

Abstract
The hippocampus is often injured in neonatal stroke. We have investigated the effect of erythropoietin (EPO) on oxygen-glucose deprived hippocampal slices and hypoxic progenitor cells. EPO improved survival of the organotypic hippocampal slices with significantly less cell death in the dentate gyrus and an increased number of proliferating cells 4-5 days after insult. Significantly fewer markers of neurogenesis were seen after the insult but when EPO was added to the culture medium, neurogenesis was sustained. When hippocampal progenitor cultures were stimulated into differentiation, more cells chose a neuronal cell fate when treated with EPO. These findings support the hypothesis that EPO not only prevents ischemia induced cell death but promotes neuronal cell fate committment in in-vitro models of neonatal stroke.
Keywords: neurogenesis, hypoxia, erythropoietin, hippocampus, differentiation, neonate
Introduction
Stroke in the neonatal period occurs in 1/4000 and is as common as in the adult (Nelson, 2007), with over 80% of the surviving newborns having neurodevelopmental sequelae (Lee et al., 2005). The pathological process of neonatal stroke is a dynamic process, evolving weeks after the event, through phases of oxidative stress, inflammation and repair (Ferriero, 2004). Neuroprotective therapies are being investigated, each targeting a specific step of the injury response (Gonzalez and Ferriero, 2008).
Many studies now confirm that neurogenesis occurs throughout the life span of mammalian and non-mammalian species (Altman, 1969; Alvarez-Buylla et al., 2002). The subventricular zone (SVZ) and the dentate gyrus (DG) of the hippocampus are the two brain regions where adult neurogenesis occurs (Gage, 2000). The role of the hippocampus in learning and memory has been known for decades (Milner, 1972) and is commonly injured in stroke (Kadam et al., 2008). Increased neurogenesis is the intrinsic response of adult brain to ischemic and traumatic injuries (Dash et al., 2001; Parent et al., 2002) with increased neural progenitor cell proliferation occuring in adult models of global and focal ischemia (Jin et al., 2001; Liu et al., 1998). Most of the surviving newly formed cells in the DG differentiate into mature neurons by 3–4 weeks after ischemia, whereas about 10–20% of the newly generated cells differentiate into astrocytes in the granule cell layer and the hippocampal hilus (Komitova et al., 2006). In the newborn brain however, oligogenesis is favored over neurogenesis after hypoxia-ischemia (Zaidi et al., 2004).
Erythropoietin (EPO) is a cytokine superfamily glycoprotein under the control of the oxygen-sensitive transcription factor hypoxia-inducible factor-1 (HIF-1). EPO–EPO receptor (EPOR) signaling is required for normal brain development (Juul, 2002). EPOR has been demonstrated on neurons, astrocytes, microglia and oligodendrocytes (Nagai et al., 2001). The basal expression of EPO is found in neurons and astrocytes, while post-ischemic EPO expression is localized to endothelial cells, microglia/macrophage-like cells, and reactive astrocytes (Bernaudin et al., 1999). In adult and neonatal animal models, EPO has a neuroprotective role when administered exogenously through anti-apoptosis, neuroregeneration and anti-inflammation (Gonzalez et al., 2007; Noguchi et al., 2007; Sola et al., 2005). EPO can stimulate neuronal progenitor cell production from pluripotent progenitor cells (Shingo et al., 2001) and can increase both neurogenesis in the subventricular zone and migration of neuronal progenitors into the ischemic cortex and striatum of neonatal rats (Iwai et al., 2007). However, there are no existing data regarding EPO effects on neuronal survival and neurogenesis in the hippocampus after hypoxia-ischemia in the neonatal period. Therefore we hypothesized that EPO would ameliorate cell death in the dentate gyrus of the hippocampus and alter cell fate determination to promote neurogenesis. Using an organotypic hippocampal slice model that preserves cell connectivity we show that EPO prevents cell death & promotes neurogenesis. Furthermore we validate the change in cell fate commitment with a hypoxia challenge to hippocampal progenitor cells, documenting neurogenesis and an increase in EPO signaling via induced EPO receptor expression.
Methods
Organotypic Hippocampal Slice Cultures (OHSC)
All animals were cared for following procedures approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco. OHSC were prepared from 7-day-old Sprague-Dawley rats (Charles River, Wilmington, MA). Rat pups were anesthetized with 2-3% isofluorane, decapitated and their hippocampi dissected out and placed in 4°C Gey's Balanced Salt Solution (UCSF Cell Culture Facility, San Francisco, CA). Next, the hippocampi were transversely sliced (400 μm) with a tissue slicer (Siskiyou Design Instruments, Grants Pass, OR), transferred onto 30 mm diameter membrane inserts (Millicell-CM; Millipore, Bedford, MA), placing 6 slices / insert. The inserts were placed into six-well culture trays adding 1.2 ml of culture medium per well. The slice culture medium consisted of 50% Minimal Essential Medium (Eagles with Earle's Balanced Salt Solution; UCSF Cell Culture Facility), 25% Earle's balanced salt solution (UCSF Cell Culture Facility), 25% heat-inactivated horse serum (UCSF Cell Culture Facility), Penicillin-Streptomycin 50 μg/ml, and the glucose 1.8 mg/ml. The OHSC were grown in an incubator in 5% CO2 / air at 37°C for 9 days, with the media changed every second day. The slices were selected with propidium iodide (PI, 2 μg/ml; Invitrogen) before further analysis to exclude those damaged during processing.
In Vitro Oxygen and Glucose Deprivation (OGD)
Before OGD, hippocampal slices were assigned randomly to different treatment groups: four groups received OGD and one of the treatments: vehicle (5 μl of 0.1M PBS), recombinant human erythropoietin (EPO; R&D Systems; 1 IU/ml of medium; the dose was established by a dose response curve with greatest effect at the selected dose, data not shown); anti-EPOR antibody (M-20, Santa Cruz; 2.5 μg/ml of medium); or wortmannin (Sigma-Aldrich; 100 nM). An additional group was not exposed to OGD and served as the control group. Anti-EPOR was used with the intention to block the EPO-EPOR signaling pathway at the receptor site. Wortmannin was chosen to block downstream EPO signaling since it is a specific PI3K inhibitor, blocking one of the signaling pathways upstream of AKT (Kilic et al., 2005).
Hippocampal slices were exposed to OGD by transferring the inserts to glass vials containing 20 ml of glucose-free artificial rat spinal fluid (aCSF), which was bubbled for at least 30 minutes prior and throughout the experiment with 95% N2 / 5% CO2. The partial pressure of oxygen of this solution was less than 2 mmHg (measured with a Clark oxygen electrode; Cameron Instrument Co., Port Aransas, TX). The vials were immersed in a water bath so that aCSF temperature was 37° ±0.2°C, monitored with a thermocouple thermometer. After 10min of OGD, slices were removed and recovered in fresh culturing medium with the added treatment (if applicable), prewarmed to 37°C. An additional group of OHSC slices was used initially as OGD-sham, where instead of aCSF normal culturing medium was used and was bubbled with 95% air / 5% CO2; we have not found any significant difference from the non-OGD control and therefore omitted this data. The aCSF was composed of NaCl (124 mM), NaHCO3 (25.7 mM), KCl (4.0 mM), MgCl2 (1.0 mM), CaCl2 (2.5 mM), KHPO (1.0 mM), with the pH adjusted to 7.30.
Cell viability after OGD
Cell viability after OGD was assessed in a separate set of experiments using PI fluorescence. At the beginning of the experiment, PI was added to the OHSC medium (5 μg/ml) for 30 min, after which the medium containing PI was replaced with Earl's basic salt solution and washed with it 4 more times. The fluorescent digital images of OHSC were taken using a Nikon Diaphot 200 inverted microscope (Nikon Corporation, Tokyo, Japan) and SPOT Jr. Digital Camera (Diagnostic Instruments Inc., Sterling Heights, MI). Excitation light wavelength was 490 nm and emission was 590 nm. All OHSC except those in the control group were then exposed to OGD, placed back in normal culture medium and treated according to the treatment groups they belonged to (described under OGD). Twenty-four hours later they were re-imaged the same way. Cells were then exposed for 1 hour to an Earle's solution containing 100 uM NaNC (sodium cyanide) and 1 mM iodoacetic acid to kill all living cells, and re-imaged for the third time.
The images were analyzed with ImageJ software (version 1.42k; U. S. National Institutes of Health, Bethesda, MD). Mean PI fluorescence was measured for the CA1, CA3 and DG regions as well as for the whole slice. Cell death was expressed as the percentage of total death using the following calculation: (PI fluorescence after treatment – PI fluorescence before tretament) / PI fluorescence after total death * 100.
Hippocampal precursor cell isolation and culture
Hippocampal precursor cells were isolated as described previously (Sall et al., 2009). Sprague-Dawley rats were separated from the dam on postnatal day 2 and decapitated. Hippocampi were immediately dissected out and placed in 10mL ice cold Hanks Basic Salt Solution without calcium. Whole hippocampi pooled from 10 animals were collected by centrifugation for 2 minutes at 300rcf (relative centrifugal force). Supernatant was removed and hippocampi were minced with a razor blade then triturated 5 times using a 200 μl pipette. Cells were then re-suspended in 10mL Hanks Basic Salt Solution for 2 minutes. After settling for 2 minutes the supernatant was transferred to a new tube. Cells were then collected by centrifugation at 300rcf for 5 minutes. Supernatant was discarded and cells were re-suspended in proliferation medium consisting of 3:1 Dubelco's Modified Eagles Medium: Ham's F12 (UCSF cell culture facility), 1% penicillin and streptomycin, 1× B-27 supplement (Invitrogen, Grand Isle, NY), 20ng/mL basic fibroblast growth factor (Chemicon, Temecula, CA), 0.75units heparin/ml (Abraxis, Schaumburg, IL). HPC were then grown in an incubator in 5% CO2 / air, at 37°C with media changed 3× per week. Non-adherent proliferating neural precursor cells were transferred to a new cell culture flask every 5 to 7 days to purify multipotent HPCs from differentiated progeny. HPC had been grown in culture for 2 to 3 weeks at the time of use.
Before plating the cells into 8 well chamber slides (Thermo Fisher Scientific, Rochester, NY), the chambers were coated with poly-L-ornithine and laminin, washed with PBS and than filled with 250 μl of culturing medium. Depending on the treatment groups, EPO was added to some wells (1 IU/ml).
In vitro hypoxia of HPC and differentiation
In vitro hypoxia was induced by placing the 8 well chamber slides carrying the HPC in a modular incubator chamber (Billups-Rothenberg, Del Mar, CA) which was flushed with 95% N2 / 5% CO2 for 15 min, decreasing the PO2 to 0 mmHg, as measured with the MiniOX I oxygen analyzer (MSA Medical Products, Pittsburgh, PA), and then sealed. The slides were left in the chamber at 37°C for 4 hours and afterwards placed back into the standard incubator conditions of 95% air / 5% CO2.
To differentiate HPC, basic fibroblast growth factor containing medium was replaced with differentiation medium consisting of Neurobasal-A (Invitrogen, Grand Isle NY), B27 supplement, 1% penicillin-streptomycin (UCSF cell culture facility, San Francisco, CA), L-glutamine (Invitrogen, Carlsbad CA), and 5% fetal bovine serum (UCSF cell culture facility, San Francisco, CA). After 24 hours, the medium was replaced with a serum free differentiation medium.
Western blot analysis (WB)
Groups of OHSC were used for Western blot analysis 72h after OGD, during which time the medium was not changed. The inserts carrying OHSC were flushed briefly with ice cold 0.1 M phosphate-buffered saline (PBS) before the slices were transferred into glass tubes containing ice cold lysis buffer composed of HEPES pH 7.9 (10mM), KCl (10mM), EDTA (0.1 mM), and EGTA (0.1 mM). Protease inhibitors and reducing agents were also used: pepstatin A (2 μg/ml), phenylmethylsulfonyl fluoride (PMSF; 0.5 mM), a protease inhibitor cocktail tablet (Roche diagnostics GmbH, Mannheim, Germany), and dithiothreitol (DTT; 1 mM). Protein concentration was determined by the Bradford method (BioRad, Richmond CA) using bovine serum albumin (BSA) as the standard. Equal amounts cytoplasmic protein (40 μg) were separated on 4 – 12% SDS polyacrylamide gels (Invitrogen Life Technologies, CA). Rainbow recombinant protein molecular weight marker (Amersham) was used as a size reference. Proteins were transferred to PVDF membranes (BioRad Laboratories, CA). The membranes were incubated in blocking buffer, 5% nonfat dry milk in TBST (20 mM Tris – HCl, pH 7.6, 137 mM NaCl, 0.1% Tween-20), at room temperature for 2 h with rotation. The membranes were then incubated with rabbit antibody against doublecortin (DCX; Cell Signaling, 1:1000) overnight at 4°C. Mouse anti-β-actin monoclonal antibody (Santa Cruz Biotechnology, 1:2000) was used as a loading control for protein quantization. Following rinses with TBST, the membranes were incubated with peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies (Santa Cruz Biotechnology, 1:2000) in blocking solution for 1 h. After rinses with TBST, signal of bound antibodies was developed by enhanced chemiluminescence (Amersham Life Science, Arlington Heights, IL). ImageJ was used to measure the optical densities of the protein signals on scans of X-ray films. The relative optical density was calculated using the optical density of DCX protein signals divided by the optical density of loading control (β-actin) for each experiment.
To evaluate the EpoR expression in HPC after OGD, the cells were kept in proliferation medium, exposed to 4h of hypoxia, and collected in lysis buffer 24 hours after start of hypoxia. Collected samples were then analyzed as above.
Immunohistochemistry
At 9 DIV the OHSC were exposed to OGD, treated according to the treatment group they belonged to, and incubated under standard conditions for 5 additional days, changing the medium on the third and fourth day. On the fourth day, 20 μM 5-bromo-2-deoxyuridine (BrdU, Sigma) per well was also added to culturing medium. After five days, cultures were washed briefly in 0.1 M ice-cold PBS and fixed in ice-cold 4% paraformaldehyde in PBS for 1 hour. After 24 h incubation with 30% sucrose, OHSC were cryosectioned (Leica CM 1850, Leica Instruments, Nussloch, Germany) and 20 μm coronal sectioned slices were used for immunodetection. DNA denaturation was achieved by treatment with 2 M HCl at 37 °C for 10 min and 500 mM boric acid pH 8.4 at room temperature for 10 minutes. Thereafter OHSC were incubated for 2 hours in blocking solution (10% goat serum, 0.3% Triton X-100 in PBS) and overnight with mouse anti-BrdU antibody (1:500; BD Biosciences) or rat anti-BrdU antibody (1:200; Abcam) and rabbit anti-β-III tubulin antibody (Tuj1; 1:2000; Covance), mouse anti-GFAP antibody (1:500; Millipore), mouse anti-ED1 antibody (1:50; Millipore) or rabbit anti-NG2 antibody (1:1000; Millipore) in blocking solution containing 2.5% goat serum at 4 °C. After rinsing with PBS, the slices were incubated for 2 h at room temperature with secondary Alexa Fluor 488 and/or 568 (Invitrogen, 1:500). After rinsing, the slices were exposed to DAPI for 5 minutes and then coverslipped with ProLong Gold (Invitrogen). Controls included omission of primary antibodies. The images were taken using a Nikon TE2000 C1 spectral confocal microscope (Nikon, Tokyo, Japan) at the Nikon Imaging Center, UCSF. To estimate the number of proliferating cells in the DG 4-5 days after OGD, the OHSC images were analyzed using ImageJ software. The mean BrdU fluorescence in the region was measured for the control, vehicle and EPO-treated groups of OHSC.
The undifferentiated HPC were placed in 8 well chamber slides, fixed and then processed using a similar protocol as for OHSC to stain for the markers Sox2 (BD Biosciences, 1:500) and Nestin (BD Biosciences, 1:500). The differentiated HPC were fixed 5 days after hypoxia and stained using anti-Tuj1 and anti-GFAP antibodies. To count the number of cells that differentiated either into Tuj1 positive or GFAP positive cells, the images were analyzed with the Metamorph Offline software (Molecular Devices, Sunnyvale, CA; version 7.0r3).
Data analysis
A statistical analysis was performed by using SPSS 16 for Mac (SPSS Inc, Chicago, IL). Numerical data were collected from at least three different experiments and are presented as mean ± standard error of the mean or standard deviation (for EPOR expression on NPC). Data from immunoblots were evaluated using one way ANOVA with Bonferroni post-hoc testing. Students t test was used for comparison of the ratio of GFAP/Tuj1 positive cells between different groups of HPC. Differences were considered significant at P < 0.05.
Results
Erythropoietin protects OHSC from cell death after OGD in the dentate gyrus
To investigate whether EPO protects OHSC from cell death after OGD we have evaluated cell death measuring PI fluorescence in three different regions of OHSC (CA1, CA3 and DG) and for the whole OHSC (Fig. 1). OGD exposure significantly increased the cell death in all regions in all treatment groups, when compared to the control group. In the OGD exposed OHSC, cell death in the CA1 regions was significantly lower in the EPO-treated group, compared to the wortmannin-treated group when EPO signaling upstream from AKT was blocked (p = 0.001). No other differences in CA1 were significantly different. There were no significant differences in cell death in the CA3 regions between groups. In the DG, EPO treatment significantly lowered cell death compared to vehicle, anti-EPOR antibody, and wortmannin-treated groups (p < 0.05, p < 0.001 and p < 0.001, respectively). Similarly, EPO treatment significantly lowered cell death of the whole OHSC compared to vehicle, anti-EPOR antibody, and wortmannin-treated groups (p < 0.05, p < 0.001 and p < 0.001, respectively). The mean percentage of cell death in the vehicle-treated group was 26.4 ± 1.0 % one day after OGD.
Figure 1.
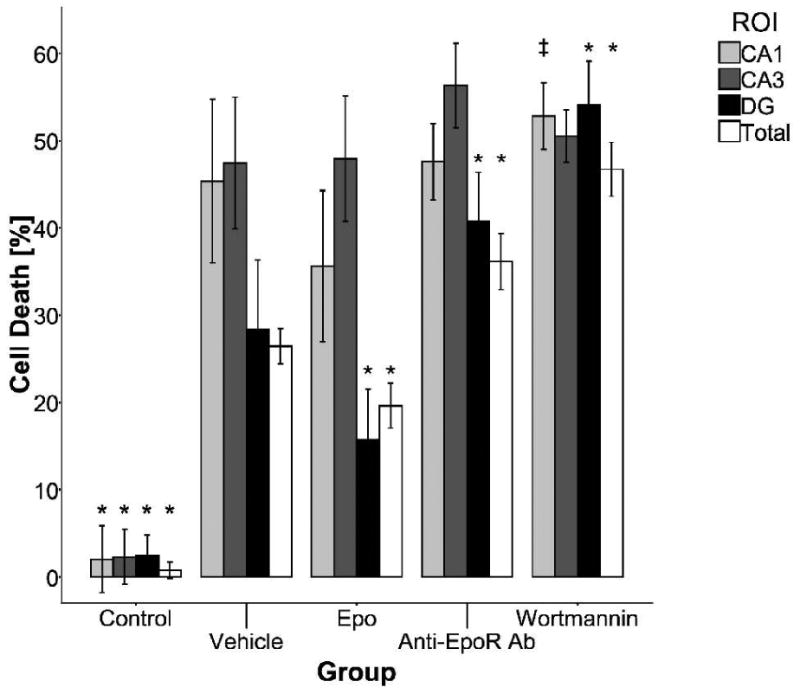
Cell death in OHSC in the different treatment groups. All groups of OHSC were exposed to OGD except the control group. Cell death was evaluated in three different regions, CA1 (light grey), CA3 (dark grey), DG (black) and for the whole OHSC (white). EPO treatment was found to be protective in the DG and when observing the OHSC as a whole. In the CA1 region, cell death was significantly lower in the EPO treated group, compared to the wortmannin treated group, but not when compared to the vehicle treated group. No significant EPO effect was found in the CA3 region. *, p < 0.05 (compared to the values of the vehicle treated group for the particular region); ‡, p < 0.05 (comparison between the EPO and wortmannin treated group in the CA1 region); bars: mean, SEM.
Erythropoietin promotes neurogenesis in OHSC after OGD
To assess neurogenesis in OHSC after OGD under different treatment conditions, we have used immunoblots to show variation in the doublecortin (DCX) expression. DCX encodes a microtubule-associated protein expressed in migrating neuroblasts and was used to demonstrate continuous neurogenesis in OHSC cultured in vitro (Chechneva et al., 2005). In our study, 72 hours post OGD the Western blot protein analysis showed the vehicle-treated group of OHSC expressed 56.6 ± 8.9 % of the DCX level found in the control group (Fig. 2; p < 0.01). EPO-treated OHSC did not express different level of DCX, compared to the control group. Treating OHSC with anti-EPOR antibody resulted in lower DCX expression, relative to the control (p < 0.001), as did treatment with wortmannin (p < 0.01).
Figure 2.
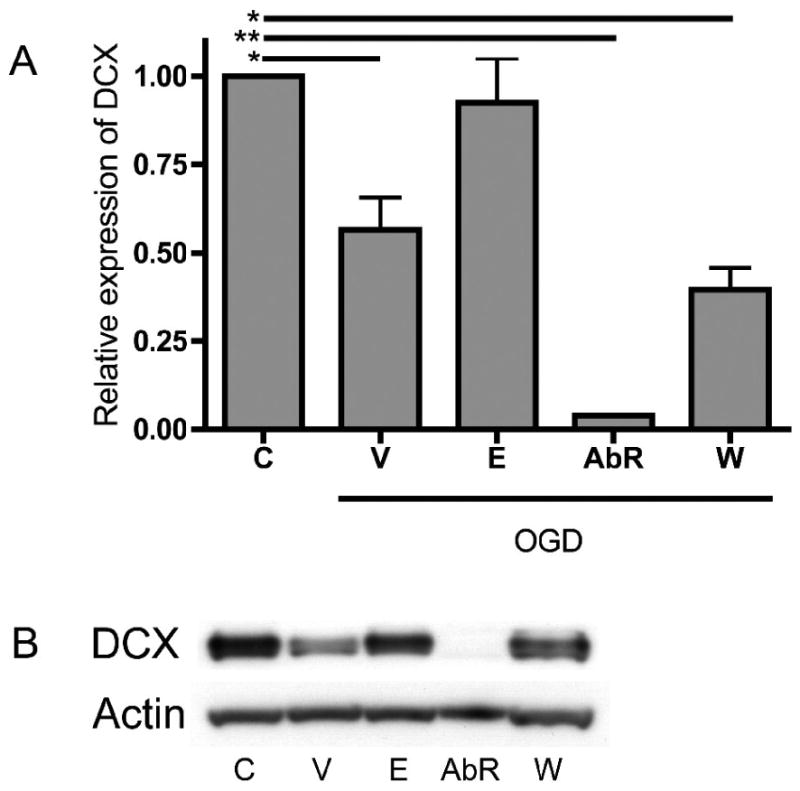
DCX expression in immunoblots. Vehicle treated group expressed significantly less DCX than the control group, but EPO treated group expressed DCX similar to the control group. Treating the OHSC with anti-EPOR almost entirely suppressed the DCX expression and treatment with wortmannin significantly decreased the DCX expression, compared to the control group (A). Western blot analysis of OHSC proteins. DCX, doublecortin (B); C, control; V, vehicle treated; E, EPO treated; AbR, anti-EPOR antibody treated; W, wortmannin treated; bars: mean, SEM; *, p < 0.01; **, p < 0.001.
Immunohistochemistry findings were in concordance with the Western blot findings. Tuj1 is a neuron-specific class III β-tubulin expressed in differentiated progenitor cells that have chosen the neuronal fate. In control group of OHSC, Tuj1 was mostly expressed in the hilus of the DG and surrounding the CA3 region (Fig. 3). After OGD, the vehicle-treated group of OHSC seemed to express less Tuj1 in the same area, although there was still some remaining Tuj1 expression found. The EPO-treated group expressed Tuj1 similarly to the control group.
Figure 3.
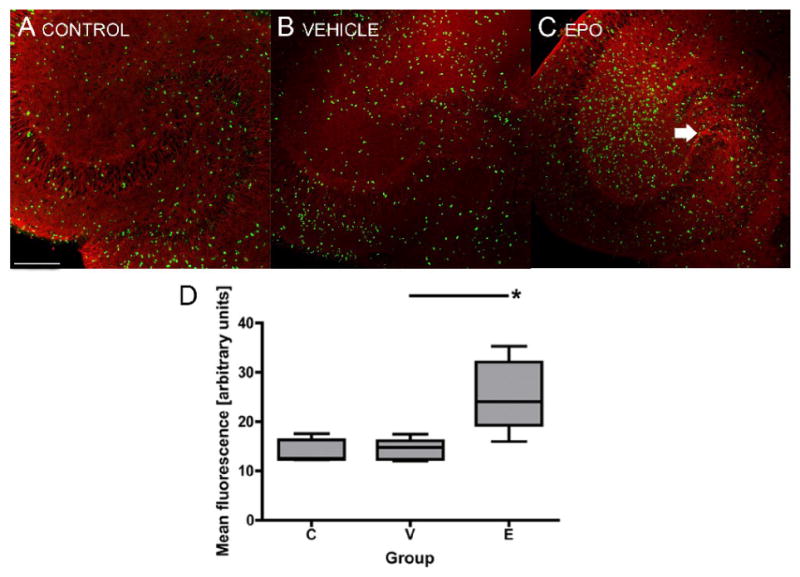
Tuj1 and BrdU expression in OHSC 5 days after OGD. The vehicle treated group (B) showed less Tuj1 expression in the hilus of the dentate gyrus and surrounding the CA3 region than the control group (A), while EPO treated group (C) showed Tuj1 expression similar to that in the control group (arrow). In EPO treated group, there were more BrdU positive cells in the dentate gyrus 5 days after OGD, compared to the control and vehicle treated groups (D). Red, Tuj1; green, BrdU; scale 200 μm; boxes: quartiles, mean, SEM; *, p < 0.01.
The number of proliferating cells in the DG 4-5 days after OGD was increased in the EPO-treated group, compared to the vehicle-treated group (p < 0.01). The vehicle-treated group and the control group did not differ in the number of proliferating cells in the DG. Less than 1% of these BrdU positive cells express markers of differentiation on day 5 (data not shown).
To outline the influence of EPO treatment on other cell types, we have also stained OHSC for astrocytic marker GFAP, microglial marker ED1, and oligodendrocytic marker NG2. In OHSC treated with EPO, there is less GFAP and ED1 immunoreactivity, but more NG2 immunoreactivity, as compared to vehicle treated OHSC (Fig. 4).
Figure 4.
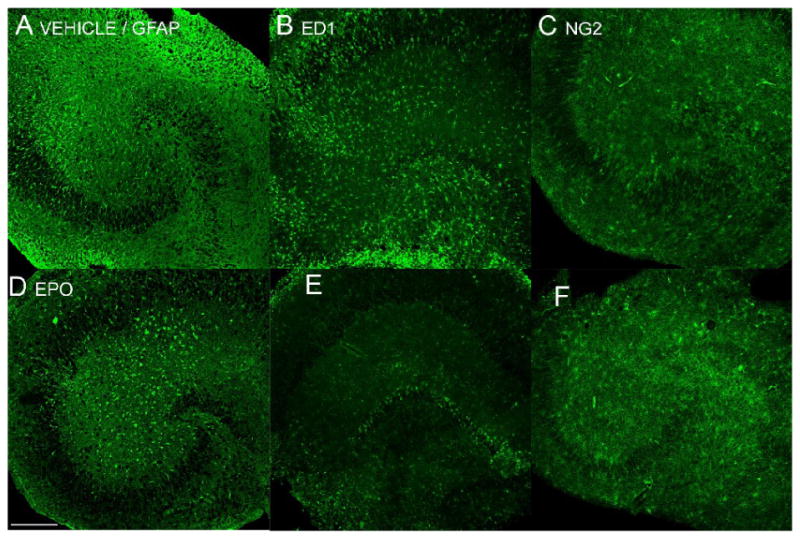
Qualitative comparison between the vehicle treated group (A, B, C) and EPO treated group (D, E, F) of OHSC in expression of markers for astrocytes (GFAP; A, D), microglia (ED1; B, E), and oligodendrocytes (NG2; C, F). EPO treated OHSC express less GFAP and ED1 immunoreactivity, while expressing more NG2 immunoreactivity 5 days after OGD. Scale 200 μm.
Erythropoietin drives hypoxia exposed HPC towards a neuronal fate
In order to better understand the effect of EPO on the cell fate commitment of hippocampal progenitors, we have used in vitro cultures of HPC. These cells expressed Sox2 and nestin, with very few positive for markers of differentiated cells such as Tuj1 or GFAP. Many of the HPC also expressed EPOR on their surface and EPOR expression was increased by a factor of 4.06 ± 0.52 compared to the control 1 day after exposure to hypoxia (Fig. 5).
Figure 5.
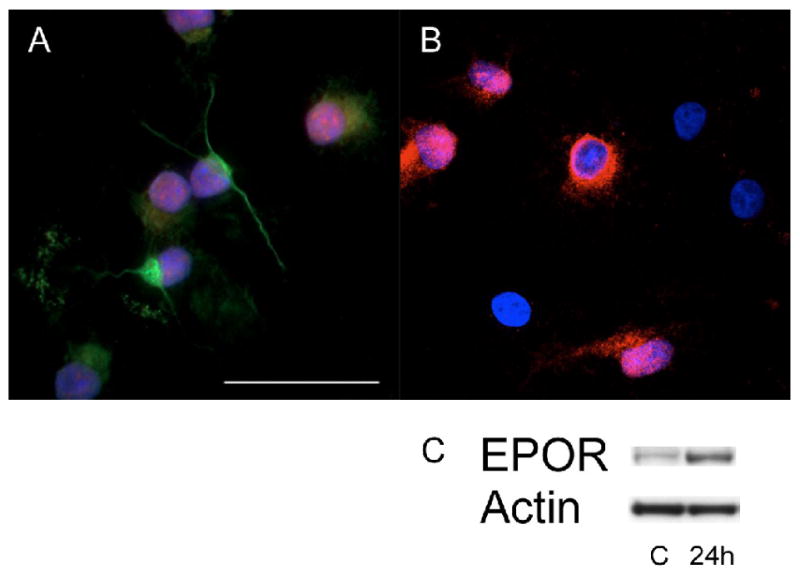
HPC expressed nestin, Sox2 (A), and EPOR (B). In an immunoassay of EpoR expression in HPC 24 hours after hypoxia, HPC expressed 4.13 ± 0.48 times more EpoR than the control group (C). A: Green, nestin; red, SOX2; blue, DAPI. B: red, EPOR; blue, DAPI; C, control; 24h, 24 hours after hypoxia; scale 50 μm.
When HPC were differentiated by bFGF withdrawal, more cells expressed Tuj1 and less expressed GFAP positive cells when EPO was added to the culture medium. Under normoxic conditions Tuj1 expression accounts for 75.53 ± 2.32 % of the total Tuj1 and GFAP expressing cells. The addition of EPO to the media increased this to 87.81 ± 1.41 % (p < 0.001) of Tuj1 and GFAP expressing cells. The relative expression of Tuj1 to GFAP was not different in normoxia vs hypoxia. However, under hypoxic conditions the addition of EPO again increased Tuj1 expressing cells from 75.41 ± 2.16 % to 82.79 ± 2.72 % (p < 0.05) of the total (Fig. 6).
Figure 6.
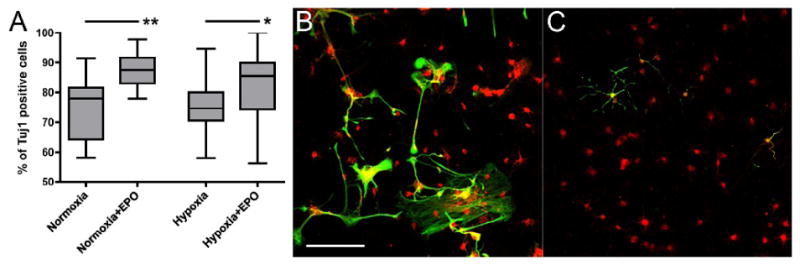
Cell fate commitment of HPC after differentiation under normoxic and hypoxic conditions. EPO treatment increased the percentage of Tuj1 positive cells under both normoxic and hypoxic conditions (A). Five days after HPC were stimulated into differentiation by bFGF withdrawal and exposed to hypoxia in the vehicle treated group, approximately 24% of cells differentiated into GFAP positive astrocytes, many of which presented abnormally looking stress fibers (B). In the EPO treated group (C), there were less GFAP positive cells (17%), compared to the vehicle treated group, while more cells expressed Tuj1, suggesting increased neural cell fate commitment. Boxes: quartiles, mean, SEM; *, p < 0.05; **, p < 0.001; red, Tuj1 positive immature neurons; green, GFAP positive astrocytes; scale 100 μm.
Discussion
This study is the first to focus on the role of EPO in hippocampal neurogenesis in a neonatal stroke model. We show that EPO treatment following OGD protects hippocampal neurons from cell death, increases the number of proliferating cells in the DG, promotes neural cell fate commitment and sustains neurogenesis in OHSC. Neural progenitor cells arising from the subgranular zone (SGZ) of the DG normally migrate into the granule cell layer (GCL) where they differentiate into new neurons and integrate into the local network (Kee et al., 2001). The physiological function of these new neurons has been linked with learning and memory (Gould et al., 1999; Shors et al., 2001). In the present study, approximately 26% of vehicle-treated OHSC cells died within 24 hours after OGD. If EPO was administered to the culture medium after OGD, cell death in OHSC was significantly decreased. This effect was most pronounced in the DG of OHSC, the primary site of hippocampal neurogenesis, although we might have underestimated cell death in this region because of increased regeneration. Montero et al. used a neonatal mouse model of OHSC, pretreated for 24 h with EPO or carbamylerythropoietin, exposed to 30 minutes of OGD and after a 24h recovery period EPO was found to be neuroprotective, but not specifically in the DG region. However, they did find carbamylerythropoietin to be protective in DG (Montero et al., 2007). Zhang et al. have studied CA1 hippocampal neurons in adult male rats and found a neuroprotective effect of EPO in the region (Zhang et al., 2006). A similar trend, although not significant, was found in our study in the CA1 region of neonatal rat hippocampi.
Increased progenitor proliferation after hypoxia has been demonstrated in several previous studies (Jin et al., 2001; Liu et al., 1998). The results of our study suggest that EPO treatment significantly increases the number of proliferating cells in the DG 4-5 days after OGD. We have avoided studying the proliferation of cells in the first three days after OGD so as to minimize the contribution of proliferating inflammatory cells. The increased proliferation in the DG might be partly due to increased cell survival in this region when EPO is administered, as discussed above, but direct effect of EPO on the progenitor cell proliferation and maintenance also likely plays a significant role (Chen et al., 2007; Gonzalez et al., 2007). Our results suggest an approximately 4-fold increase in the EPOR expression in the progenitors 1 day after exposure to hypoxia, which likely increases the responsiveness of progenitors to the effects of EPO. Indeed, in neurons, optimal neuroprotection by EPO was found to require elevated expression of its receptor (Sanchez et al., 2009). In adult gerbils, Liu et al. have found a peak in neurogenesis 9 days after transient global ischemia (Liu et al., 1998). In our neonatal rat model, we did not find an increased progenitor cell proliferation in the DG in the vehicle-treated group of OHSC 4-5 days after OGD which suggests that EPO may be necessary to increase progenitor proliferation in the early phase. Several studies have found that in OHSC markers of neurogenesis can be detected in the DG and can be stimulated by epidermal growth factor application (Kamada et al., 2004; Raineteau et al., 2004). Granule cell neurogenesis peaks at P7 (Danzer, 2008), the age at which we have sacrificed the animals to make the OHSC. In our study, under normal culture conditions the OHSC expressed markers of neurogenesis (DCX, Tuj1) 9 DIV and beyond. Exposure of OHSC to OGD decreased the DCX and Tuj1 expression, suggesting disrupted differentiation of progenitors into neurons. However, treating OHSC with EPO sustained the markers of neurogenesis in levels similar to those seen in the controls. On the contrary, when OHSC were treated with anti-EPOR antibody, the markers of neurogenesis were reduced to minimal levels. This finding supports our hypothesis that EPO-EPOR signaling is crucial for neurogenesis after exposure to OGD. Lewczuk et al. have shown in cultured hippocampal neurons, an increase in EPO and EPOR mRNA after exposure to hypoxia (Lewczuk et al., 2000). Shingo et al. have found a similar increase in EPO mRNA in neural stem cells after exposure to hypoxia. These data support our findings that link EPO directly to neurogenesis. The mechanism of EPO action on neurogenesis might involve the AKT pathway, but other pathways may also be involved in EPO promoted neurogenesis, and even pathways that do not require EPO-EPOR interaction (van der Kooij et al., 2008). We did not attempt to quantify the effect of EPO on other cell types, but EPO seems to influence other cell types as well. Treating OHSC with EPO after OGD appears to decrease the population of astrocytes and microglia while increasing the population of oligodendrocytes. These data further support the hypothesis that EPO can repair the brain after an hypoxic-ischemic insult.
Although preserved DCX expression in the EPO-treated group was at least partly due to improved survival of the preexisting differentiating neurons, we have been able to demonstrate that EPO, besides increasing the number of proliferating cells in the DG, promotes neuronal cell fate commitment of HPC under both normoxic and hypoxic conditions. When differentiation of HPC was initiated by FGF withdrawal, most of the cells differentiated into immature neurons expressing Tuj1, while approximately 24% of cells differentiated into GFAP positive astrocytes. When EPO was added to the medium, the percentage of Tuj1 positive cells increased significantly, and the percentage of GFAP positive cells dropped from 24% to 12% in the normoxic group and from 25% to 17% in the hypoxic group of cells. In the hypoxic setting, many of the astrocytes showed also a more abnormal morphology, presenting abnormal stress fibers. Komitova et al. (Komitova et al., 2006) have found that after hypoxia-ischemia most of the surviving newly formed DG cells differentiate into NeuN or calbindin positive mature neurons by 3–4 weeks after ischemia, whereas about 10–20% of the newly generated cells differentiate into GFAP positive astrocytes in the GCL and the hippocampal hilus. Neurobasal media is known to suppress the growth of both oligodendrocytes and astrocytes, therefore the absolute percentages of cells that have chosen the neuronal or astrocytic lineage might not resemble the actual in vivo differentiation. However, we believe that we were still able to show that adding EPO to the medium promotes neuronal cell fate.
In summary, our data show that EPO mediates neuroprotection in neonatal hippocampus after ischemia by salvaging neurons after injury and inducing repair by stimulating neurogenesis. In a clinical setting, neonatal stroke is not always easily recognizable and the currently limited neuroprotective strategies are often not available immediately after the injury. Therapies modulating later phases of stroke evolution, i.e. promoting innate self-repair mechanisms, such as neurogenesis, are therefore of immense value as the neurons born postnataly may support brain plasticity. Although some controversy exists whether more neurogenesis is always better (Scharfman and Hen, 2007), neurogenesis might prove to be important from a therapeutic and prognostic point of view. The results of our study support the use of EPO as a possible promoter of neurogenesis after neonatal stroke. Exact timing of EPO administration and its effect on cognitive function has yet to be determined.
Acknowledgments
This work was supported by the Slovenian Research Agency grant Nr. Z3-9060-0312-06 and the Fulbright Scholarship (DO), the P50NS35902 and March of Dimes Grant Nr. 6-FY2006-465 (DMF), Foundation for Anesthesia education and research (JWS). The authors would also like to thank Jason Leong for much assistance with cell culture.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altman J. Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J Comp Neurol. 1969;137:433–57. doi: 10.1002/cne.901370404. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, et al. Identification of neural stem cells in the adult vertebrate brain. Brain Res Bull. 2002;57:751–8. doi: 10.1016/s0361-9230(01)00770-5. [DOI] [PubMed] [Google Scholar]
- Bernaudin M, et al. A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab. 1999;19:643–51. doi: 10.1097/00004647-199906000-00007. [DOI] [PubMed] [Google Scholar]
- Chechneva O, et al. Identification and characterization of two neurogenic zones in interface organotypic hippocampal slice cultures. Neuroscience. 2005;136:343–55. doi: 10.1016/j.neuroscience.2005.07.058. [DOI] [PubMed] [Google Scholar]
- Chen ZY, et al. Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J Biol Chem. 2007;282:25875–83. doi: 10.1074/jbc.M701988200. [DOI] [PubMed] [Google Scholar]
- Danzer SC. Postnatal and adult neurogenesis in the development of human disease. Neuroscientist. 2008;14:446–58. doi: 10.1177/1073858408317008. [DOI] [PubMed] [Google Scholar]
- Dash PK, et al. Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J Neurosci Res. 2001;63:313–9. doi: 10.1002/1097-4547(20010215)63:4<313::AID-JNR1025>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351:1985–95. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–8. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Gonzalez FF, Ferriero DM. Therapeutics for neonatal brain injury. Pharmacol Ther. 2008;120:43–53. doi: 10.1016/j.pharmthera.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Gonzalez FF, et al. Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Dev Neurosci. 2007;29:321–30. doi: 10.1159/000105473. [DOI] [PubMed] [Google Scholar]
- Gould E, et al. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260–5. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]
- Iwai M, et al. Erythropoietin promotes neuronal replacement through revascularization and neurogenesis after neonatal hypoxia/ischemia in rats. Stroke. 2007;38:2795–803. doi: 10.1161/STROKEAHA.107.483008. [DOI] [PubMed] [Google Scholar]
- Jin K, et al. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci U S A. 2001;98:4710–5. doi: 10.1073/pnas.081011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juul S. Erythropoietin in the central nervous system, and its use to prevent hypoxic-ischemic brain damage. Acta Paediatr Suppl. 2002;91:36–42. doi: 10.1111/j.1651-2227.2002.tb02904.x. [DOI] [PubMed] [Google Scholar]
- Kadam SD, et al. Neurogenesis and neuronal commitment following ischemia in a new mouse model for neonatal stroke. Brain Res. 2008 doi: 10.1016/j.brainres.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada M, et al. Intrinsic and spontaneous neurogenesis in the postnatal slice culture of rat hippocampus. Eur J Neurosci. 2004;20:2499–508. doi: 10.1111/j.1460-9568.2004.03721.x. [DOI] [PubMed] [Google Scholar]
- Kee NJ, et al. Enhanced neurogenesis after transient global ischemia in the dentate gyrus of the rat. Exp Brain Res. 2001;136:313–20. doi: 10.1007/s002210000591. [DOI] [PubMed] [Google Scholar]
- Kilic E, et al. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. Faseb J. 2005;19:2026–8. doi: 10.1096/fj.05-3941fje. [DOI] [PubMed] [Google Scholar]
- Komitova M, et al. Enriched environment after focal cortical ischemia enhances the generation of astroglia and NG2 positive polydendrocytes in adult rat neocortex. Exp Neurol. 2006;199:113–21. doi: 10.1016/j.expneurol.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Lee J, et al. Predictors of outcome in perinatal arterial stroke: a population-based study. Ann Neurol. 2005;58:303–8. doi: 10.1002/ana.20557. [DOI] [PubMed] [Google Scholar]
- Lewczuk P, et al. Survival of hippocampal neurons in culture upon hypoxia: effect of erythropoietin. Neuroreport. 2000;11:3485–8. doi: 10.1097/00001756-200011090-00017. [DOI] [PubMed] [Google Scholar]
- Liu J, et al. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci. 1998;18:7768–78. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner B. Disorders of learning and memory after temporal lobe lesions in man. Clin Neurosurg. 1972;19:421–46. doi: 10.1093/neurosurgery/19.cn_suppl_1.421. [DOI] [PubMed] [Google Scholar]
- Montero M, et al. Comparison of neuroprotective effects of erythropoietin (EPO) and carbamylerythropoietin (CEPO) against ischemia-like oxygen-glucose deprivation (OGD) and NMDA excitotoxicity in mouse hippocampal slice cultures. Exp Neurol. 2007;204:106–17. doi: 10.1016/j.expneurol.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Nagai A, et al. Erythropoietin and erythropoietin receptors in human CNS neurons, astrocytes, microglia, and oligodendrocytes grown in culture. J Neuropathol Exp Neurol. 2001;60:386–92. doi: 10.1093/jnen/60.4.386. [DOI] [PubMed] [Google Scholar]
- Nelson KB. Perinatal ischemic stroke. Stroke. 2007;38:742–5. doi: 10.1161/01.STR.0000247921.97794.5e. [DOI] [PubMed] [Google Scholar]
- Noguchi CT, et al. Role of erythropoietin in the brain. Crit Rev Oncol Hematol. 2007;64:159–71. doi: 10.1016/j.critrevonc.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, et al. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol. 2002;52:802–13. doi: 10.1002/ana.10393. [DOI] [PubMed] [Google Scholar]
- Raineteau O, et al. Neurogenesis in hippocampal slice cultures. Mol Cell Neurosci. 2004;26:241–50. doi: 10.1016/j.mcn.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Sall JW, et al. Isoflurane inhibits growth but does not cause cell death in hippocampal neural precursor cells grown in culture. Anesthesiology. 2009;110:826–33. doi: 10.1097/ALN.0b013e31819b62e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, et al. Optimal neuroprotection by erythropoietin requires elevated expression of its receptor in neurons. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0901840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Hen R. Neuroscience. Is more neurogenesis always better? Science. 2007;315:336–8. doi: 10.1126/science.1138711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingo T, et al. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci. 2001;21:9733–43. doi: 10.1523/JNEUROSCI.21-24-09733.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, et al. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–6. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Sola A, et al. Potential for protection and repair following injury to the developing brain: a role for erythropoietin? Pediatr Res. 2005;57:110R–117R. doi: 10.1203/01.PDR.0000159571.50758.39. [DOI] [PubMed] [Google Scholar]
- van der Kooij MA, et al. Neuroprotective properties and mechanisms of erythropoietin in in vitro and in vivo experimental models for hypoxia/ischemia. Brain Res Rev. 2008;59:22–33. doi: 10.1016/j.brainresrev.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Zaidi AU, et al. New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia. 2004;46:380–90. doi: 10.1002/glia.20013. [DOI] [PubMed] [Google Scholar]
- Zhang F, et al. Erythropoietin protects CA1 neurons against global cerebral ischemia in rat: potential signaling mechanisms. J Neurosci Res. 2006;83:1241–51. doi: 10.1002/jnr.20816. [DOI] [PubMed] [Google Scholar]