) is an aspartic protease that is involved in the lysosomal digestion of the outer segments (Rakoczy et al., 1997). An age-related increase in the inert indigestible substrate inactive catD, or procathepsin D (proCatD) in RPE cells, is postulated to cause vision impairment by impairing photoreceptor outer segment proteolysis (Wiederanders and Oelke, 1984; Zhang et al., 2002). To further study the role of catD in vivo, a homozygous (mcd/mcd) transgenic mouse model was made that expresses a form of catD that lacks the catD cleavage site (Zhang et al., 2002). The retinal phenotypes of these mice were described by Rakoczy and colleagues (Rakoczy et al., 2002).
On fundoscopic exam at 9 months of age, RPE hypopigmentation, hyperpigmentation, and drusenoid lesions were present in most mcd/mcd mice, while no lesions were present in wild-type mice. After 10 months of age, RPE cells in mcd/mcd mice were found to have focal attenuation, hypertrophy, and hypopigmentation. By 18 months of age, minimal Bruch’s membrane thickening, drusen-like sub-RPE deposits, and extensive BlamD and BlinD were observed. An increase in apoptotic photoreceptors in mcd/mcd mice was noted when compared to age-matched wild-type controls, and progressive thinning of the ONL layer of photoreceptors was present at 12 months with disorganization of the INL. ERG abnormalities (reduced a-wave and b-wave amplitudes) were present in transgenic mcd/mcd mice at 12 months of age. No changes in the choriocapillaris were noted (Rakoczy et al., 2002).
To investigate whether the observed phenotype in the mcd/mcd mice is a direct result of the amount of proCatD, a transgenic mouse (mcd2/mcd2) was made with additional deletions in the catD cleavage side such that mcd2/mcd2 mice cannot make any active catD (Zhang et al., 2005). The mcd2/mcd2 mice experienced earlier signs of retinal degeneration than the mcd2/mcd2 mice. Three month-old mcd2/mcd2 mice showed focal areas of RPE disorganization, clumping, proliferation, and pleomorphism with attenuation, depigmentation, and atrophy. These early findings progressed with age, and by 12 months old almost all the animals developed RPE proliferation. These RPE findings, collectively referred to as “pigment mottling,” are accompanied by progressive photoreceptor outer segment disturbances and photoreceptor atrophy by 12 months. The basal lamina and Bruch’s membrane were unaffected. Tight junctions and basal polarity of the RPE nucleus was lost in the mcd2/mcd2 mice (Zhang et al., 2005).
3.1.2.3.3. Cp−/−Heph−/Y murine model
Iron is an essential cofactor for many enzymes, but ferrous iron (Fe2+) can cause oxidative damage via the Fenton reaction. If Fe2+ causes oxidative damage in the macula, it may contribute to AMD pathogenesis (Hahn et al., 2003; Hahn et al., 2004). In iron overload states, retinal degeneration and photoreceptor toxicity have been observed (Doly et al., 1986). Compared to normal eyes, AMD eyes have a statistically significant increase in total iron in the RPE and Bruch’s membrane and in AMD lesions (Hahn et al., 2003). Ceruloplasmin (Cp) is a multicopper feroxidase that, when secreted, circulates in the blood without crossing the blood-brain barrier (Patel and David, 1997). Cp is expressed in human and mouse retinas in the inner nuclear layer (Klomp and Gitlin, 1996; Klomp et al., 1996), and Cp expression increases in Müller glia after photic injury (Chen et al., 2003). Cp is necessary for iron export from cells, and Cp−/− mice have mild iron overload with increased susceptibility to oxidative stress (Patel et al., 2002). Hephaestin (Heph) is a multicopper feroxidase that is 50% identical to Cp and facilitates iron export. Heph is naturally mutated in sex-linked anemia (sla) mice, which have low Heph ferroxidase levels (Vulpe et al., 1999; Chen et al., 2004). Mice with a combined deficiency in Cp and Heph (Cp−/−Heph−/Y mice) were made and found to have age-related iron accumulation, secondary increases in ferritin, and retinal degeneration with AMD-like features (Hahn et al., 2004).
By 5–6 months of age, the Cp−/−Heph−/Y mice had increased iron levels, and the highest iron levels were in the RPE and photoreceptor OS. Iron-laden electron dense vesicles, likely lysosomes or lysosome-melanosome fusion vesicles, were visible in the RPE cells of 5 month-old Cp−/−Heph−/Y mice only. At 6–9 months old, Cp−/−Heph−/Y mice had severe retinal degeneration with RPE hypopigmentation, hypertrophy, hyperplasia, and necrosis. Hypertrophic RPE cells were full of lipofuscin, phagosomes, and lysosomes with partially digested photoreceptor outer segment membranes. This RPE hypertrophy is greater than what has been observed in AMD retinas (Hahn et al., 2004; Hadziahmetovic et al., 2008). Cp−/−Heph−/Y mice had no clinically observable drusen. Focal subretinal deposits with banded structures and wide-spaced collagen, similar to BlamD, were observed in the 9 month-old Cp−/−Heph−/Y mice. Local photoreceptor degeneration and thinning was found in the ONL along with inner segment vacuolization (Hahn et al., 2004). In 12 month-old Cp−/−Heph−/Y mice, 90% of RPE cells were hypertrophic, and subretinal macrophage infiltration was evident. The complement system is activated in Cp−/−Heph−/Y mice. Evidence of focal CNV was also present in 75% of 12–13 month-old mice and 50% of 7–9 month-old mice (Hahn et al., 2004; Hadziahmetovic et al., 2008).
3.1.2.3.4. Murine models of aberrant cholesterol metabolism
Cholesterol deposition in Bruch’s membrane is one of the most common age-related changes in the human retina (Pauleikhoff et al., 1990). The deposition of lipid- and proteoglycan-rich extracellular material is a common risk factor for atherosclerosis and AMD (Snow and Seddon, 1999). Mice with higher plasma LDL levels via transgenic methods and/or high fat and cholesterol diets have more circulating lipoproteins and are more prone to both atherosclerosis and degeneration of Bruch’s membrane, a key factor in AMD pathogenesis (Sallo et al., 2009; Rudolf et al., 2004). Degenerative changes in Bruch’s membrane have been recognized by several groups in association with elevated serum lipid levels in both wild-type mice and transgenic mice in advanced age (Cousins et al., 2003; Miceli et al., 2000; Cousins et al., 2002; Dithmar et al., 2000; Rudolf et al., 2004). Laser photodisruption of the RPE in mice fed a high fat diet accumulate BlamD, indicating that compromise to RPE lipid metabolism in a hypercholesterolemic mouse leads to BlamD (Dithmar et al., 2001).
3.1.2.3.4.1. ApoE transgenic murine models
Apolipoprotein E (apoE) is a polymorphic CNS apolipoprotein that is important for plasma lipid metabolism, cholesterol and lipid mobilization, and neuronal cell membrane maintenance and repair (Pitas et al., 1987; Mahley, 1988). ApoE has been found in drusen and BlamD (Klaver et al., 1998). ApoE−/− (Apolipoprotein E deficient) mice have elevated total plasma cholesterol and VLDL. Starting at 2 months of age, these mice form vacuolated electrolucent vescicles in Bruch’s membrane, similar to BlinD, that increase with age (Dithmar et al., 2000). Several genetic association studies show that APOE polymorphisms provide a significant risk for AMD: the APOE ε4 allele confers a decreased AMD risk; the APOE ε2 allele is associated with a slightly increased AMD risk compared to APOE ε3 allele homozygotes (Klaver et al., 1998; Baird et al., 2004; Bojanowski et al., 2006). These findings contrast with studies that show a positive association between Alzheimer’s disease, atherosclerosis, multiple sclerosis, stroke and the APOE ε4 allele (Kalaria, 1997; Weller and Nicoll, 2003; Enzinger et al., 2004).
To compare the apoE2, apoE3, and apoE4 alleles, transgenic (TR) mice models were made expressing one of the three human APOE isoforms (Sullivan et al., 1997). The apoE2, apoE3, and apoE4 TR mice were fed a low or high fat diet, and their retinal pathology was compared and summarized (Malek et al., 2005). The young (12–13 week-old) apoE TR mice (irrespective of the fat content in their diet) had a normal ocular phenotype. The aged (65–127 week-old) apoE3 TR mice on a high-fat diet were ultrastructurally normal except for minor RPE vacuolization. Aged apoE2 TR mice on a high-fat diet had a more severe phenotype with RPE vacuolization, mottling, hyperpigmentation and hypopigmentation, disorganized basal infoldings, sub-RPE deposits, and Bruch’s membrane thickening (Malek et al., 2005).
Aged apoE4 TR mice on a high-fat diet had the most severe degenerative phenotype with RPE hyperpigmentation, hypopigmentation, atrophy, disorganization of basal infoldings with electron-dense deposits, many electron-dense sub-RPE basal deposits, lipid-rich “drusenoid” deposits, disorganization with varying thickness of Bruch’s membrane, and CNV of varying severity (Malek et al., 2005). In studies of patients with Alzheimer’s disease and the APOE4 allele, there is increased deposition of Aβ; in the apoE4 TR mouse, CFH and Aβ form a complex in sub-RPE deposits, which may cause local inflammation leading to the progression of the disesase seen in the apoE4 TR mouse model (Wellington, 2004; Strohmeyer et al., 2002; Malek et al., 2005; Bruban et al., 2009).
Another ApoE transgenic mouse model is the APO*E3-Leiden mouse, a mouse that produces a dysfunctional form of human APOE3 and has hyperlipoproteinemia (van den Maagdenberg et al., 1993). After 9 months on a high fat/cholesterol diet, 100% of APO*E3 Leiden mice eyes have BlamD, while 33% of APO*E3 Leiden mice on a normal diet have BlamD (Kliffen et al., 2000). Interestingly, more BlamD were found in atherosclerotic APO*E3 Leiden mice compared to non-atherosclerotic APO*E3 Leiden mice (Kliffen et al., 2000), supporting existing epidemiologic data linking AMD and atherosclerosis (Vingerling et al., 1996; Klein et al., 1999). BlamDs are the main pathologic finding in this model, along with an increase and elongation of the RPE basal infoldings (Kliffen et al., 2000). Further electron microscopy is needed to analyze the ultrastructure of the RPE and Bruch’s membrane in this model.
3.1.2.3.4.2. Apo B100 transgenic murine model
Insertion of the apoB-100 gene into mice creates a sharp increase in plasma LDL after a high-fat diet, a lipid profile similar to what has been seen in humans (Bjelik et al., 2006; Purcell-Huynh et al., 1995). The apoB100 transgenic mice have higher serum total and LDL cholesterol and a statistically significant cholesterol-dependent increase in Bruch’s membrane thickness (Sallo et al., 2009). The apoB-100 transgenic mice fed with a high cholesterol diet develop BlamD (Sallo et al., 2009; Espinosa-Heidmann et al., 2004; Fujihara et al., 2009) and highly specific BlinD (Fujihara et al., 2009; Curcio and Millican, 1999) with outer collagenous zone deposits and loss of basal infoldings (Fujihara et al., 2009). When young apoB-100 mice were placed on a high-fat diet and exposed to acute photo-oxidative stress in the form of blue-green laser light or cigarette smoke, sub-RPE deposits, specifically BlamD, developed at two months of age. Additionally, diffuse Bruch’s membrane thickening with granular deposits, choriocapillaris hypertrophy, and reduplication of the basement membrane was observed (Espinosa-Heidmann et al., 2004; Espinosa-Heidmann et al., 2006). The RPE of apoB-100 transgenic mice on high-fat diets have misaligned, disorganized, amorphous material between the RPE plasma and basement membrane, and atrophic basal processes with occasional vacuolization. No signs of photoreceptor damage or atrophy were found in the apoB-100 transgenic mouse (Sallo et al., 2009).
3.2. Immunologically manipulated carboxyethylpyrrole (CEP) immunized mouse
Ample evidence has implicated AMD as an immunologically-mediated disease (Patel and Chan, 2008). One hypothesis is that a signal from the outer retina initiates immune involvement in AMD. One potential signal is carboxyethylpyrrole (CEP), an adduct from the oxidation fragment of deocosahexaenoic acid (DHA), an easily oxidizable long-chain polyunsaturated fatty acid that is abundant in the outer retina (Anderson, 1970; Gu et al., 2003). CEP forms when an oxidation fragment of DHA covalently interacts with an ε-lysyl amino group in a tissue protein (Gu et al., 2003; Crabb et al., 2002). CEP, a hapten, acts as a biomarker of oxidative stress, a well-known factor in AMD pathogenesis (Snow and Seddon, 1999). In an AMD patient’s outer retina and plasma, there are more CEP-modified proteins when compared to age-matched controls (Crabb et al., 2002). CEP is also pro-angiogenic via a VEGF-independent pathway (Ebrahem et al., 2006).
Serum albumin is one of the major proteins modified with CEP, and one group used this concept to immunize mice with CEP-modified mouse serum albumin, generating a strong immunologic response against endogenous CEP adducts in the outer retina (Hollyfield et al., 2008; Crabb et al., 2002). The severity of retinal pathology correlated directly with the CEP-antibody titer. The RPE cells in CEP-immunized mice had vesiculation, pyknosis, lysis, and areas of RPE loss with overlying swollen photoreceptors. Fundoscopy of immunized mice revealed patchy, reticular changes that were not present in controls (Hollyfield et al., 2008). Compared to age-matched controls, CEP-immunized mice have significant BlamD accumulation throughout the retina. Bruch’s membrane was significantly thicker (about 3 fold) in immunized mice. Immunized mice had C3d, a C3b degradation product in Bruch’s membrane, signifying that an intact immune system responds to CEP immunization with sub-RPE complement deposition, a well-known feature of AMD (Hollyfield et al., 2008; Patel and Chan, 2008). CEP-immunization has also been shown to exacerbate angiogenesis in mice with laser-induced breaks in Bruch’s membrane, a finding that is reversible with administration of a monoclonal anti-CEP IgM antibody (Ebrahem et al., 2006).
3.3. Natural mice strains with features of AMD
Several naturally occurring mouse strains have shown features of retinal degeneration (Chang et al., 2002). Retinal degeneration is a broad term for conditions that cause irreversible vision loss, and over 140 genes have been identified in human retinal degenerations. The first identified retinal degeneration model was RdsRd2, an autosomal dominant slow retinal degeneration identified in inbred mice that accumulated an insertion of foreign DNA into an exon of Rds (Sanyal and Hawkins, 1986; van Nie et al., 1978). While many of the mouse models have been used to study retinitis pigmentosa, certain mouse models of retinal degeneration help provide insight into the molecular pathology of photoreceptor loss in AMD (Chang et al., 2002; Rakoczy et al., 2006). An accelerated aging model, the Senescence-Accelerated Mouse (SAM) family, is a collection of mouse strains with naturally occurring premature aging and associated pathologies (Takeda, 1997). As AMD is an age-related phenomenon, the retinas of these mice may provide insight into the causes of naturally occurring alterations in the aging human retina.
3.3.1. arrd2/arrd2 (Mdm gene mutation) murine model
A murine model of a naturally occurring, severe, late-onset, age-related, retinal degeneration (arrd2) has been identified to have a nonsense mutation in the Mdm1 gene on mouse chromosome 10 that is inherited in an autosomal recessive manner (Chang et al., 2008). Analysis of a cohort of AMD patients with the human ortholog of MDM1 on chromosome 12q did not reveal an association between MDM1 and AMD, though this location has previously been reported to be an AMD susceptibility locus (Chang et al., 2008; Iyengar et al., 2004; Fisher et al., 2005). At 4 months of age, the arrd2/arrd2 mice had early photoreceptor outer segment degeneration, increased phagosomes and debris in the RPE, normal ONL, swollen synaptic complexes, and large mitochondria. At 6 months, the arrd2/arrd2 mice had subnormal rod and cone ERG responses and an undetectable ERG response at 22 months. At 9 months of age, the arrd2/arrd2 mice had fragmented photoreceptor outer segments, abundant phagosomes with loss of the apical processes in the RPE, thinning of the outer plexiform layer (INL and ONL), swollen and necrotic synaptic terminals, and mitochondrial edema. No basal deposits or Bruch’s membrane changes were noted in arrd2/arrd2 mice. In this model, cone degeneration occurs slower than rod degeneration. The arrd2/arrd2 mice also have retinal vascular attenuation, RPE hypo-pigmentation and atrophy at 14 months. This phenotype has features similar to human progressive rod-cone degeneration and AMD (Chader, 2002; Klein, 2007).
3.3.2. Mfrprd6 retinal degeneration murine model
One of the naturally occurring retinal degeneration and retinitis pigmentosa models, rd6, is inherited in an autosomal recessive manner and maps to mouse chromosome 9 with a human homolog on chromosome 11q23 (Hawes et al., 2000). A splice-donor mutation with the loss of an exon in a gene encoding membrane-type frizzled protein (Mfrp) was identified in the rd6 mouse (Kameya et al., 2002). Mfrp, expressed at high levels exclusively in the RPE and ciliary epithelium and not the neural retina, has strong homology to the cysteine-rich domain of frizzled, a protein that is an important member of the Wnt family of proteins (Kameya et al., 2002; Xu and Nusse, 1998). Aberrations in the Wnt/frizzled signaling pathway have been implicated in photoreceptor degeneration in retinitis pigmentosa patients, and abnormal Wnt/frizzled signaling in the RPE may be responsible for photoreceptor degeneration (Jones et al., 2000; Kameya et al., 2002). The homozygous Mfrprd6 mice have pan-retinal drusen-like deposits on fundoscopy at 8 weeks of age that progress to advanced retinal degeneration by 8 months of age, and these lesions are associated with macrophage invasion of the sub-retinal space and retinal dysplasia (Hawes et al., 2000). By 3 months of age, Mfrprd6 RPE cells accumulate lipofuscin-like material and pigment. The photoreceptor layer degenerates to 33% of its original thickness by 4.5 months of age, 15% of normal by 7 months of age, and becomes 1 cell layer thick by 24 months of age (Kameya et al., 2002). Homozygous Mfrprd6 mice have diminished ERG responses beginning at 1 month of age, supportive of a slow progressive retinal rod and cone dysfunction that culminates in extinguished ERG activity by 70 weeks of age. No aberrations in Bruch’s membrane have been reported, but further analysis of the retinal ultrastructure is necessary.
3.3.3. Nr2e3rd7 retinal degeneration murine model
The retinal degeneration 7 mouse has a mutation in the Nr2e3 gene responsible for Goldmann Favre disease, or human enhanced S-cone syndrome (ESCS) (Haider et al., 2000; Chang et al., 1998). The inheritance pattern is autosomal recessive, and Nr2e3 is mapped to mouse chromosome 9 and human chromosome 15q23. The Nr2e3rd7 mouse has an exon 4,5 deletion in the Nr2e3 gene, causing a frameshift mutation that results in a premature stop codon (Akhmedov et al., 2000). Nr2e3 is part of the cellular and nuclear receptor family for steroid hormones and is involved in cone cell proliferation, maintenance of photoreceptor topography and intercellular interactions, and expression of rod photoreceptor cell-specific genes (Fuller, 1991; Haider et al., 2001; Cheng et al., 2004). At one month of age, large evenly-spaced retinal lesions are visible throughout the retina of homozygous Nr2e3rd7 mice by fundoscopy (Hawes et al., 1999). Histologic analysis of the photoreceptor ONL demonstrates retinal dysplasia with whorls and rosettes, which decrease by 5 months of age and disappear when the photoreceptor layer flattens to 5-cell layers thick. There is additional retinal vascular attenuation and mottled retinal pigmentation at 16 months of age (Chang et al., 1998; Haider et al., 2001; Akhmedov et al., 2000). ERGs of homozygous Nr2e3rd7 mice show progressive reduction of both rod and cone signals starting at 5 months of age, and wave amplitudes were 50% of normal by 16 months of age (Akhmedov et al., 2000). The retinal ultrastructure demonstrates disorganized photoreceptor outer segments and apical microvilli retardation in the RPE cells (Won et al., 2008).
3.3.4. cpfl3 (Gnat2 gene mutation) cone degeneration murine model
Most AMD cases have regional cone photoreceptor loss (Sarks, 1976). Additionally, cone dysfunction is an important clinical predictor of AMD severity (Hogg and Chakravarthy, 2006). Shelley and colleagues recently showed that, in AMD, cones lose their synaptic connections before dying, and signs of photoreceptor degeneration are found in cones before rods. Rod photoreceptor cell death also expedites cone cell death (Shelley et al., 2009). Therefore, studying models of cone photoreceptor cell dysfunction is important in understanding AMD pathogenesis and testing therapeutic options.
A mouse with a naturally occurring mutation in cone photoreceptor function loss-3 (cpfl3) is being used a model for rod monochromism, or achromatopsia, an autosomal recessive disease in which there is a loss of functional retinal cone photoreceptors, resulting in a disease that presents in infancy with horizontal nystagmus, photophobia, color blindness, and poor visual acuity (Aligianis et al., 2002). A mutation in exon 6 of Gnat2 results in a missense mutation of the α-subunit of transducin, preventing photoreceptor hyperpolarization in the light transduction cascade, resulting loss of function in the structurally normal cones in cpfl3/cpfl3 mice. Fundoscopy of cpfl3/cpfl3 mice shows no retinal lesions and only dilated retinal vessels at 8 months of age. Gnat2cpfl3 mice have normal rod responses on ERG with an abnormal, significantly reduced, photopic response that was extinguished by 9 months of age. Histopathology demonstrates increased vacuolization and photoreceptor outer segment shortening with increased age (Chang et al., 2006). However, the ocular ultrastructure has not yet been described. While this model does not have the Bruch’s membrane, RPE, and choriocapillaris changes seen in AMD patients, it demonstrates the cone pathology and dysfunction that can be present in the AMD macula.
3.3.5. SAM (senescence-accelerated mouse) model strains
Aging, or senescence, has been shown to be associated with photoreceptor loss in humans and in murine models (Gao and Hollyfield, 1992; Katz and Robison, 1986). The Senescence-Accelerated Mouse (SAM) family of 12 murine lines demonstrates accelerated senescence that has been developed by selective inbreeding of the AKR/J strain (Takeda et al., 1994). The SAM mice have many characteristic pathologic phenotypes that develop after a period of normal development, including lordokyphosis, hair loss, skin lesions, amyloidosis, and early death. There are nine senescence-prone inbred strain mice SAMP(1–9) and four senescence-resistant inbred mice SAMR(1–4) (Takeda et al., 1994). The SAMP mice are short-lived and have double the aging features when compared to the SAMR mice at 8 months of age (Takeda, 1997; Takeda et al., 1994).
Each SAM strain has a distinct strain-specific phenotype and genetic identity, though mice in the same family (SAMP or SAMR) have similar biochemical and immunogenic markers (Kitado, 1994). Genetic studies of the SAM mice reveal that several endogenous provirus genes, polytropic murine leukemia virus 25 (Pmv-25), modified polytropic murine leukemia virus 17 (Mpmv-17), and modified polytropic murine leukemia virus 29 (Mpmv-29), are found exclusively in the SAMP strains, and polytropic murine leukemia virus 35 (Pmv-35) is found exclusively in the SAMR strains (Kitado, 1994).
The SAMP8 mice have a shortened life-span, learning and memory deficits, features of AMD, and no signs of brain atrophy (Kawamata et al., 1997). As AMD is an age-related phenomenon, another strain of SAM mice with a longer life-span, SAMR1 mice, have age-related retinal pathology reminiscent of AMD (Sarks, 1976; Majji et al., 2000). Neuronal cell loss was investigated in SAMP1 and SAMR1 mice at various time points compared to control mice. By 25 months of age, SAMR1 mice had near complete loss of the photoreceptor layer peripherally and significant loss of the photoreceptor and ganglion cells centrally (Shoji et al., 1998). The SAMP1 strain has been shown to have age-related changes in the RPE and Bruch’s membrane (Ogata et al., 1992; Takada et al., 1994; Takada et al., 1993). The RPE cells in SAM mice have swelling of basal infoldings and accumulation of lipofuscin granules that increase with age (Ogata et al., 1992). In SAMP8 mice, early disruption of the RPE basal microvilli begins at 8 months of age and progresses to severe RPE degeneration by 12 months of age (Majji et al., 2000). In Bruch’s membrane, fragmentation between the elastic and outer collagenous layers occurs with age (Ogata et al., 1992). As the SAMP1 and SAMP8 mice age, deposition of fine fibrils in Bruch’s membrane increases, resulting in a four-fold increase in thickness of Bruch’s membrane by 8 months of age (Takada et al., 1993; Majji et al., 2000). BlamD, amorphous deposits in the sub-RPE space on the RPE side of Bruch’s membrane, were also found (Majji et al., 2000). There is also an age-related increase in the thickness of the choriocapillaris basement membrane, which was found to have anti-type IV collagen antibodies by gold particle labeling (Takada et al., 1993; Takada et al., 1994). Intra-Bruch’s membrane choroidal neovascularization was also seen in 40% of the SAMP8 mice older than 11 months of age (Majji et al., 2000).
4. Summary
The molecular pathology of dry AMD has demonstrated a role of oxidative stress, inflammation, and metabolic dysregulation (Ding et al. 2009). Pathologic hallmarks of dry AMD include drusen accumulation (BlamD and BlinD), Bruch’s membrane thickening, RPE lipofuscin accumulation followed by degeneration, and photoreceptor degeneration and atrophy. Although mice do not have a macula and lipofuscin extrusion follows a different process, mice have been the models for retinal disease because the mouse retina closely resembles the human nasal and peripheral retina. The described murine models of dry AMD have drusenoid lesions and RPE atrophy on fundoscopy, ERG abnormalities, A2E accumulation, sub-RPE basal deposits, and Bruch’s membrane thickening (Table 2). One of the key advantages of murine models is the ability to relatively easily manipulate mice genetically, surgically, pharmacologically and via changes in their diet and environment.
Table 2.
Retinal pathology of dry AMD models. This table presents a summary of the clinical, biochemical, and retinal pathologic findings in dry AMD murine models.
| No. | Mouse model | Clinical information | Biochemical data |
Pathologic information | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age of onset | Clinical exam |
Type of lesion | ERG changes | A2E level | RPE changes | Basal deposits |
BM changes | Photoreceptor atrophy |
CNV | ||
| 1 | abcr−/− | 44 w | not xamined | - | reduced a-wave mplitude | elevated | lipofuscin, melanosomes vacuolization, | none | thickening | shortening of the PR OS | none |
| 2 | ELOVL4-mutant | 7 mo | retinal lesions | RPE atrophy | reduced b-wave responses | elevated | lipofuscin, debris, central retinal RPE atrophy | present | none | OS disk disorganization, geographic atrophy | none |
| 3 | Efemp1R345W/R345W | 4 mo | no lesions | none | normal a-waves and b-waves | not measured | vacuolization, loss of basal infoldings | BlamD | with amorphous debris | None | none |
| 4 | Timp3S156C/S156C | 8 mo | no lesions | none | normal b-waves | not measured | basal layer disruption | present | thickening | None | present |
| 5 | cfh−/− | 2 yrs | retinal lesions | hyperfluorescent | reduced a-wave and b-wave amplitudes | not measured | lipofuscin, melanosomes, disorganized organelles | decreased | thinning | disorganized PR OS | |
| 6 | Ccl2−/− | 9 mo | retinal lesions | drusenoid | not performed | elevated | vacuolated, lipofuscin, menalosomes | present | thickening | pyknotic PR, geographic atrophy | present |
| 7 | Ccr2−/− | 9 mo | retinal lesions | drusenoid | not performed | elevated | hypopigmentation, loss of basal infolding | present | thickening | ONL atrophy, geographic atrophy | present |
| 8 | Cx3cr1−/− | 12 mo | retinal lesions | drusenoid | not performed | not measured | intracellular lipid deposits and phagosomes | present | none | outer retinal thinning, progressive degeneration | only after laser injury |
| 9 | Ccl2−/−/Cx3cr1−/− | 4–6 w | retinal lesions | drusenoid | reduced a-wave and b-wave amplitudes | elevated | hypopigmentation, vacuolization, lipofuscin, melanosome loss --> degeneration | present | thickening | OS disorganization and atrophy | present |
| 10 | Sod1−/− | 7 mo | retinal lesions | drusenoid | not performed | not measured | vacuolization, degeneration | present | thickening | ISand OS loss | present |
| 11 | Sod2 knockdown | 4 mo | not examined | - | reduced a-wave and b-wave amplitudes | elevated | lipofuscin, hypopigmentation, basal lamina deterioration, vacuolization, mitochondrial abnormalities | BlamD | thickening | disorganized PR OS, inner retinal thinning | none |
| 12 | neprilysin −/− | 27 mo | not examined | - | not performed | not measured | vacuolization, loss of tight and adherens junctions, distorted basal infolding, degeneration | BlamD and BlinD | none | none | none; VEGF upregulation, PEDF downregulation |
| 13 | mcd/mcd mice | 12 mo | retinal lesions | RPE pigmentary changes and drusenoid deposits | reduced a-wave and b-wave amplitudes | not measured | hypertrophy, hypopigmentation, and attenuation | BlamD and BlinD | none | ONL thinning, INL disorganization | none |
| 14 | Cp−/−/Heph−/Y | 5–6 mo | no lesions | none | not performed | not measured | lipofuscin, lysosomes, endosomes, and phagosomes, hypopigmentation; RPE hypertrophy, hyperplasia, necrosis | present | vacuolization | focal areas of PR loss, progressing to eventual loss of IS, OS, and ONL thinning | present |
| 15 | ApoE−/− | 2 mo | not examined | none | not performed | not measured | normal | BlinD | vacuolization | none | none |
| 16 | ApoE4 TR | 65 wk-127 wks | not examined | drusenoid | not performed | not measured | vacuolization, hyperpigmentation, hypopigmentation, atrophy, disorganized basal infoldings none detected; | BlamD | thickening | thinning of the ONL | present |
| 17 | APO*E3-Leiden | 9 mo | not examined | - | not performed | not measured | however, unable to assess | BlamD | thickening | none | none |
| 18 | APO B100 | 3 mo | not examined | - | not performed | not measured | occasional vacuolization | BlamD | thickening | none | none |
| 19 | CEP-immunized mouse | 3 mo after immunization | retinal lesions | patchy reticular changes | not performed | not measured | vesiculation, pyknosis, lysis, and areas of RPE loss | BlamD | thickening | swollen PR | only after laser injury |
| 20 | arrd2/arrd2 | 4 mo | retinal lesions | RPE hyper- and hypopigmentation vessel attenuation | reduced amplitude, ultimately extinguished waves | not measured | phagosomes, loss of apical processes, hypopigmentation, atrophy | none | none | INL and ONL thinning, necrotic synapses, OS fragmentation | none |
| 21 | Mfrprd6 | 3 mo | retinal lesions | drusenoid | reduced amplitude, ultimately extinguished waves | not measured | lipofuscin | none | none | PR degeneration in all layers | none |
| 22 | Nr2e3rd7 | 1 mo | retinal lesions | drusenoid | reduced a-wave and b-wave amplitudes | not measured | apical villi retardation | none | none | ONL dysplasia with whorls and rosettes, progressive thinning | none |
| 23 | cpfl3 | 8 mo | no lesions | none | reduced photopic response | not measured | none | none | none | vacuolization, PR OS shortening | none |
| 24 | SAMP8 | 8 mo | not examined | - | not performed | not measured | lipofuscin, swelling of basal infoldings, microvilli disruption, severe degeneration | BlamD | thickening, fragmentation | none | present |
Abbreviations: RPE, retinal pigment epithelium; CNV, choroidal neovascularization; ERG, electroretinography; BM, Bruch’s membrane; mo, month; A2E, N-retinylidene-N-retinylethanolamine; BlamD, basal laminar deposits; BlinD, basal linear deposits; PR, photoreceptors; OS, outer segment; ONL, outer nuclear layer; IS, inner segment; VEGF, vascular endothelial growth factor; PEDF, pigment epithelium-derived factor
The aforementioned dry AMD murine models fall into three categories: genetically engineered mice, immunologically manipulated mice, and naturally occurring mouse strains. Most murine models are genetically engineered to manipulate genes that cause macular degeneration-like disease in humans or are relevant to AMD pathogenesis. Inflammatory genes Cfh, Ccl2, Ccr2, and Cx3cr1, oxidative stress associated genes Sod1 and Sod2, and metabolic pathway genes Neprilysin, mcd, Cp, Heph, ApoE and ApoB100 have been manipulated to create models with features of dry AMD (Figure 3). The immunologically manipulated mouse model was immunized against CEP, an endogenous biomarker of oxidative stress, and its phenotype consisted of immune dysregulation and complement deposition as seen in AMD. The naturally occurring mouse models include retinal degeneration models with mutations in Mdm1, Mfrp, Nr2e3, and Gnat2 with significant photoreceptor pathology and the senescence-accelerated mouse (SAM) models with systemic and retinal age-related pathology, including Bruch’s membrane thickening, basal deposits, and CNV. These models contrast with the murine models of wet AMD that are mostly induced mechanically.
Figure 3.

Diagrams of classic ultrastructural changes in the photoreceptor, RPE, Bruch’s membrane and choroid in different mouse AMD models (IS, photoreceptor inner segment; OS, photoreceptor outer segment; RPE, retinal pigment epithelial cells; BM, Bruch’s membrane; Cr, Choroid; BlamD, basal laminar deposits; CNV, choroidal neovascularization)
5. Future directions and challenges
AMD is a challenging disease to study because of its complex genetics, late onset, and confounding environmental risk factors. As with other human diseases, finding a model system for AMD has been a demanding task. Working with primates, the only mammals with a macula, may have scientific advantages. Aged primates have macular degeneration that presents in the late 20s with soft drusen and an immunophenotype of drusen very similar to that of humans (Umeda et al., 2005). Monkey models of both late-onset macular degeneration and early-onset macular degeneration are bred in Japan, and the genetics of affected monkeys is being studied vigorously with the hope of identifying new AMD-related genes (Umeda et al., 2005). Primate research is often difficult, given a limited supply of animals, economic constraints, the slow course of disease, and ethical challenges in making genetically manipulated monkeys.
Mice, while very amenable to genetic manipulation, diverged from humans about 65 million years ago. The study of immune responses in mice has brought great insight to human immunology. Despite the obvious differences in size and lifespan, sequencing of mouse and human genomes reveals great conservation of function and only about 300 unique genes (Waterston et al., 2002). The general structure of the immune system in mice and humans is also very similar, though discrepancies in both the innate and adaptive immune system exist (Haley, 2003; Mestas and Hughes, 2004). While many biologic paradigms translate between the species, the trend toward more specific and sophisticated therapeutics makes it very important to understand the limitations of the extrapolation of mouse research to human disease.
In spite of the great advances made in mouse immunology in the past fifty years, few observations made in the murine system have made a clinical impact. Therapies that have been shown to work in mice have failed in human trials, and therapies that do not work in mice have shown success in humans (Shepherd and Sridhar, 2003; Panitch et al., 1987). Several reasons for these discrepancies exist. Inbred mouse strains are excellent for experimentation; however, results seen in these mice may be different than those seen in genetically diverse outbred strains. An outbred mouse population would be more similar to a human population, but these mice are neither easily available nor amenable to large-scale scientific experimentation. On the other hand, there are successful therapies for human disease that arose from mouse models. Indeed, the usage of cyclosporine, rapamycin, or daclizumab for uveitis is based on its effectiveness in experimental autoimmune uveitis (Nussenblatt, 2002; Martin et al., 1995; Nussenblatt et al., 1983).
An important role for mouse models in AMD research is to serve as a preclinical test of safety and efficacy of new therapeutic options. Recently, human embryonic stem cell (hESC)-derived RPE cells were subretinally transplanted into the ELOVL4-mutant mouse and rats with retinal degeneration (RSC rats). These mice were followed for a prolonged period (>220 days) and shown to recover visual function and photoreceptor integrity without complications (Lu et al., 2009). We have also reported that high omega-3 diet ameliorates retinal AMD-like lesions in Ccl2−/−/Cx3cr1−/− mice (Tuo et al., 2009). Mouse models with pathology that is similar to AMD at the ultrastructural level allow for robust preclinical studies of novel AMD therapeutics.
Mouse models will likely play a critical role in the future of AMD research, as they may answer many specific questions at the molecular level, potentially providing further insight into the pathogenesis of this disease. Data from mouse experiments has generated significant basic scientific progress and has set the stage for several new clinical trials for dry AMD. A detailed understanding of the differences between mouse and human biology and pathology will help guide and improve the efficacy of future research therapeutic trials. Ultimately, meaningful advances in AMD research are made when the distance from the “bedside to the bench” shrinks. With greater access to human samples for basic research, scientists and physicians will be able to further understand the pathophysiology of AMD in humans and make meaningful mouse models and therapies based on clinical and scientific insights.
Acknowledgments
The NEI Intramural Research Program provided support. Dr. Robert B. Nussenblatt brought his insight into the challenges of AMD research in mice and men.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhmedov NB, Piriev NI, Chang B, Rapoport AL, Hawes NL, Nishina PM, Nusinowitz S, Heckenlively JR, Roderick TH, Kozak CA, Danciger M, Davisson MT, Farber DB. A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc Natl Acad Sci U S A. 2000;97:5551–6. doi: 10.1073/pnas.97.10.5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aligianis IA, Forshew T, Johnson S. Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the alpha subunit of cone transducin (GNAT2) J Med Genet. 2002;39:656–60. doi: 10.1136/jmg.39.9.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- Alsenz J, Schulz TF, Lambris JD, Sim RB, Dierich MP. Structural and functional analysis of the complement component factor H with the use of different enzymes and monoclonal antibodies to factor H. Biochem J. 1985;232:841–50. doi: 10.1042/bj2320841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambasudhan R, Wang X, Jablonski MM, Thompson DA, Lagali PS, Wong PW, Sieving PA, Ayyagari R. Atrophic macular degeneration mutations in ELOVL4 result in the intracellular misrouting of the protein. Genomics. 2004;83:615–25. doi: 10.1016/j.ygeno.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–7. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- Anderson DH, Mullins RF, Hageman GS, LVJ A role for local inflammation in the formation of drusen in the aging eye. American Journal of Ophthalmology. 2002;134:411–31. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- Anderson DH, Talaga KC, Rivest AJ, Barron E, Hageman GS, Johnson LV. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res. 2004;78:243–56. doi: 10.1016/j.exer.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Anderson RE. Lipids of ocular tissues. IV. A comparison of the phospholipids from the retina of six mammalian species. Exp Eye Res. 1970;10:339–44. doi: 10.1016/s0014-4835(70)80046-x. [DOI] [PubMed] [Google Scholar]
- Baird PN, Guida E, Chu DT, Vu HT, Guymer RH. The epsilon2 and epsilon4 alleles of the apolipoprotein gene are associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45:1311–5. doi: 10.1167/iovs.03-1121. [DOI] [PubMed] [Google Scholar]
- Behndig A, Svensson B, Marklund SL, Karlsson K. Superoxide dismutase isoenzymes in the human eye. Invest Ophthalmol Vis Sci. 1998;39:471–5. [PubMed] [Google Scholar]
- Bjelik A, Bereczki E, Gonda S, Juhasz A, Rimanoczy A, Zana M, Csont T. Human apoB overexpression and a high-cholesterol diet differently modify the brain APP metabolism in the transgenic mouse model of atherosclerosis. Neurochem Int. 2006;49:393–400. doi: 10.1016/j.neuint.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Boekhoorn SS, Vingerling JR, Witteman JC, Hofman A, De Jong PT. C-reactive protein level and risk of aging macula disorder: The Rotterdam Study. Arch Ophthalmol. 2007;125:1396–401. doi: 10.1001/archopht.125.10.1396. [DOI] [PubMed] [Google Scholar]
- Bojanowski CM, Shen D, Chew EY, Ning B, Csaky KG, Green WR, Chan CC, Tuo J. An apolipoprotein E variant may protect against age-related macular degeneration through cytokine regulation. Environ Mol Mutagen. 2006;47:594–602. doi: 10.1002/em.20233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruban J, Glotin AL, Dinet V, Chalour N, Sennlaub F, Jonet L, An N, Faussat AM, Mascarelli F. Amyloid-beta(1–42) alters structure and function of retinal pigmented epithelial cells. Aging Cell. 2009;8:162–77. doi: 10.1111/j.1474-9726.2009.00456.x. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson LALMm. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 1979;188:245–62. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- Chader GJ. Animal models in research on retinal degenerations: past progress and future hope. Vision Res. 2002;42:393–9. doi: 10.1016/s0042-6989(01)00212-7. [DOI] [PubMed] [Google Scholar]
- Chan CC, Ross RJ, Shen D, Ding X, Majumdar Z, Bojanowski CM, Zhou M, Salem N, Bonner R, Tuo J. Ccl2/Cx3cr1-deficient mice: an animal model for age-related macular degeneration. Ophthalmic Res. 2008;40:124–8. doi: 10.1159/000119862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CC, Tuo J, Bojanowski CM, Csaky KG, Green WR. Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol Histopathol. 2005;20:857–63. doi: 10.14670/hh-20.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Dacey MS, Hawes NL, Hitchcock PF, Milam AH, Atmaca-Sonmez P, Nusinowitz S, Heckenlively JR. Cone Photoreceptor Function Loss-3, a Novel Mouse Model of Achromotopsia Due to a Mutation in Gnat2. Invest Ophthalmol Vis Sci. 2006;47:5017–21. doi: 10.1167/iovs.05-1468. [DOI] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002;42:517–25. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Hurd RE, Wang J, Howell D, Davisson MT, Roderick TH, Nusinowitz S, Heckenlively JR. Mouse models of ocular diseases. Vis Neurosci. 2005;22:587–93. doi: 10.1017/S0952523805225075. [DOI] [PubMed] [Google Scholar]
- Chang B, Heckenlively JR, Hawes NL, Davisson MT. A new mouse model of retinal dysplasia and degeneration (rd7) Invest Ophthalmol Vis Sci. 1998;39(Suppl) [Google Scholar]
- Chang B, Mandal MN, Chavali VR, Hawes NL, Khan NW, Hurd RE, Smith RS, Davisson ML, Kopplin L, Klein BE, Klein R, Iyengar SK, Heckenlively JR, Ayyagari R. Age-related retinal degeneration (arrd2) in a novel mouse model due to a nonsense mutation in the Mdm1 gene. Hum Mol Genet. 2008;17:3929–41. doi: 10.1093/hmg/ddn295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Attieh ZK, Su T, Syed BA, Gao H, Alaeddine RM, Fox TC, Usta J, Naylor CE, Evans RW, Mckie AT, Anderson GJ, Vulpe CD. Hephaestin is a ferroxidase that maintains partial activity in sex-linked anemia mice. Blood. 2004;103:3933–9. doi: 10.1182/blood-2003-09-3139. [DOI] [PubMed] [Google Scholar]
- Chen L, Dentchev T, Wong R, Hahn P, Wen R, Bennett J, Dunaief JL. Increased expression of ceruloplasmin in the retina following photic injury. Mol Vis. 2003;9:151–8. [PubMed] [Google Scholar]
- Cheng H, Khanna H, Oh EC, Hicks D, Mitton KP, Swaroop A. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum Mol Genet. 2004;13:1563–75. doi: 10.1093/hmg/ddh173. [DOI] [PubMed] [Google Scholar]
- Cherepanoff S, Mitchell P, Wang JJ, Gillies MC. Retinal autoantibody profile in early age-related macular degeneration: preliminary findings from the Blue Mountains Eye Study. Clin Exp Ophthalmol. 2006;34:590–95. doi: 10.1111/j.1442-9071.2006.01281.x. [DOI] [PubMed] [Google Scholar]
- Chong NHV, Alexander RA, Gin T, Bird AC, Luthert PJ. TIMP-3, collagen, and elastin immunohistochemistry and histopathology of Sorsby’s fundus dystrophy. Invest Ophthalmol Vis Sci. 2000;41:898–902. [PubMed] [Google Scholar]
- Coffey PJ, Gias C, Mcdermott CJ, Lundh P, Pickering MC, Sethi C, Bird A, Fitzke FW, Maass A, Chen LL, Holder GE, Luthert PJ, Salt TE, Moss SE, Greenwood J. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc Natl Acad Sci U S A. 2007;104:16651–6. doi: 10.1073/pnas.0705079104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HR, Chan CC, Ferris FL, Chew EY. Age-related macular degeneration. Lancet. 2008;372:1835–45. doi: 10.1016/S0140-6736(08)61759-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T. A pathological report upon a case of Doyne’s choroiditis (‘honeycomb’ or ‘family choroidits’) Ophthalmoscope. 1913;11:537–38. [Google Scholar]
- Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, Lavalette S, Houssier M, Jonet L, Picard E, Debre P, Sirinyan M, Deterre P, Ferroukhi T, Cohen SY, Chauvaud D, Jeanny JC, Chemtob S, Behar-Cohen F, Sennlaub F. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–8. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon N, O’colmain B, Klaver CC, Klein R, Munoz B. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–85. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- Cousins SW, Espinosa-Heidmann DG, Alexandridou A, Sall J, Dubovy S, Csaky K. The role of aging, high fat diet and blue light exposure in an experimental mouse model for basal laminar deposit formation. Exp Eye Res. 2002;75:543–53. doi: 10.1006/exer.2002.2047. [DOI] [PubMed] [Google Scholar]
- Cousins SW, Marin-Castano ME, Espinosa-Heidmann DG, Alexandridou A, Striker L, Elliot S. Female gender, estrogen loss, and Sub-RPE deposit formation in aged mice. Invest Ophthalmol Vis Sci. 2003;44:1221–9. doi: 10.1167/iovs.02-0285. [DOI] [PubMed] [Google Scholar]
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–7. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshanks KJ, Klein R, Klein BE. Sunlight and age-related macular degeneration. The Beaver Dam Eye Study. Arch Ophthalmol. 1993;111:514–8. doi: 10.1001/archopht.1993.01090040106042. [DOI] [PubMed] [Google Scholar]
- Curcio CA, Medeiros NE, Millican CL. Photoreceptor loss in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37:1236–49. [PubMed] [Google Scholar]
- Curcio CA, Millican CL. Basal linear deposit and large drusen are specific for early age-related maculopathy. Arch Ophthalmol. 1999;117:329–39. doi: 10.1001/archopht.117.3.329. [DOI] [PubMed] [Google Scholar]
- Curcio CA, Millican CL, Bailey T, Kruth HS. Accumulation of cholesterol with age in human Bruch’s membrane. Invest Ophthalmol Vis Sci. 2001;42:265–74. [PubMed] [Google Scholar]
- Dastgheib K, Green WR. Granulomatous reaction to Bruch’s membrane in age-related macular degeneration. Arch Ophthalmol. 1994;112:813–8. doi: 10.1001/archopht.1994.01090180111045. [DOI] [PubMed] [Google Scholar]
- Dentchev T, Milam AH, Lee VM, Trojanowski JQ, Dunaief JL. Amyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Mol Vis. 2003;9:184–90. [PubMed] [Google Scholar]
- Ding X, Patel M, Chan CC. Molecular pathology of age-related macular degeneration. Progress in Retinal and Eye Research. 2009;28:1–18. doi: 10.1016/j.preteyeres.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dithmar S, Curcio CA, Le NA, Brown S, Grossniklaus HE. Ultrastructural changes in Bruch’s membrane of apolipoprotein E-deficient mice. Invest Ophthalmol Vis Sci. 2000;41:2035–42. [PubMed] [Google Scholar]
- Dithmar S, Sharara NA, Curcio CA, Le NA, Zhang Y, Brown S, Grossniklaus HE. Murine high-fat diet and laser photochemical model of basal deposits in Bruch membrane. Arch Ophthalmol. 2001;119:1643–9. doi: 10.1001/archopht.119.11.1643. [DOI] [PubMed] [Google Scholar]
- Doly M, Bonhomme B, Vennat JC. Experimental study of the retinal toxicity of hemoglobinic iron. Ophthalmic Res. 1986;18:21–7. doi: 10.1159/000265409. [DOI] [PubMed] [Google Scholar]
- Ebrahem Q, Renganathan K, Sears J, Vasanji A, Gu X, Lu L, Salomon RG, Crabb JW, Anand-Apte B. Carboxyethylpyrrole oxidative protein modifications stimulate neovascularization: Implications for age-related macular degeneration. Proc Natl Acad Sci U S A. 2006;103:13480–4. doi: 10.1073/pnas.0601552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Enzinger C, Ropele S, Smith S, Strasser-Fuchs S, Poltrum B, Schmidt H, Matthews PM, Fazekas F. Accelerated evolution of brain atrophy and “black holes” in MS patients with APOE-epsilon 4. Ann Neurol. 2004;55:563–9. doi: 10.1002/ana.20027. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Sall J, Hernandez EP, Cousins SW. Basal laminar deposit formation in APO B100 transgenic mice: complex interactions between dietary fat, blue light, and vitamin E. Invest Ophthalmol Vis Sci. 2004;45:260–6. doi: 10.1167/iovs.03-0910. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suner IJ, Catanuto P, Hernandez EP, Marin-Castano ME, Cousins SW. Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Invest Ophthalmol Vis Sci. 2006;47:729–37. doi: 10.1167/iovs.05-0719. [DOI] [PubMed] [Google Scholar]
- Ethen CM, Reilly C, Feng X, Olsen TW, Ferrington DA. The proteome of central and peripheral retina with progression of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:2280–90. doi: 10.1167/iovs.05-1395. [DOI] [PubMed] [Google Scholar]
- Farkas TG, Sylvester VDA. The histochemistry of drusen. Am J Ophthalmol. 1971;71:1206–15. doi: 10.1016/0002-9394(71)90964-0. [DOI] [PubMed] [Google Scholar]
- Feeney-Burns L, Eldred GE. The fate of the phagosome: conversion to “age pigment” and impact in human retinal pigment epithelium. Trans Ophthalmol Soc UK. 1983;103:416–21. [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Fisher SA, Abecasis GR, Yashar BM, Zareparsi S, Swaroop A, Iyengar SK, Klein BE, Klein R, Lee KE, Majewski J, Schultz DW, Klein ML, Seddon JM, Santangelo SL, Weeks DE, Conley YP, Mah TS, Schmidt S, Haines JL, Pericak-Vance MA, Gorin MB, Schulz HL, Pardi F, Lewis CM, Weber BH. Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet. 2005;14:2257–64. doi: 10.1093/hmg/ddi230. [DOI] [PubMed] [Google Scholar]
- Fogarasi M, Janssen A, Weber BH, Stohr H. Molecular dissection of TIMP3 mutation S156C associated with Sorsby fundus dystrophy. Matrix Biol. 2008;27:381–92. doi: 10.1016/j.matbio.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Francis PJ, Zhang H, Dewan A, Hoh J, Klein ML. Joint effects of polymorphisms in the HTRA1, LOC387715/ARMS2, and CFH genes on AMD in a Caucasian population. Mol Vis. 2008;14:1395–400. [PMC free article] [PubMed] [Google Scholar]
- Friedman DS, O’colmain Bj, Muñoz B, Tomany Sc, Mccarty C, De Jong Pt, Nemesure B, Mitchell P, Kempen J, The Eye Diseases Prevalence Research Group Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–72. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- Fu L, Garland D, Yang Z, Shukla D, Rajendran A, Pearson E, Stone EM, Zhang K, Pierce EA. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum Mol Genet. 2007;16:2411–22. doi: 10.1093/hmg/ddm198. [DOI] [PubMed] [Google Scholar]
- Fujihara M, Bartels E, Nielsen LB, Handa JT. A human apoB100 transgenic mouse expresses human apoB100 in the RPE and develops features of early AMD. Exp Eye Res. 2009;88:1115–23. doi: 10.1016/j.exer.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller P. The steroid receptor superfamily: mechanism of diversity. FASEB J. 1991;5:3092–99. doi: 10.1096/fasebj.5.15.1743440. [DOI] [PubMed] [Google Scholar]
- Gao H, Hollyfield JG. Aging of the human retina. Differential loss of neurons and retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1992;33:1–17. [PubMed] [Google Scholar]
- Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration—emerging pathogenetic and therapeutic concepts. Annals of Medicine. 2006;38:450–71. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosbell AD, Stefanovic N, Scurr LL, Pete J, Kola I, Favilla I, De Haan JB. Retinal light damage: structural and functional effects of the antioxidant glutathione peroxidase-1. Invest Ophthalmol Vis Sci. 2006;47:2613–22. doi: 10.1167/iovs.05-0962. [DOI] [PubMed] [Google Scholar]
- Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999;5 [PubMed] [Google Scholar]
- Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman lecture. Ophthalmology. 1993;100:1519–35. doi: 10.1016/s0161-6420(93)31466-1. [DOI] [PubMed] [Google Scholar]
- Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW, Salomon RG. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem. 2003;278:42027–35. doi: 10.1074/jbc.M305460200. [DOI] [PubMed] [Google Scholar]
- Hadziahmetovic M, Dentchev T, Song Y, Haddad N, He X, Hahn P, Pratico D, Wen R, Harris ZL, Lambris JD, Beard J, Dunaief JL. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest Ophthalmol Vis Sci. 2008;49:2728–36. doi: 10.1167/iovs.07-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezi F, Grimm C, Simmen BC, Wenzel A, Remé CE. Molecular ophthalmology: an update on animal models for retinal degenerations and dystrophies. Br J Ophthalmol. 2000;84:922–27. doi: 10.1136/bjo.84.8.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn P, Milam AH, Dunaief JL. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch’s membrane. Arch Ophthalmol. 2003;121:1099–105. doi: 10.1001/archopht.121.8.1099. [DOI] [PubMed] [Google Scholar]
- Hahn P, Qian Y, Dentchev T, Chen L, Beard J, Harris ZL, Dunaief JL. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc Natl Acad Sci U S A. 2004;101:13850–5. doi: 10.1073/pnas.0405146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider NB, Jacobson SG, Cideciyan AV, Swiderski R, Streb LM, Searby C, Beck G, Hockey R, Hanna DB, Gorman S. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet. 2000;24:127–31. doi: 10.1038/72777. [DOI] [PubMed] [Google Scholar]
- Haider NB, Naggert JK, Nishina PM. Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum Mol Genet. 2001;10:1619–26. doi: 10.1093/hmg/10.16.1619. [DOI] [PubMed] [Google Scholar]
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- Haley PJ. Species differences in the structure and function of the immune system. Toxicology. 2003;188:49–71. doi: 10.1016/s0300-483x(03)00043-x. [DOI] [PubMed] [Google Scholar]
- Hawes NL, Chang B, Hageman GS, Nusinowitz S, Nishina PM, Schneider BS, Smith RS, Roderick TH, Davisson MT, Heckenlively JR. Retinal degeneration 6 (rd6): a new mouse model for human retinitis punctata albescens. ARVO. 2000;41:3149–57. [PubMed] [Google Scholar]
- Hawes NL, Smith RS, Chang B, Davisson ML, Heckenlively JR, John SW. Mouse fundus photography and angiography: a catalogue of normal and mutant phenotypes. Mol Vis. 1999;15 [PubMed] [Google Scholar]
- Hayasaka S. Lysosomal enzymes in ocular tissues and diseases. Surv Ophthalmol. 1983;27:245–58. doi: 10.1016/0039-6257(83)90125-x. [DOI] [PubMed] [Google Scholar]
- He X, Hahn P, Iacovelli J, Wong R, King C, Bhisitkul R, Massaro-Giordano M, Dunaief JL. Iron homeostasis and toxicity in retinal degeneration. Prog Retin Eye Res. 2007;26:649–73. doi: 10.1016/j.preteyeres.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckenlively JR, Chang B, Erway LC, Peng C, Hawes NL, Hageman GS, Roderick TH. Mouse model for Usher syndrome: linkage mapping suggests homology to Usher type I reported at human chromosome 11p15. Proc Natl Acad Sci U S A. 1995;92:11100–4. doi: 10.1073/pnas.92.24.11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzlich AA, Tuo J, Chan CC. Peroxisome proliferator-activated receptor and age-related macular degeneration. PPAR Res. 2008;2008:389–507. doi: 10.1155/2008/389507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan MJ, Alvarado J. Studies on the human macula: IV. Aging changes in Bruch’s membrane. Arch Ophthalmol. 1967;77:410–20. doi: 10.1001/archopht.1967.00980020412022. [DOI] [PubMed] [Google Scholar]
- Hogg RE, Chakravarthy U. Visual function and dysfunction in early and late age related maculopathy. Prog Retin Eye Res. 2006;25:249–76. doi: 10.1016/j.preteyeres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–8. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson J, Tay W. Symmetrical central chorioretinal disease occurring in senile persons. R London Ophthalmol Hosp Rep. 1875;8:231–44. [Google Scholar]
- Imamura Y, Noda S, Hashizume K, Shinoda K, Yamaguchi M, Uchiyama S, Shimizu T, Mizushima Y, Shirasawa T, Tsubota K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc Natl Acad Sci U S A. 2006;103:11282–7. doi: 10.1073/pnas.0602131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Takaki Y, Fukami S, Tsubuki S, Saido TC. Region-specific reduction of A beta-degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J Neurosci Res. 2002;70:493–500. doi: 10.1002/jnr.10390. [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–2. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- Iyengar SK, Song D, Klein BE, Klein R, Schick JH, Humphrey J, Millard C, Liptak R, Russo K, Jun G, Lee KE, Fijal B, Elston RC. Dissection of genomewide-scan data in extended families reveals a major locus and oligogenic susceptibility for age-related macular degeneration. Am J Hum Genet. 2004;74:20–39. doi: 10.1086/380912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt NB, Javitt JC. The retinal oxysterol pathway: a unifying hypothesis for the cause of age-related macular degeneration. Curr Opin Ophthalmol. 2009;20:151–7. doi: 10.1097/ICU.0b013e32832af468. [DOI] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. The Journal of Neuroscience. 1998;18:8936–46. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachim SC, Bruns K, Lackner KJ, Pfeiffer N, Grus FH. Analysis of IgG antibody patterns against retinal antigens and antibodies to alpha-crystallin, GFAP, and alpha-enolase in sera of patients with “wet” age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2007;245:619–26. doi: 10.1007/s00417-006-0429-9. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer’s A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:11830–5. doi: 10.1073/pnas.192203399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PT, Betts KE, Radeke MJ, Hageman GS, Anderson DH, Johnson LV. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci USA. 2006;103:17456–61. doi: 10.1073/pnas.0606234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SE, Jomary C, Grist J, Stewart HJ, Neal MJ. Modulated expression of secreted frizzled-related proteins in human retinal degeneration. Neuroreport. 2000;11:3963–67. doi: 10.1097/00001756-200012180-00012. [DOI] [PubMed] [Google Scholar]
- Justilien V, Pang JJ, Renganathan K, Zhan X, Crabb JW, Kim SR, Sparrow JR, Hauswirth WW, Lewin AS. SOD2 knockdown mouse model of early AMD. Invest Ophthalmol Vis Sci. 2007;48:4407–20. doi: 10.1167/iovs.07-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaria RN. Arteriosclerosis, apolipoprotein E, and Alzheimer’s disease. Lancet. 1997;349:1174. doi: 10.1016/s0140-6736(05)63052-8. [DOI] [PubMed] [Google Scholar]
- Kameya S, Hawes NL, Chang B, Heckenlively JR, Naggert JK, Nishina PM. Mfrp, a gene encoding a frizzled related protein, is mutated in the mouse retinal degeneration 6. Hum Mol Genet. 2002;11:1879–86. doi: 10.1093/hmg/11.16.1879. [DOI] [PubMed] [Google Scholar]
- Karan G, Lillo C, Yang Z, Cameron DJ, Locke KG, Zhao Y, Thirumalaichary S, Li C, Birch DG, Vollmer-Snarr HR, Williams DS, Zhang K. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: a model for macular degeneration. Proc Natl Acad Sci U S A. 2005;102:4164–9. doi: 10.1073/pnas.0407698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ML, Robison WG., Jr Evidence of cell loss from the rat retina during senescence. Exp Eye Res. 1986;42:293–304. doi: 10.1016/0014-4835(86)90022-9. [DOI] [PubMed] [Google Scholar]
- Kawamata T, Akiguchi I, Yagi H. Neuropathological studies on strains of senescense-accelerated mice (SAM) with age related deficits in learning and memory. Exp Gerontol. 1997;1997:161–69. doi: 10.1016/s0531-5565(96)00063-0. [DOI] [PubMed] [Google Scholar]
- Keeler CE. The inheritance of a retinal abnormality in white mice. Proc Natl Acad Sci. 1924;10:329–33. doi: 10.1073/pnas.10.7.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Sadda S, Humayun MS, De Juan E, Jr, Melia BM, Green WR. Morphometric analysis of the macula in eyes with geographic atrophy due to age-related macular degeneration. Retina. 2002;22:464–70. doi: 10.1097/00006982-200208000-00011. [DOI] [PubMed] [Google Scholar]
- Kitado H, Higuchi K, Takeda T. Molecular Genetic Characterization of the Senescence-Accelerated Mouse (SAM) Strains. Journal of Gerontology. 1994;49:B247–B54. doi: 10.1093/geronj/49.6.b247. [DOI] [PubMed] [Google Scholar]
- Klaver CC, Kliffen M, Van Duijn CM, Hofman A, Cruts M, Grobbee DE, Van Broeckhoven C, De Jong PT. Genetic association of apolipoprotein E with age-related macular degeneration. Am J Hum Genet. 1998;63:200–6. doi: 10.1086/301901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ML, Schultz DW, Edwards A, Matise TC, Rust K, Berselli CB, Trzupek K, Weleber RG, Ott J, Wirtz MK, Acott TS. Age-related macular degeneration. Clinical features in a large family and linkage to chromosome 1q. Arch Ophthalmol. 1998;116:1082–88. doi: 10.1001/archopht.116.8.1082. [DOI] [PubMed] [Google Scholar]
- Klein R. Overview of progress in the epidemiology of age-related macular degeneration. Ophthalmic Epidemiol. 2007;14:184–7. doi: 10.1080/09286580701344381. [DOI] [PubMed] [Google Scholar]
- Klein R, Clegg L, Cooper LS, Hubbard LD, Klein BE, King WN, Folsom AR. Prevalence of age-related maculopathy in the Atherosclerosis Risk in Communities Study. Arch Ophthalmol. 1999;117:1203–10. doi: 10.1001/archopht.117.9.1203. [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, Sangiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–89. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliffen M, Lutgens E, Daemen MJ, De Muinck ED, Mooy CM, De Jong PT. The APO(*)E3-Leiden mouse as an animal model for basal laminar deposit. Br J Ophthalmol. 2000;84:1415–9. doi: 10.1136/bjo.84.12.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klomp LW, Farhangrazi ZS, Dugan LL, Gitlin JD. Ceruloplasmin gene expression in the murine central nervous system. J Clin Invest. 1996;98:207–15. doi: 10.1172/JCI118768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klomp LW, Gitlin JD. Expression of the ceruloplasmin gene in the human retina and brain: implications for a pathogenic model in aceruloplasminemia. Hum Mol Genet. 1996;5:1989–96. doi: 10.1093/hmg/5.12.1989. [DOI] [PubMed] [Google Scholar]
- Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA. 1997;94:12053–58. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leys A, Vanrenterghem Y, Van Damme B, Snyers B, Pirson Y, Leys M. Fundus changes in membranoproliferative glomerulonephritis type II. A fluorescein angiographic study of 23 patients. Graefes Arch Clin Exp Ophthalmol. 1991;229:406–10. doi: 10.1007/BF00166300. [DOI] [PubMed] [Google Scholar]
- Li D, Sun F, Wang K. Protein profile of aging and its retardation by caloric restriction in neural retina. Biochem Biophys Res Commun. 2004;318:253–8. doi: 10.1016/j.bbrc.2004.04.022. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–81. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Lu B, Malcuit C, Wang S, Girman S, Francis P, Lemieux L, Lanza R, Lund R. Long-term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cells. 2009;27:2126–35. doi: 10.1002/stem.149. [DOI] [PubMed] [Google Scholar]
- Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–8. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–30. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Majji AB, Cao J, Chang KY, Hayashi A, Aggarwal S, Grebe RR, De Juan EJ. Age-related retinal pigment epithelium and Bruch’s membrane degeneration in senescence-accelerated mouse. Invest Ophthalmol Vis Sci. 2000;41:3936–42. [PubMed] [Google Scholar]
- Malek G, Johnson LV, Mace BE, Saloupis P, Schmechel DE, Rickman DW, Toth CA, Sullivan PM, Bowes Rickman C. Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci U S A. 2005;102:11900–5. doi: 10.1073/pnas.0503015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maller J, George S, Purcell S, Fagerness J, Altshuler D, Daly MJ, Seddon JM. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055–59. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- Marmorstein AD, Marmorstein LY. The challenge of modeling macular degeneration in mice. TRENDS in Genetics. 2007;23:225–31. doi: 10.1016/j.tig.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Marmorstein LY, Mclaughlin PJ, Peachey NS, Sasaki T, Marmorstein AD. Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Hum Mol Genet. 2007;16:2423–32. doi: 10.1093/hmg/ddm199. [DOI] [PubMed] [Google Scholar]
- Martin DF, Debarge LR, Nussenblatt RB, Chan CC, Roberge FG. Synergistic effect of rapamycin and cyclosporin A in the treatment of experimental autoimmune uveoretinitis. J Immunol. 1995;154:922–7. [PubMed] [Google Scholar]
- Mata NL, Tzekov RT, Liu X, Weng J, Birch DG, Travis G. Delayed Dark-Adaptation and Lipofuscin Accumulation in abcr+/− Mice: Implications for Involvement of ABCR in Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 2001;42:1685–90. [PubMed] [Google Scholar]
- Matrisian LM. The matrix-degrading metalloproteinases. Bioessays. 1992;14:455–63. doi: 10.1002/bies.950140705. [DOI] [PubMed] [Google Scholar]
- Mcgeer EG, Klegeris A, Mcgeer PL. Inflammation, the complement system and the diseases of aging. Neurobiol Aging. 2005;26(Suppl 1):94–7. doi: 10.1016/j.neurobiolaging.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Mclaughlin PJ, Bakall B, Choi J, Liu Z, Sasaki T, Davis EC, Marmorstein AD, Marmorstein LY. Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Hum Molec Genet. 2007;16:3059–70. doi: 10.1093/hmg/ddm264. [DOI] [PubMed] [Google Scholar]
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–63. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- Miceli MV, Newsome DA, Tate DJ, Jr, Sarphie TG. Pathologic changes in the retinal pigment epithelium and Bruch’s membrane of fat-fed atherogenic mice. Curr Eye Res. 2000;20:8–16. [PubMed] [Google Scholar]
- Mishima H, Hasebe H. Some observations in the fine structure of age changes of the mouse retinal pigment epithelium. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1978;209:1–9. doi: 10.1007/BF00419157. [DOI] [PubMed] [Google Scholar]
- Mishima H, Kondo K. Extrusion of lysosomal bodies from apical mouse retinal pigment epithelium. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1981;216:209–17. doi: 10.1007/BF00408162. [DOI] [PubMed] [Google Scholar]
- Moore DJ, Hussain AA, Marshall J. Age-related variation in the hydraulic conductivity of Bruch’s membrane. Invest Ophthalmol Vis Sci. 1995;36:1290–97. [PubMed] [Google Scholar]
- Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–46. [PubMed] [Google Scholar]
- Neuner B, Komm A, Wellmann J, Dietzel M, Pauleikhoff D, Walter J, Busch M, Hense H-W. Smoking history and the incidence of age-related macular degeneration—Results from the Muenster Aging and Retina Study (MARS) cohort and systematic review and meta-analysis of observational longitudinal studies. Addict Behav. 2009;34:938–47. doi: 10.1016/j.addbeh.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Nussenblatt RB. Bench to bedside: new approaches to the immunotherapy of uveitic disease. Int Rev Immunol. 2002;21:273–89. doi: 10.1080/08830180212067. [DOI] [PubMed] [Google Scholar]
- Nussenblatt RB, Salinas-Carmona M, Waksman BH, Gery I. Cyclosporin A: alterations of the cellular immune response in S-antigen-induced experimental autoimmune uveitis. Int Arch Allergy Appl Immunol. 1983;70:289–94. doi: 10.1159/000233339. [DOI] [PubMed] [Google Scholar]
- Ogata N, Ohkuma H, Kanai K, Nango K, Takada Y, Uyama M. [Histological changes in the retinal pigment epithelium and Bruch’s membrane in senescence accelerated mouse] Nippon Ganka Gakkai Zasshi. 1992;96:180–9. [PubMed] [Google Scholar]
- Organization, W.H. Magnitude and causes of visual impairment. 2009. Vol. Fact Sheet No 282. [Google Scholar]
- Panda-Jonas S, Jonas JB, Jakobczyk-Zmija M. Retinal photoreceptor density decreases with age. Ophthalmology. 1995;102:1853–9. doi: 10.1016/s0161-6420(95)30784-1. [DOI] [PubMed] [Google Scholar]
- Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;1:893–5. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- Patel BN, David S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem. 1997;272:20185–90. doi: 10.1074/jbc.272.32.20185. [DOI] [PubMed] [Google Scholar]
- Patel BN, Dunn RJ, Jeong SY, Zhu Q, Julien JP, David S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J Neurosci. 2002;22:6578–86. doi: 10.1523/JNEUROSCI.22-15-06578.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Chan CC. Immunopathological aspects of age-related macular degeneration. Semin Immunopathol. 2008;30:97–110. doi: 10.1007/s00281-008-0112-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, Ohbayashi M, Nugent AK, Ramchand K, Toda M, Chau KY, Bunce C, Webster A, Bird AC, Ono SJ, Chong V. Circulating anti-retinal antibodies as immune markers in age-related macular degeneration. Immunology. 2005;115:422–30. doi: 10.1111/j.1365-2567.2005.02173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauleikhoff D, Harper CA, Marshall J, Bird AC. Aging changes in Bruch’s membrane. A histochemical and morphologic study. Ophthalmology. 1990;97:171–8. [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 1987;262:14352–60. [PubMed] [Google Scholar]
- Purcell-Huynh DA, Farese RV, Jr, Johnson DF, Flynn LM, Pierotti V, Newland DL, Linton MF, Sanan DA, Young SG. Transgenic mice expressing high levels of human apolipoprotein B develop severe atherosclerotic lesions in response to a high-fat diet. J Clin Invest. 1995;95:2246–57. doi: 10.1172/JCI117915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines MF, Duvall-Young J, Short CD. Fundus changes in mesangiocapillary glomerulonephritis type II: vitreous fluorophotometry. Br J Ophthalmol. 1989;73:411–31. doi: 10.1136/bjo.73.11.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoczy EP, Meaghan JT, Yu Nusinowitz S, Chang B, JRH Mouse models of age-related macular degeneration. Experimental Eye Research. 2006;82:741–52. doi: 10.1016/j.exer.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Rakoczy PE, Lai CM, Baines M, Di Grandi S, Fitton JH, Constable IJ. Modulation of cathepsin D activity in retinal pigment epithelial cells. Biochem J. 1997;324(Pt 3):935–40. doi: 10.1042/bj3240935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoczy PE, Zhang D, Robertson T, Barnett NL, Papadimitriou J, Constable IJ, Lai CM. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am J Pathol. 2002;161:1515–24. doi: 10.1016/S0002-9440(10)64427-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz-Prag D, Ayyagari R, Fariss RN, Mandal MN, Vasireddy V, Majchrzak S, Webber AL, Bush RA, Salem N, Jr, Petrukhin K, Sieving PA. Haploinsufficiency is not the key mechanism of pathogenesis in a heterozygous Elovl4 knockout mouse model of STGD3 disease. Invest Ophthalmol Vis Sci. 2006;47:3603–11. doi: 10.1167/iovs.05-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque RS, Rosales AA, Jingjing L, Agarwal N, Al-Ubaidi MR. Retina-derived microglial cells induce photoreceptor cell death in vitro. Brain Res. 1999;836:110–19. doi: 10.1016/s0006-8993(99)01625-x. [DOI] [PubMed] [Google Scholar]
- Ross RJ, Zhou M, Shen D, Fariss RN, Ding X, Bojanowski CM, Tuo J, Chan CC. Immunological protein expression profile in Ccl2/Cx3cr1 deficient mice with lesions similar to age-related macular degeneration. Exp Eye Res. 2008;86:675–83. doi: 10.1016/j.exer.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf M, Ivandic B, Winkler J, Schmidt-Erfurth U. [Accumulation of lipid particles in Bruch’s membrane of LDL receptor knockout mice as a model of age-related macular degeneration] Ophthalmologe. 2004;101:715–9. doi: 10.1007/s00347-003-0942-8. [DOI] [PubMed] [Google Scholar]
- Saido TC. Alzheimer’s disease as proteolytic disorders: anabolism and catabolism of beta-amyloid. Neurobiol Aging. 1998;19:S69–75. doi: 10.1016/s0197-4580(98)00033-5. [DOI] [PubMed] [Google Scholar]
- Sallo FB, Bereczki E, Csont T, Luthert PJ, Munro P, Ferdinandy P, Santha M, Lengyel I. Bruch’s membrane changes in transgenic mice overexpressing the human biglycan and apolipoprotein b-100 genes. Exp Eye Res. 2009;89:178–86. doi: 10.1016/j.exer.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Sandbach JM, Coscun PE, Grossniklaus HE, Kokoszka JE, Newman NJ, Wallace DC. Ocular pathology in mitochondrial superoxide dismutase (Sod2)-deficient mice. Invest Ophthalmol Vis Sci. 2001;42:2173–8. [PubMed] [Google Scholar]
- Sanyal S, Hawkins RK. Development and degeneration of retina in rds mutant mice: effects of light on the rate of degeneration in albino and pigmented homozygous and heterozygous mutant and normal mice. Vision Res. 1986;26:1177–85. doi: 10.1016/0042-6989(86)90099-4. [DOI] [PubMed] [Google Scholar]
- Sarks SH. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol. 1976;60:324–41. doi: 10.1136/bjo.60.5.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Cote J, Page WF, Aggen SH, Neale MC. The US twin study of age-related macular degeneration: relative roles of genetic and environmental influences. Arch Ophthalmol. 2005;123:321–7. doi: 10.1001/archopht.123.3.321. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Gensler G, Milton RC, Klein ML, Rifai N. Association between C-reactive protein and age-related macular degeneration. JAMA. 2004;291:704–10. doi: 10.1001/jama.291.6.704. [DOI] [PubMed] [Google Scholar]
- Shelley EJ, Madigan MC, Natoli R, Penfold PL, Provis JM. Cone degeneration in aging and age-related macular degeneration. Arch Ophthalmol. 2009;127:483–92. doi: 10.1001/archophthalmol.2008.622. [DOI] [PubMed] [Google Scholar]
- Shepherd FA, Sridhar SS. Angiogenesis inhibitors under study for the treatment of lung cancer. Lung Cancer. 2003;41(Suppl 1):S63–72. doi: 10.1016/s0169-5002(03)00144-2. [DOI] [PubMed] [Google Scholar]
- Shoji M, Okada M, Ohta A, Higuchi K, Hosokawa M, Honda Y. A morphological and morphometrical study of the retina in aging SAM mice. Ophthalmic Res. 1998;30:172–9. doi: 10.1159/000055471. [DOI] [PubMed] [Google Scholar]
- Sickel W. Electrical and metabolic manifestations of receptor and higherorder neuron activity in vertebrate retina. Adv Exp Med Biol. 1972;24:101–18. doi: 10.1007/978-1-4684-8231-7_11. [DOI] [PubMed] [Google Scholar]
- Snow KK, Seddon JM. Do age-related macular degeneration and cardiovascular disease share common antecedents? Ophthalmic Epidemiol. 1999;6:125–43. doi: 10.1076/opep.6.2.125.1558. [DOI] [PubMed] [Google Scholar]
- Sorsby A, Mason MEJ, Gardner N. A fundus dystrophy with unusual features. Br J Ophthalmol. 1949;33 doi: 10.1136/bjo.33.2.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spraul CW, Grossniklaus HE. Characteristics of Drusen and Bruch’s membrane in postmortem eyes with age-related macular degeneration. Arch Ophthalmol. 1997;115:267–73. doi: 10.1001/archopht.1997.01100150269022. [DOI] [PubMed] [Google Scholar]
- Stargardt K. Uber familiare, progressive degeneration in der maculagegend des auges. Graefes Arch Ophthalmol. 1909;71:534–50. [Google Scholar]
- Starita C, Hussain AA, Pagliarini S, Marshall J. Hydrodynamics of ageing Bruch’s membrane: implications for macular disease. Exp Eye Res. 1996;62:565–72. doi: 10.1006/exer.1996.0066. [DOI] [PubMed] [Google Scholar]
- Stone EM, Braun TA, Russell SR, Kuehn MH, Lotery AJ, Moore PA, Eastman CG, Casavant TL, Sheffield VC. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346–53. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- Stone EM, Fingert JH, Alward WLM, Nguyen TD, Polansky JR. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275:668–70. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- Stone EM, Lotery AJ, Munier FL, Heon E, Piguet B, Guymer RH, Vandenburgh K, Cousin P, Nishimura D, Swiderski RE. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- Stone EM, Nichols BE, Kimura AE, Weingeist TA, Drack A, Sheffield VC. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch Ophthalmol. 1994;112:765–72. doi: 10.1001/archopht.1994.01090180063036. [DOI] [PubMed] [Google Scholar]
- Strohmeyer R, Ramirez M, Cole GJ, Mueller K, Rogers J. Association of factor H of the alternative pathway of complement with agrin and complement receptor 3 in the Alzheimer’s disease brain. J Neuroimmunol. 2002;131:135–46. doi: 10.1016/s0165-5728(02)00272-2. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–80. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- Swaroop A, Branham KE, Chen W, Abecasis G. Genetic susceptibility to age-related macular degeneration: a paradigm for dissecting complex disease traits. Hum Mol Genet. 2007;16:R174–82. doi: 10.1093/hmg/ddm212. [DOI] [PubMed] [Google Scholar]
- Szel A, Rohlich P, Caffe AR, Juliusson B, Aguirre G, Van Veen T. Unique topographic separation of two spectral classes of cones in the mouse retina. J Comp Neurol. 1992;325:327–42. doi: 10.1002/cne.903250302. [DOI] [PubMed] [Google Scholar]
- Takada Y, Ogata N, Ohkuma H, Uyama M. [Age-related changes in Bruch’s membrane of the senescence accelerated mouse] Nippon Ganka Gakkai Zasshi. 1993;97:595–601. [PubMed] [Google Scholar]
- Takada Y, Uyama M, Ohkuma H, Ogata N, Matsushima M, Deguchi J, Sugasawa K. [Immunohistological study in Bruch’s membrane of senescence accelerated mouse] Nippon Ganka Gakkai Zasshi. 1994;98:955–61. [PubMed] [Google Scholar]
- Takeda T, Hosokawa M, Higuchi K, Hosono M, Akiguchi I, Katoh H. A novel murine model of aging, Senescence-Accelerated Mouse (SAM) Arch Gerontol Geriatr. 1994;19:185–92. doi: 10.1016/0167-4943(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Takeda T, Hosokawa M, Higuchi K. Senescence-accelerated mouse (SAM): A novel murine model of senescence. Experimental Gerontology. 1997;32:105–09. doi: 10.1016/s0531-5565(96)00036-8. [DOI] [PubMed] [Google Scholar]
- Timpl R, Sasaki T, Kostka G, Chu ML. Fibulins: a versatile family of extracellular matrix proteins. Nat Rev Mol Cell Biol. 2003;4:479–89. doi: 10.1038/nrm1130. [DOI] [PubMed] [Google Scholar]
- Tree M. Familial hyaline dystrophy in the fundus oculi or Doyne’s family honeycomb “choroiditis”. Br J Ophthalmol. 1937;21:65–91. doi: 10.1136/bjo.21.2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi CSK, Egashira K, Qiao H, Hisatomi T, Nakao S, Ishibashi M, Charo If, Sakamoto T, Murata T, Ishibashi T. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J Leukoc Biol. 2003;74:25–32. doi: 10.1189/jlb.0902436. [DOI] [PubMed] [Google Scholar]
- Tuo J, Bojanowski CM, Zhou M, Shen D, Ross RJ, Rosenberg KI, Cameron DJ, Yin C, Kowalak JA, Zhuang Z, Zhang K, Chan CC. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:3827–36. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo J, Smith BC, Bojanowski CM, Meleth AD, Gery I, Csaky KG, Chew EY, Chan CC. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–99s. doi: 10.1096/fj.04-1862fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchihara T, Akiyama H, Kondo H, Ikeda K. Activated microglial cells are colocalized with perivascular deposits of amyloid-beta protein in Alzheimer’s disease brain. Stroke. 1997;28:1948–50. doi: 10.1161/01.str.28.10.1948. [DOI] [PubMed] [Google Scholar]
- Umeda S, Ayyagari R, Allikmets R, Suzuki MT, Karoukis AJ, Ambasudhan R, Zernant J, Okamoto H, Ono F, Terao K, Mizota A, Yoshikawa Y, Tanaka Y, Iwata T. Early-onset macular degeneration with drusen in a cynomolgus monkey (Macaca fascicularis) pedigree: exclusion of 13 candidate genes and loci. Invest Ophthalmol Vis Sci. 2005;46:683–91. doi: 10.1167/iovs.04-1031. [DOI] [PubMed] [Google Scholar]
- Umeda S, Suzuki MT, Okamoto H, Ono F, Mizota A, Terao K, Yoshikawa Y, Tanaka Y, Iwata T. Molecular composition of drusen and possible involvement of anti-retinal autoimmunity in two different forms of macular degeneration in cynomolgus monkey (Macaca fascicularis) FASEB J. 2005;19:1683–5. doi: 10.1096/fj.04-3525fje. [DOI] [PubMed] [Google Scholar]
- Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–93. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- Van Den Maagdenberg AM, Hofker MH, Krimpenfort PJ, De Bruijn I, Van Vlijmen B, Van Der Boom H, Havekes LM, Frants RR. Transgenic mice carrying the apolipoprotein E3-Leiden gene exhibit hyperlipoproteinemia. J Biol Chem. 1993;268:10540–5. [PubMed] [Google Scholar]
- Van Der Schaft TL, Mooy CM, De Bruijn WC, Oron FG, Mulder PG, De Jong PT. Histologic features of the early stages of age-related macular degeneration. A statistical analysis. Ophthalmology. 1992;99:278–86. doi: 10.1016/s0161-6420(92)31982-7. [DOI] [PubMed] [Google Scholar]
- Van Nie R, Ivanyi D, Demant P. A new H-2-linked mutation, rds, causing retinal degeneration in the mouse. Tissue Antigens. 1978;12:106–8. doi: 10.1111/j.1399-0039.1978.tb01305.x. [DOI] [PubMed] [Google Scholar]
- Vasireddy V, Jablonski MM, Khan NW, Wang XF, Sahu P, Sparrow JR, Ayyagari R. Elovl4 5-bp deletion knock-in mouse model for Stargardt-like macular degeneration demonstrates accumulation of ELOVL4 and lipofuscin. Exp Eye Res. 2009 doi: 10.1016/j.exer.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasireddy V, Jablonski MM, Mandal MNA, Raz-Prag D, Wang XF, Nizol L, Iannaccone A, Musch DC, Bush RA, Salem N., Jr Elovl4 5-bp-deletion knock-in mice develop progressive photoreceptor degeneration. ARVO. 2006;47:45–58. doi: 10.1167/iovs.06-0353. [DOI] [PubMed] [Google Scholar]
- Vasireddy V, Vijayasarathy C, Huang J, Wang XF, Jablonski MM, Petty HR, Sieving PA, Ayyagari R. Stargardt-like macular dystrophy protein ELOVL4 exerts a dominant negative effect by recruiting wild-type protein into aggresomes. Mol Vis. 2005;11:665–76. [PubMed] [Google Scholar]
- Vingerling JR, Hofman A, Grobbee DE, De Jong PT. Age-related macular degeneration and smoking. The Rotterdam Study. Arch Ophthalmol. 1996;114:1193–6. doi: 10.1001/archopht.1996.01100140393005. [DOI] [PubMed] [Google Scholar]
- Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–9. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, Mccarthy M, Mccombie WR, Mclaren S, Mclay K, Mcpherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O’connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Weber BH, Lin B, White K, Kohler K, Soboleva G, Herterich S, Seeliger MW, Jaissle GB, Grimm C, Reme C, Wenzel A, Asan E, Schrewe H. A mouse model for Sorsby fundus dystrophy. Invest Ophthalmol Vis Sci. 2002;43:2732–40. [PubMed] [Google Scholar]
- Weber BHF, Vogt G, Pruett RC, Stohr H, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet. 1994;8:352–56. doi: 10.1038/ng1294-352. [DOI] [PubMed] [Google Scholar]
- Weller RO, Nicoll JA. Cerebral amyloid angiopathy: pathogenesis and effects on the ageing and Alzheimer brain. Neurol Res. 2003;25:611–6. doi: 10.1179/016164103101202057. [DOI] [PubMed] [Google Scholar]
- Wellington CL. Cholesterol at the crossroads: Alzheimer’s disease and lipid metabolism. Clin Genet. 2004;66:1–16. doi: 10.1111/j.0009-9163.2004.00280.x. [DOI] [PubMed] [Google Scholar]
- Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005;24:275–306. doi: 10.1016/j.preteyeres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Wiederanders B, Oelke B. Accumulation of inactive cathepsin D in old rats. Mech Ageing Dev. 1984;24:265–71. doi: 10.1016/0047-6374(84)90112-x. [DOI] [PubMed] [Google Scholar]
- Wikler KW, Rakic P. Distribution of photoreceptor subtypes in the retina of diurnal and nocturnal primates. J Neurosci. 1990;10:3390–401. doi: 10.1523/JNEUROSCI.10-10-03390.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Ghiso J, Frangione B. Biology of A beta amyloid in Alzheimer’s disease. Neurobiol Dis. 1997;4:313–28. doi: 10.1006/nbdi.1997.0147. [DOI] [PubMed] [Google Scholar]
- Wolfensberger TJ. The Retinal Pigment Epithelium. Oxford University Press; 1998. Toxicology of the retinal pigment epithelium; pp. 621–47. [Google Scholar]
- Won J, Smith RS, Peachey NS, Wu J, Hicks WL, Naggert JK, Nishina PM. Membrane frizzled-related protein is necessary for the normal development and maintenance of photoreceptor outer segments. Vis Neurosci. 2008;25:563–74. doi: 10.1017/S0952523808080723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TY, Loon S-C, Saw S-M. The epidemiology of age related eye diseases in Asia. Br J Ophthalmol. 2006;90:506–11. doi: 10.1136/bjo.2005.083733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Chen M, Forrester JV. Para-inflammation in the ageing retina. Prog Retin Eye Res. 2009;28:348–68. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Xu H, Chen M, Manivannan A, Lois N, Forrester JV. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell. 2008;7:58–68. doi: 10.1111/j.1474-9726.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- Xu YK, Nusse R. The Frizzled CRD domain is conserved in diverse proteins including several receptor tyrosine kinases. Curr Biol. 1998;8:R405–R06. doi: 10.1016/s0960-9822(98)70262-3. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Ohno-Matsui K, Ichinose S, Sato T, Iwata N, Saido TC, Hisatomi T, Mochizuki M, Morita I. The potential role of amyloid beta in the pathogenesis of age-related macular degeneration. J Clin Invest. 2005;115:2793–800. doi: 10.1172/JCI24635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol. 1969;42:392–403. doi: 10.1083/jcb.42.2.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack DJ, Dean M, Molday RS, Nathans J, Redmond TM, Stone EM, Swaroop A, Valle D, Weber BH. What can we learn about age-related macular degeneration from other retinal diseases? Mol Vis. 1999;5:30. [PubMed] [Google Scholar]
- Zhang D, Brankov M, Makhija MT, Robertson T, Helmerhorst E, Papadimitriou JM, Rakoczy PE. Correlation between inactive cathepsin D expression and retinal changes in mcd2/mcd2 transgenic mice. Invest Ophthalmol Vis Sci. 2005;46:3031–8. doi: 10.1167/iovs.04-1510. [DOI] [PubMed] [Google Scholar]
- Zhang D, Lai MC, Constable IJ, Rakoczy PE. A model for a blinding eye disease of the aged. Biogerontology. 2002;3:61–6. doi: 10.1023/a:1015259413857. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML, Allikmets R, Zack DJ, Kakuk LE, Lagali PS, Wong PW, Macdonald IM, Sieving PA, Figueroa DJ, Austin CP, Gould RJ, Ayyagari R, Petrukhin K. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]