

Abstract
In this study we used colony forming unit (CFU) assays to demonstrate rapid suppression (within 6 h) of lymphoid (CFU-preB) and myeloid (CFU-GM) progenitor cells in DMBA treated mice. The duration of these changes were consistent with the blood levels of DMBA and its metabolites that were achieved by either IP or oral DMBA administration. CFU-GM and CFU-preB activities returned to control levels by 2 and 7 days after oral DMBA exposure, respectively, but remained suppressed through 7 days after IP DMBA administration. The continued presence of low levels of DMBA in the bloodstream following IP administration was associated with sustained suppression of CFU-preB, total bone marrow lymphoid cells and peripheral blood lymphocytes. The changes noted above were not observed in Cyp1b1 null mice, demonstrating the need for local DMBA metabolism in the bone marrow by Cyp1b1 to impair bone marrow CFU-preB and CFU-GM. Furthermore, these data provide evidence that myeloid lineage cells are restored more quickly than lymphoid lineage cells after DMBA exposure.
Keywords: DMBA, PAHs, Cyp1b1, Hematopoiesis, Bone marrow, Blood, CFU assays
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous environmental contaminants that are carcinogenic and immunosuppressive. Both toxicological and epidemiological data have linked exposure to PAHs with various forms of cancer (lung, skin, breast, esophageal, bladder), cardiovascular diseases, asthma/immunological effects, neurological effects, reproductive and developmental effects. 7, 12-dimethylbenz(a)anthracene (DMBA) and benzo(a)pyrene (BP) are frequently used as model polycyclic aromatic hydrocarbons due to their potent carcinogenic and immunosuppressive activities (Dean et al. 1986; Ward et al. 1984). BP is among the most abundant of PAHs in the environment, and is formed by multiple combustion processes (Poland et al. 1982; Wilson et al. 1984). In contrast, DMBA is a synthetic PAH in which the methyl substitution greatly enhances carcinogenicity and toxicity (Smithgall et al. 1988).
Previous studies show that IP administration of DMBA to mice results in a substantial hypocellularity of the bone marrow at 48 h after exposure (Heidel et al., 2000; Page et al., 2003; 2004; Galvan et al., 2005; 2006). This response was dependent on local metabolism of the DMBA by Cyp1b1 that is expressed in the bone marrow, spleen, thymus and peripheral blood leukocytes, but not the liver parenchyma (Bhattacharyya et al., 1995; Baron et al., 1998; Heidel et al., 2000; Shimada et al., 2003; Galvan et al, 2005; Uno et al., 2006). The reduction in bone marrow cellularity was evident in both the lymphoid (B cell) and myeloid (largely granulocyte) populations (Heidel et al., 2000; Galvan et al., 2006). Although these previous studies clearly identified the adverse effects of DMBA on bone marrow hematopoiesis, they did not examine whether exposure to DMBA changes the ability of lineage-specific progenitor cells to proliferate and differentiate into mature bone marrow cell populations (Igarashi et al., 2006; Kincade 1994; Pelayo et al., 2005; 2006). Nor did they examine whether changes in bone marrow cell numbers and populations were reflected in demonstrable changes in peripheral blood leukocyte populations. Thus, there is a need to provide insights into how DMBA exposure alters maturation and proliferation of bone marrow progenitor cells into the major mature cell populations (B cells and granulocytes) that then leave the bone marrow and enter the circulation.
The present study addresses how the route of administration (oral vs. IP) influences the temporal effects of DMBA on hematopoietic progenitor cells in bone marrow and mature leukocytes in the blood. The process of hematopoiesis can be recapitulated in vitro using colony forming assays that measure bone marrow progenitor cell proliferation and differentiation into lymphoid-lineage (CFU-preB) and myeloid-lineage (CFU-GM) colonies in agarose supplemented with carefully defined growth factors (Igarashi et al., 2006; Nagasawa, 2006; 2007; Pelayo et al., 2005; Smithgall, 1998). Mice exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (Murante and Gasiewicz, 2000; Thurmond and Gasiewicz, 2000; Thurmond et al., 2000) exhibited alterations in CFU activities that are thought to reflect the differences in bone marrow cell populations that occur after 2, 3, 7, 8 – tetrachlorodibenzo-p-dioxin (TCDD) exposure. However, such studies are lacking for animals exposed to DMBA or other polycyclic aromatic hydrocarbons. This is a substantive knowledge gap that must be addressed if we are to understand how exposure to DMBA, and by inference other PAHs, affects hematopoiesis.
In addition, previous studies of the adverse effects of DMBA or benzo[a]pyrene on hematopoiesis in general focused on a single time point after administration that coincided with the maximal reduction in bone marrow cellularity (Galvan et al., 2003; 2006; Uno et al., 2006). They did not provide a temporal analysis of whether impairment of hematopoiesis in the bone marrow was sustained, or returned to normal levels, after exposure. It is well recognized that administration of DMBA by the oral or IP routes will result in substantial temporal differences in blood levels of DMBA and its metabolites. In essence, oral delivery of DMBA results in a relatively short-lived acute exposure to DMBA and its metabolites, largely because of first pass metabolism in the liver. As we report here, IP or oral delivery of DMBA result in similar early blood levels of DMBA, which is followed by a second phase of sustained blood levels that are not seen after oral DMBA administration. The purpose of this study was to use the differences in the kinetics of metabolism and bloodstream distribution of DMBA following a single oral or IP DMBA treatment for an integrated temporal analysis of how the route of administration and bloodstream distribution of DMBA metabolites affects bone marrow hematopoiesis.
In this study, we used colony forming assays to investigate ex vivo the link between DMBA-mediated changes in bone marrow cellularity and alteration in lineage-specific bone marrow hematopoietic progenitor cell activity. We integrate the bone marrow and mature blood cell responses with a temporal analysis of DMBA clearance from blood following a single oral or IP administration. The data presented here show that DMBA metabolites, generated by Cyp1b1 metabolism in bone marrow, rapidly disrupt and retard the developmental program of hematopoiesis. Bone marrow cells play a significant role in hematopoiesis, angiogenesis, and immune responses to tissue injury and chemical effects in the bone marrow therefore may have broad systemic implications on toxicity, carcinogenicity, and overall health.
2. Materials and Methods
2.1. Reagents and Antibodies
7, 12-dimethylbenz(a)anthracene (DMBA) was purchased from AccuStandard, Inc (New Haven, CT) and dissolved in olive oil at a concentration of 5 mg/ml. RPMI 1640 was purchased from Sigma Chemical and was supplemented with 5% FBS (v/v) (Atlanta Biologicals), 50 IU penicillin/ml, and 50 μg streptomycin/ml (w/v). The following monoclonal antibodies were purchased from BD Pharmingen (Franklin Lakes, NJ): CD45/B220-phycoerythrin (PE) and Gr-1-fluorescein isothiocyanate (FITC). Methocult methylcellulose media for clonogenic assays were purchased from StemCell Technologies, Inc (Vancouver, Canada). The CFU-PreB media (M3630) contained FBS, 2-mercaptoethanol, L-glutamine, and recombinant human IL-7 and the CFU-GM media (M3534) contained FBS, BSA, recombinant human insulin, human transferring, 2-mercaptoethanol, L-glutamine, recombinant mouse stem cell factor, recombinant mouse IL-3, and recombinant human IL-6.
2.2. Animals and treatments
C57BL/6 mice (6 weeks old) were purchased from the Jackson Laboratories (Bar Harbor, ME). Cyp1b1-/- mice (C57BL/6 background) were bred in our animal care facility as described previously (Buters et al., 1999). Animals were housed at the AAALAC certified University of Wisconsin Madison School of Veterinary Medicine Animal Care Unit and used in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Groups of female C57BL/6 mice were randomly selected and injected IP or oral gavaged with DMBA (10 or 50 mg/kg) in olive oil (0.2 ml). This dose of DMBA (50 mg/kg) is similar to those used to initiate tumors in rodent carcinogenicity studies (Medina et al, 1974; Buters et al, 1999; 2003) and also consistent or lower than those used in numerous previously published investigations (Uno et al, 2004; 2006; Gao et al, 2005; 2007; Heidel et al, 2000; Galvan et al, 2003; 2005; 2006). Time-matched vehicle control animals were injected with an equivalent volume of olive oil. All mice were euthanized with CO2 asphyxiation at selected time points in accordance with UW Madison IACUC approved protocol and NIH Guide for the Care and Use of laboratory Animals.
2.3. Detection of DMBA blood levels
Mice were euthanized with CO2 asphyxiation. Immediately following euthanization, blood samples were collected by cardiac puncture of C57BL/6 mice at 1.5, 3, 6, 24, 48, and 168 h post DMBA treatment. Sonication was applied to whole blood (10-100 μl) and diluted in 0.2 ml of 0.5 M NaCl. Following sonication, the blood samples were brought to a total volume of 1.0 ml with NaCl. Dibenzo(a,l)pyrene (DBP) (50 μl, 0.5 μM) was added to each sample as an extraction control. DMBA and metabolites were extracted by the addition of ethylacetate: acetone (2:1), with extensive vortexing, followed by centrifugation. The extracted material was dried under nitrogen gas and resuspended in a 20 μl MeOH immediately prior to reverse phase HPLC analysis. DMBA and metabolites were separated on a Beckman C18 Ultrasphere column with a gradient of 50-100% MeOH over the first 30 min, followed by sustained 100% MeOH for 25 min. The levels of DMBA and metabolites were quantified by UV (254 nm) and fluorescence detection (Excitation and emission wavelengths of 273/395 nm, 5, 6- and 8, 9+10, 11-dihydrodiols; 273/470 nm, 7-OH DMBA; 273/415 nm, DMBA and DBP). DMBA levels were quantified relative to known quantities of purified standard.
2.4. Bone marrow cell isolation
DMBA-treated and time-matched vehicle control wild type mice were euthanized by CO2 asphyxiation at 6, 24, 48, 72, and 168 h post-treatment. In some experiments, DMBA treated and control Cyp1b1-null mice were included and also euthanized by CO2 asphyxiation at 6, 48, and 168 h after treatment. To isolate bone marrow cells, the femurs were dissected free of muscle tissue and the ends of the bones removed with a surgical blade. Cells were flushed from the femurs with 2 ml of RPMI culture medium using a syringe equipped with a 25-gauge needle. The bone marrow cells were dispersed into single cell suspensions by successive passage through 22- and 25-guage needles. Following centrifugation, red blood cells were lysed in ACK buffer (150 mM NH4Cl, 10 mM KHCO3, and 100 mM Na2EDTA pH 7.3). Viable white cells were enumerated in a hemocytometer by their exclusion of 0.05% Trypan Blue. The remaining cells were then used in flow cytometry or mouse colony forming cell assays for hematopoietic progenitor cells.
2.5. Flow cytometry staining and analysis
Freshly isolated cells were suspended at 1 × 106 cells per 100 μl media. Aliquots of cells (1 × 106) were maintained on ice for 20 min with 0.5 μg purified rat anti-mouse CD16/CD32 (Fcγ III/II Receptor) (Mouse BD Fc Block, Caltag) to block Fc receptors. The cells were then incubated with 1 μg of anti-CD45/B220-PE or anti-Gr-1 FITC (to detect B lymphocytes and neutrophils, respectively) for 30 min on ice and washed twice with 200 μl of wash buffer (1% PBS). Cells were re-suspended in 200 μl of wash buffer and fixed with 2% paraformaldehyde. Fixed cells were stored in the dark at 4°C, acquired and analyzed within 3 days. One hundred thousand events were acquired for each sample with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Populations of B lymphocytes and myeloid cells were gated as described previously (Thurmond and Gasiewicz, 2000). Data were analyzed using Flow Jo 6.4 software.
2.6. Bone marrow cell colony forming unit (CFU) assays
Freshly isolated bone marrow cells were re-suspended at concentrations of 1.0 × 105 cells per ml (CFU-preB assay) or 8 × 104 cells per ml (CFU-GM assay) in RPMI-1640 tissue culture medium supplemented with FBS and penicillin/streptomycin. Colony forming (CFU) assays were performed following protocols developed by manufacturer (StemCell Technologies Inc, Vancouver, BC, Canada). Briefly, an aliquot (300 μl) of re-suspended bone marrow cells was added to 2.2 ml of CFU-preB or CFU-GM methylcellulose (Methocult) media (StemCell Technologies, Canada), vortexed, and allowed to stand for 5 min for bubbles to dissipate. The bone marrow cells suspended in the Methocult media were then dispensed into duplicate pre-tested culture dishes (StemCell Technologies,) using a syringe and blunt-end needle. Bone marrow cells were incubated for 7 to 14 days in a humidified incubator at 37°C and 5% CO2. Myeloid and lymphoid colonies were evaluated and counted using an inverted microscope and gridded scoring dishes.
2.8. Complete blood counts with differential analysis
Immediately following euthanasia by CO2 asphyxiation, blood was collected by cardiac puncture and placed in a microtainer tube containing lyophilized K2EDTA (BD Diagnostics, Franklin Lakes, NJ). Whole blood was kept at room temperature until analyzed within 3 h of collection. For analysis, approximately 210 μL of whole blood was transferred to a second microtainer tube and a complete blood count (CBC) with differential analysis was performed using the ADVIA 120 hematology system, in the clinical pathology unit of the UW-Madison Veterinary Medical Teaching Hospital.
2.9. Data Analysis
Statistical analysis, unless otherwise defined, was performed using analysis of variance followed by Tukey's post hoc test using Prism 5 software (GraphPad Software, San Diego, CA). Differences between treatment groups were considered significant when p < 0.05.
3. Results
3.1. Clearance of DMBA from the blood following oral or IP administration
Because of our interest in relating changes in bone marrow cell CFU-preB and CFU-GM activities with blood levels of DMBA and its metabolites, we first performed an analysis of these metabolites in mice treated orally or IP with a single bolus of DMBA. We reasoned that oral treatment would result in a relatively short duration of bone marrow exposure to DMBA compared to IP treated mice. Surprisingly, the two treatments provided remarkably similar peaks of blood DMBA at 1.5 to 3 hours (8 to 12 μM), followed by a 4 to 6 fold decrease by 6 hours (Fig. 1A). At 6 h, the DMBA level was somewhat higher following IP administration, and declined only two fold further by 48h. In contrast, DMBA blood levels following oral administration continued to drop precipitously and were scarcely detectable at 24 hours. Interestingly, elimination of DMBA after IP treatment demonstrated log-linear first order kinetics, with low levels of DMBA detected in samples collected as late as 168 h after IP administration (data not shown). DMBA metabolites detected in the bloodstream (8, 9 dihydrodiols and 10, 11 dihydrodiols and 7-OH DMBA) were similar at 6 h after oral or IP administration of DMBA (Fig. 1B). Low levels of the 3, 4 dihydrodiol were detectable, which represent metabolites that escape conjugation.
Fig 1. Comparisons of DMBA blood levels and major metabolites after oral gavage or IP administration.
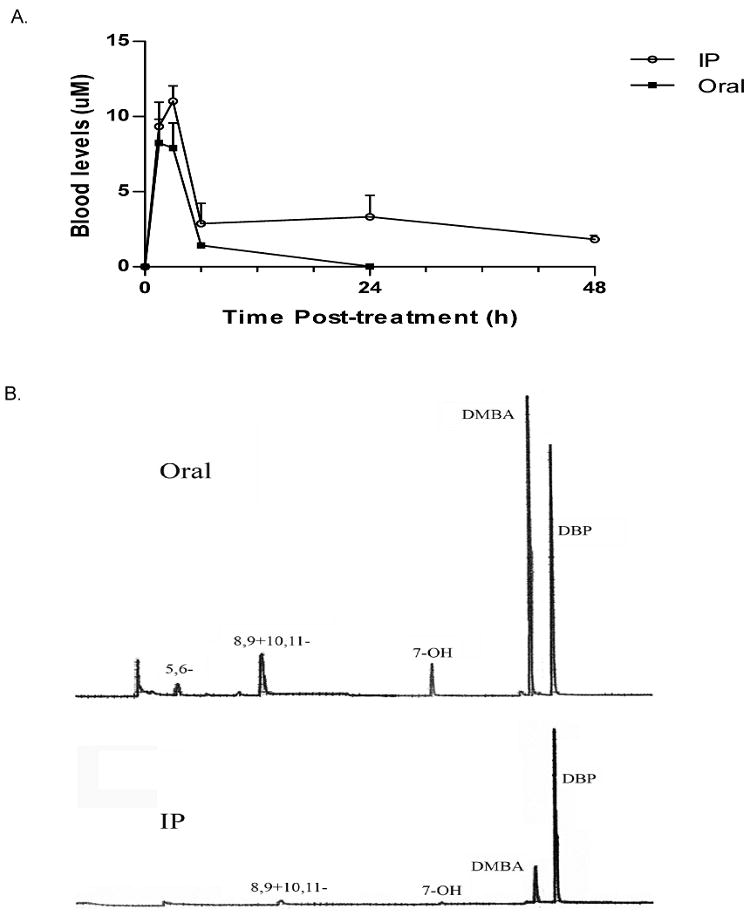
A) Circulating DMBA levels in blood collected by cardiac puncture at the indicated time points following oral or IP DMBA treatment (50 mg/kg). Each time point represents the mean (± SEM) of 2 to 3 different mice. B) Representative HPLC profiles of DMBA metabolites in the blood at 6 h after oral gavage (upper panel) or IP administration (lower panel). Major metabolites identified are: 5, 6-dihydrodiol, 8, 9-+10, 11-dihydrodiol, 7-OH DMBA, DMBA, DBP.
3.2. Effects of oral and IP DMBA on bone marrow progenitor cell populations
Colony forming unit (CFU) assays provide a reproducible method to recapitulate hematopoiesis and quantify maturation and proliferation of bone marrow progenitor cells into lineage specific lymphoid progenitors (CFU-preB) and granulocyte progenitors (CFU-GM) ex vivo (Funk et al., 1994; Kincade, 1994; Lotem and Sachs, 2002; Nagasawa, 2006). We reasoned that DMBA might decrease numbers of bone marrow progenitors, or diminish their proliferation, before a reduction in bone marrow cell numbers would be observed. This proved to be the case. We observed at least 50 percent reduction in CFU-preB and CFU-GM numbers within 6 h after either oral or IP DMBA treatments (Fig. 2 A-D). These data suggest that both lineages of bone marrow progenitor cells were similarly susceptible to DMBA metabolites produced during the early peak blood levels of DMBA.
Fig 2. Reduction in bone marrow (BM) colony forming units (CFU-preB and CFU-GM) after IP or oral DMBA treatment.
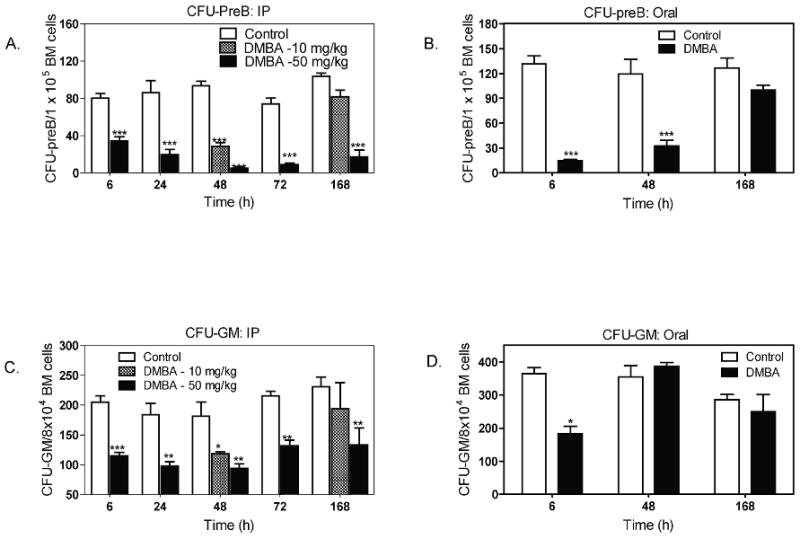
Mice received DMBA (10 or 50 mg/Kg) IP (A and C) or by oral gavage (50 mg/Kg) (B and D); control mice received the same volume (0.2 ml) of oil vehicle. At the indicated times, mice were euthanized and bone marrow cell suspensions prepared as described in the Methods. These were then used to prepare progenitor lymphoid cell (CFU-preB) and progenitor myeloid cell (CFU-GM) colony forming assays. Results are illustrated as the mean ± SEM of 4 to 6 animals per group. *p < 0.05, **p< 0.01, ***p< 0.001 as compared to control mice.
Because DMBA blood levels diminish rapidly after oral administration of DMBA, this route should illustrate whether the effects of DMBA on bone marrow progenitor cells are sustained in the absence of elevated blood levels of DMBA. We found that CFU-preB numbers were suppressed at 6 and 48 h after oral DMBA, but were almost fully restored to control levels by 168 h (Fig. 2B). In contrast, we observed greater suppression of CFU-preB after IP (Fig. 2A) than oral (Fig. 2B) DMBA treatment (50 mg/kg) at 168 hr (80% versus 20% reduction, respectively). Likewise CFU-GM remained suppressed through 168 h after IP DMBA treatment (50 mg/kg), whereas CFU-GM numbers had returned to untreated control levels by 48 h after oral DMBA (Fig. 2D).
Although examined only at 48 h and 168 h, mice given a 5 fold lower dose of DMBA IP (10mg/Kg), exhibited 60-70 % suppression of CFU-preB and CFU-GM at 48 h, that was nearly fully restored to control levels at 168 h (Fig. 2A,C). These latter responses are consistent with those seen after oral treatment with DMBA at 50 mg/kg (Fig. 2B, D).
3.3. Reduction in bone marrow cellularity follows decreased progenitor cell activity
IP administration of DMBA (50 mg/kg) resulted in an approximately 60% reduction in total numbers of bone marrow cells at 48 to 72 h, that recovered somewhat by 168 h (Fig. 3A). Oral DMBA produced a similar reduction in bone marrow cells at 48 h, with complete restoration of cell numbers at 168 h (Fig. 3B). When mice were treated IP with a fivefold lower dose of DMBA (10 mg/kg), we observed a reduction in cellularity at 48 h, and restoration to normal cell numbers at 168 h, that was similar to that exhibited by mice treated orally with 50 mg/kg DMBA. Flow cytometry analysis of bone marrow cells revealed differences between B220+ cells (pro-B and pre-B through mature B cells) and Gr-1+ granulocytes (Fig. 3C-F). After IP treatment, B220+ and Gr-1+ cells were both decreased by 50 percent or more at 48 h. However, B220+ cells decreased progressively in number through 168 h (>90 percent reduction) (Fig. 3C-D), whereas Gr-1+ myeloid cells (neutrophils and monocytes) were increased at 72 h and recovered to near control numbers by 168 h. As a result, myeloid cells became the predominant bone marrow population at 72 to 168 h (Fig. 3E-F). This switch to a relatively greater proportion of myeloid cells in the bone marrow parallels the swifter recovery of CFU-GM than CFU-preB activity after DMBA treatment (Fig. 2C-D). A similar trend was seen after oral DMBA treatment, where B220+ cells but not Gr-1+ cells remained reduced in number at 168 h.
Fig 3. Decreased bone marrow (BM) cellularity in mice following IP or oral DMBA treatment.
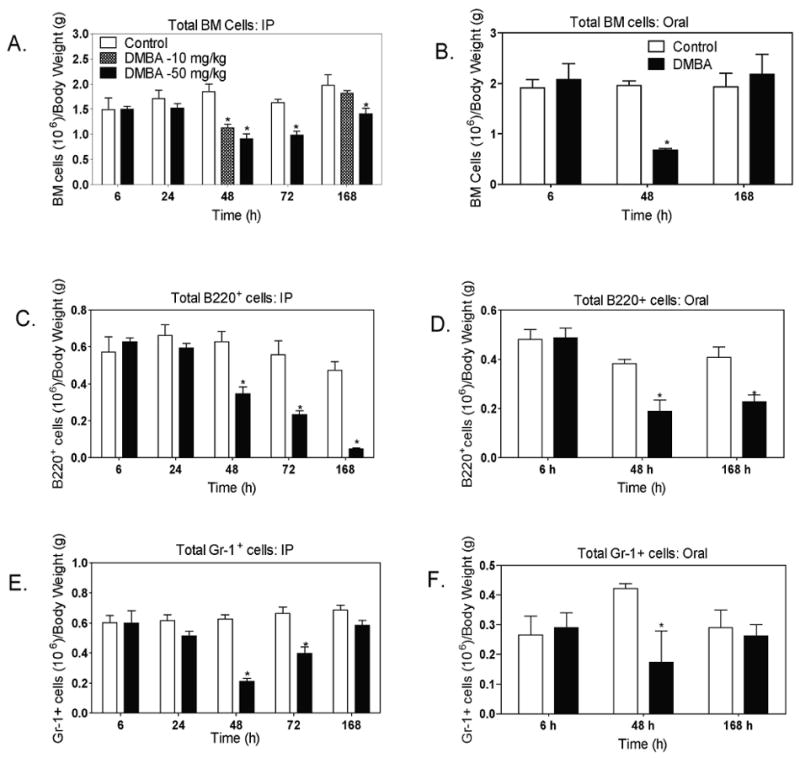
Mice received DMBA (10 or 50 mg/Kg) IP (A, C, and E) or by oral gavage (50 mg/Kg) (B, D, and F); control mice received the same volume (0.2 ml) of oil vehicle. At the indicated times, mice were euthanized. Bone marrow cell suspensions prepared as described in the Methods, and evaluated for total number of bone marrow cells (A and B); B220+ B-lymphocyte lineage cells (C and D)) and Gr-1+ myeloid cells (E and F). Total numbers of B220+ and Gr-1+ cells were estimated by multiplying the percentage of cells in that population by the total number of bone marrow cells for each mouse. Results are illustrated as the mean ± SEM of 4 to 6 animals per group. *p < 0.05, as compared to control mice. Data in some panels also include mice treated IP with 10mg/kg DMBA.
3.4. Effects of DMBA on peripheral blood cells
We were interested in assessing whether the changes in CFU activities and numbers of bone marrow cells would be reflected in numbers of peripheral blood leukocytes (WBC). We found that total numbers of WBC, as well as numbers of neutrophils and lymphocytes, were each reduced by 50% within 6 h after IP or oral DMBA (50 mg/kg) (Table 1, Fig. 4A - D). It was surprising that this leucopenia occurs at a time that precedes any obvious reduction in bone marrow cellularity. The reduction in total WBC and lymphocytes was sustained through 168 h in mice treated IP or orally. In contrast, numbers of peripheral blood neutrophils were reduced in IP DMBA treated mice at 6 h, but recovered to a level greater than control mice at later time points (48 to 168 hr) (Fig 4E). A similar 1.5 fold increase in numbers of neutrophils was seen at 168 hours after oral DMBA administration (Fig. 4F). The increase in peripheral blood neutrophils and decrease in lymphocytes at 168 h after DMBA administration are consistent with the switch in relative proportions of Gr-1+ myeloid cells and B 220+ lymphocytes in the bone marrow at 168 hours (Fig. 3C-F). This in turn follows the more rapid recovery of myeloid progenitor cells (CFU-GM) than lymphoid progenitor cells (CFU-preB) (Fig. 2A-D).
Table 1.
Peripheral blood cell differential cell counts (× 103 per μl) for Cyp1b1 knockout and wild type mice treated IP with DMBA or oil vehicle
| Endpoint | Time (h) | Wild type: control | Wild type: DMBA | Cyp1b1 knockout: Control | Cyp1b1 knockout: DMBA |
|---|---|---|---|---|---|
| Lymphocytes | 6 | 6.35 ± 0.58 | 3.29 ± 0.52* | 5.24 ± 0.43 | 5.01 ± 0.25 |
| 168 | 8.25 ± 0.61 | 3.36 ± 0.16* | 7.30 ± 0.85 | 7.19 ± 1.16 | |
| Neutrophils | 6 | 1.70 ± 0.22 | 0.90 ± 0.09* | 1.65 ± 0.23 | 2.56 ± 0.21* |
| 168 | 0.61 ± 0.10 | 1.20 ± 0.41* | 0.75 ± 0.10 | 5.71 ± 0.57* | |
| Monocytes | 6 | 0.2 ± 0.01 | 0.13 ± 0.03* | 0.32 ± 0.03 | 0.42 ± 0.02* |
| 168 | 0.22 ± 0.02 | 0.04 ± 0.00* | 0.23 ± 0.02 | 0.26 ± 0.06 |
Mice were injected IP with 50 mg/kg DMBA in 0.2 ml oil vehicle; controls received oil alone. Mice were euthanized at 6 or 168 h and blood collected by cardiac puncture. Total white blood cell and differential counts were performed as described in the methods.
Denotes p< 0.05 compared to vehicle control mice.
Fig 4. Reduction in peripheral blood leukocyte numbers following IP or oral DMBA treatment.
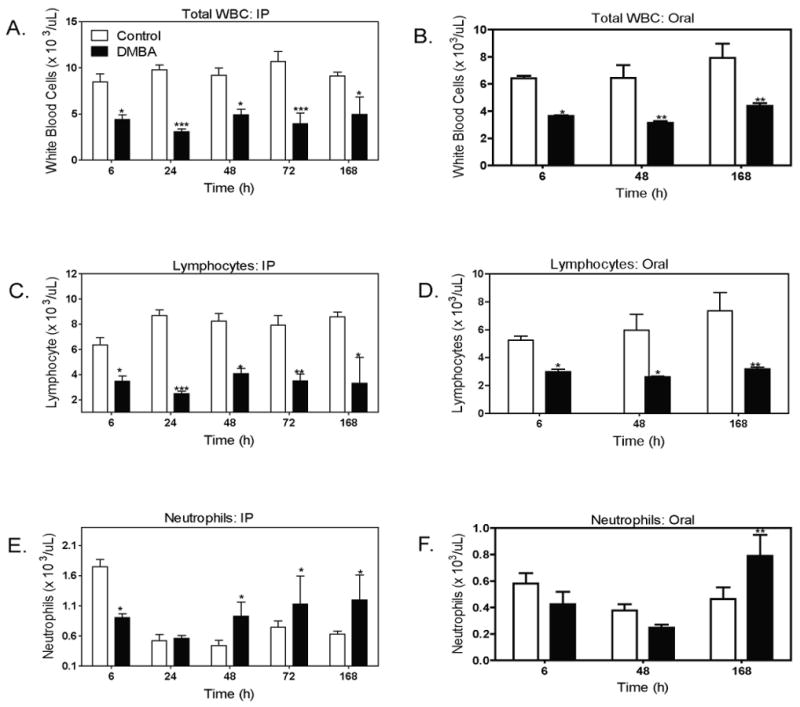
Mice were injected IP (A, C, E) or by oral gavage (B, D, F) with DMBA (50 mg/kg); control mice received oil vehicle. At the indicated time points, mice were euthanized and blood collected by cardiac puncture using EDTA as anticoagulant. Blood was then analyzed for total WBC numbers (A and B); total lymphocytes (C and D); neutrophils (E and F) using an ADVIA 120 hematology system. All cell counts are expressed as (cells × 103/μL). Results illustrate the mean ± SEM of 4 to 6 animals per group. *p < 0.05, **p< 0.01, ***p< 0.001 as compared to controls.
It is important to note that neither IP nor oral DMBA treatment caused any significant changes in red blood cell numbers, hematocrit or platelet numbers at any time point (data not shown). These findings indicate that DMBA selectively targets white blood cell rather than red cell or thrombocyte precursors in the bone marrow.
3.6. Cyp1b1-/- mice are resistant to the effects of DMBA
We have previously shown that DMBA does not cause bone marrow hypocellularity in Cyp1b1 -/- mice (Heidel et al., 2000; Galvan et al., 2005). Others have similarly reported no reduction in cellularity in the spleen or numbers of peripheral blood leukocytes in DMBA treated Cyp1b1 -/- mice (Gao et al., 2005; 2007; 2008). In the present study, Cyp1b1 -/- mice displayed no decrease in bone marrow cellularity (Fig. 5A), impairment of CFU-preB (Fig. 5B), nor (CFU-GM) activities (Fig. 5C) when treated IP with DMBA (50 mg/kg). We observed a paradoxical response to DMBA by peripheral blood cells in Cyp1b1 null mice (Fig. 5D, Table 1). As expected, peripheral blood lymphocytes were not suppressed by DMBA treatment in Cyp1b1 null mice. However, total peripheral blood cells were increased in DMBA treated Cyp1b1 null mice, largely as a result of a 7-fold increase in peripheral blood neutrophils (Table 1).
Fig 5. Cyp1b1-/- mice are resistant to the effects of DMBA.
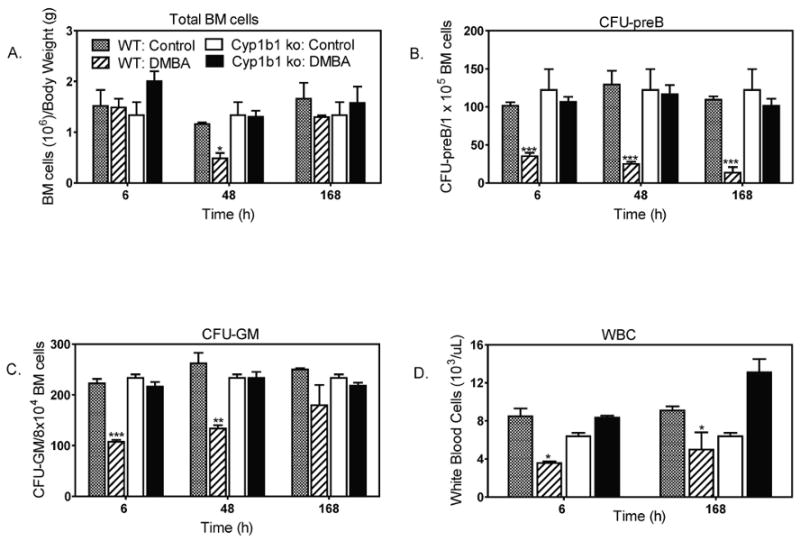
Wild type and Cyp1b1-/- mice were injected IP with DMBA (50 mg/kg) or vehicle. Results illustrate the mean ± SEM of 3 to 4 mice per group. Effects of DMBA on: (A) total bone marrow cellularity; (B) bone marrow progenitor lymphoid cells (CFU-preB); (C) Bone marrow progenitor myeloid cells (CFU-GM); (D) total WBC counts. *p < 0.05, **p< 0.01, ***p< 0.001 as compared to controls.
4. Discussion
In this study, we used colony forming unit (CFU) assays to interrogate mouse bone marrow cells for the effects of DMBA on hematopoiesis. We provide what we believe is the first report that DMBA treatment results in a rapid and sustained reduction in CFU-preB and CFU-GM activities. These data demonstrate that exposure to DMBA and it metabolites in vivo impairs the maturation and proliferation of lymphoid and myeloid progenitor cells. It is noteworthy that these differences were observed before any demonstrable reduction in bone marrow cellularity was evident. This observation speaks to a temporal link between early impairment (6 h) in the maturation and proliferation of bone marrow progenitor cells (reduction in CFU-preB and CFU-GM) and the subsequent reduction in numbers of lymphoid and myeloid lineage cells in the bone marrow at later time points (i.e. 48 h and later). It was also apparent that lymphoid and myeloid lineage cells displayed selective differences in their responses to DMBA. The diminution in CFU-preB and numbers of bone marrow B220+ cells was sustained longer than the transient suppression in CFU-GM and bone marrow Gr-1+ granulocytes. This difference was more apparent in mice that were given DMBA IP, rather than by oral gavage.
We infer that the prolonged reduction in progenitor cell activities (CFU-preB and CFU-GM) following IP DMBA administration reflects the slower clearance and sustained blood levels of DMBA after IP than oral administration (Fig. 1A). Similar ratios and proportions of circulating DMBA dihydrodiol metabolites were identified for both routes of administration (Fig. 1B). The more rapid clearance of orally administered DMBA allowed us to resolve the relative contributions of an early transient peak blood level of DMBA (similar for both oral and IP treated mice) versus the sustained DMBA blood levels seen after IP treatment. The greater and longer suppression of bone marrow lymphoid than myeloid cells likely results from restoration of myeloid progenitor cells (CFU-GM) as DMBA blood levels diminish, rather than differences in the initial susceptibility of CFU-preB and CFU-GM to disruption by DMBA treatment.
Differences in the transfer of DMBA to the liver after oral or IP administration probably accounts for the differing blood levels between the two routes of administration that were observed at later time points. Oral administration leads to a more direct transfer of polycyclic aromatic hydrocarbons (PAHs) from the intestine to the liver via the portal vein. Previous reports by Uno et al. (2004; 2006) for benzo(a)pyrene (BP) demonstrate the importance of first pass metabolism of orally delivered BP in the liver. However, in the BP studies mice were assessed after repeated oral administration of far higher doses (125 mg/kg/day, for 18 days) than the single dose of DMBA (50 mg/kg) used in the present study. The blood levels of DMBA following oral administration that we observed exhibited a comparable time course to that reported for BP (Uno et al., 2006) Orally administered DMBA reached peak blood levels at 1.5-3h (8 μM) and was substantially cleared from the blood by 6 hours. IP administration resulted in an early peak blood level of DMBA (10 to 12 μM) that was somewhat greater than the peak for orally administered DMBA, and then slowly declined (estimated halftime of approximately 48 hours) to a level approximately 30 percent of the peak blood level. We infer that the early peak DMBA blood levels in IP treated mice represents that proportion of the DMBA that rapidly entered peritoneal lymphatics and blood vessels, while the sustained blood levels represent that proportion of the lipophilic DMBA that partitioned into abdominal fat and then was slowly released. The importance of blood levels of DMBA was further demonstrated by decreasing the IP DMBA dose by fivefold (10 mg/Kg). This lower dose resulted in reductions in progenitor cell activities (CFU-preB and CFU-GM) (Fig. 2) and bone marrow cellularity (Fig. 3) that were comparable to those seen in orally treated mice.
The acute responses (6 h) of lymphoid and myeloid progenitors (CFU-preB and CFU-GM) were similar for oral and IP treated mice. These observations are consistent with the similar areas under the curve (AUC) for DMBA blood levels during the first 6 hours after administration. The progenitor cell responses are remarkably rapid in view of the time needed to form the likely toxicant, DMBA 3, 4 dihydrodiol epoxide, and to complete the biological processes that disrupt progenitor cells. The losses in bone marrow lymphoid (B220+) and myeloid (Gr1+) cell populations at 48 hours (Fig. 3 C-F) correspond closely with the earlier reductions in progenitor cell activities at 6 hours (Fig. 2A-D).
Peripheral blood lymphocytes responded as rapidly to DMBA as did bone marrow progenitor cells and remained depleted through 168 hours in both oral and IP treated mice. This finding suggests that the effects of DMBA on peripheral blood leukocytes involve factors beyond bone marrow progenitor cell activities. Interestingly, peripheral blood neutrophils rebounded to numbers 50 percent above time-matched control mice at 168 h. This was true for both IP and orally treated mice. This neutrophilia reflects the relative preponderance of myeloid versus lymphoid cells in the bone marrow at 72 to 168 h after DMBA treatment. Changes in peripheral blood cell populations may also be influenced by trafficking of peripheral blood cells from the bone marrow, spleen, lymphatics and other tissues, and by turnover of mature cells. Lymphopenia may reflect adverse effects of DMBA on other lymphoid organs, as reported previously (Gao et al., 2005; Gao et al., 2007).
We have previously shown that Cyp1b1 is required for DMBA-mediated suppression of bone marrow cellularity (Heidel et al., 2000; Galvan et al., 2005). Here we show that the acute loss of progenitor cells (CFU-preB and CFU-GM) and peripheral blood leukocytes are each dependent on Cyp1b1 (Fig. 5 and Table 1). Previous studies implicated both Cyp1b1 and epoxide hydrolase in DMBA mediated cell losses in the spleen and peripheral blood (Miyata et al., 1999; Gao et al. 2005; 2007; 2008). Cyp1b1 is expressed in most hematopoietic cells including lymphocytes, neutrophils and macrophages, but not in liver (Heidel et al 2000; Galvan et al 2005, Ward et al., 2004; Uno et al. 2006). Cyp1b1 is also appreciably expressed in mouse vascular endothelial cells (Tang et al., 2009). A role for both Cyp1b1 and epoxide hydrolase in bone marrow CFU activities is consistent with the suppression of DMBA 3, 4 dihydrodiol 1, 2 epoxide adducts in bone marrow by Cyp1b1 deletion (Galvan et al., 2006). We have reported minimal involvement of Cyp1b1 in producing circulating DMBA 3, 4 dihydrodiol (Halberg et al., 2008). This finding supports our hypothesis that the large differences in bone marrow cellularity between Cyp1b1 null and WT mice results from local metabolism of DMBA by Cyp1b1 at these sites (Galvan et al., 2003; Halberg et al., 2008). The acute effect of DMBA on peripheral blood cell numbers could arise in several ways. These include: 1) rapid effects on the spleen, where Cyp1b1 is highly expressed (Uno et al., 2006) and is involved in DMBA-mediated toxicity (Gao et al., 2005); 2) direct effects mediated by Cyp1b1 expression in peripheral blood lymphocytes, neutrophils or mononuclear phagocytes (Ward et al., 2004; Uno et al., 2006); 3) or through peripheral activation of lymphatics due to Cyp1b1-mediated peripheral toxicity (Buters et al., 1999; Buters et al., 2003).
The suppression of bone marrow CFU-preB and CFU-GM that we observed is more rapid than the previously reported suppression of lymphoid progenitors by TCDD (Murante and Gasiewicz, 2000; Singh et al., 2009). However, it should be noted that TCDD-mediated suppression results from AhR activation, rather than metabolism, and was not apparent until 24 h post TCDD treatment. The prolonged impairment of bone marrow progenitor cell responses (CFU-preB and CFU-GM) that we observed after IP DMBA treatment likely arises from either: 1) the slow turnover of bone marrow stem cells or early progenitor cells, therein retaining for several days the insult caused by DMBA metabolites; or 2) Cyp1b1 metabolism of DMBA within the bone marrow stroma, which triggers a sustained stress response that alters the paracrine regulation of bone marrow progenitor cells (Allan et al., 2003; Funk et al., 1994; Lotem and Sachs, 2002). In the first scenario, perhaps DMBA metabolites inhibit proliferation or differentiation of hematopoietic stem cells (HSCs), resulting in impaired maturation into committed lymphoid progenitor and committed myeloid progenitor cells. These committed lineage cells in turn may be subject to proliferation arrest, apoptosis (Mann et al., 1999; 2001; van Grevenynghe et al., 2003; 2004; 2006) or diminished chemokine regulation and homing to target sites (Lecureur et al., 2005; Monteiro et al., 2007; N'diaye et al., 2006; Nie et al., 2008; Pelayo et al., 2005; 2006). Any or all of these events could result in lower bone marrow CFU responses. In support of the second scenario, we have shown that TNF receptor signaling and p53-associated stress responses are also essential participants in DMBA-mediated loss of bone marrow cellularity (Page et al., 2003; Page et al., 2004).
In summary, the present study demonstrated that bone marrow lymphoid (CFU-preB) and myeloid (CFU-GM) progenitor cell activities are rapidly disrupted by DMBA. Analysis of bloodstream distribution of DMBA and its metabolites demonstrated that suppression of lymphoid progenitors (CFU-preB) was prolonged in mice with the sustained low blood levels of DMBA that occurred following IP administration. Conversely, the shorter lived DMBA blood levels following oral administration resulted in more rapid recovery of myeloid progenitors. We also document that DMBA metabolism by Cyp1b1 is required for suppression of CFU-preB, CFU-GM and bone marrow cell numbers. Our data provide further evidence that local metabolism of DMBA by Cyp1b1 in the bone marrow, rather than metabolism by Cyp1a1 in the liver (Uno et al., 2006), is critically important for the adverse effects of DMBA on bone marrow and peripheral blood leukocyte populations.
Acknowledgments
We thank Nancy Faith, Dr. Jeremy Sullivan and Dr. Robert Sahaghian for assistance in animal experiments, and Dr. Yumi Nakayama and Dr. M. Suresh for assistance with flow cytometry analysis. We appreciate the histopathological analysis of tissues from DMBA-treated mice by Dr. Deepa Rao (NIEHS, Raleigh NC).
Funding: This work was supported by US Public Health Service grant RO1 DK072749 (C.R.J) and the Walter and Martha Renk Endowed Laboratory for Food Safety (C.J.C)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allan LL, Mann KK, Matulka RA, Ryu HY, Schlezinger JJ, Sherr DH. Bone marrow stromal-B cell interactions in polycyclic aromatic hydrocarbon-induced pro/pre-B cell apoptosis. Toxicol Sci. 2003;76:357–365. doi: 10.1093/toxsci/kfg239. [DOI] [PubMed] [Google Scholar]
- Baron JM, Zwadlo-Klarwasser G, Jugert F, Hamann W, Rubben A, Mukhtar H, Merk HF. Cytochrome P450 1B1: a major P450 isoenzyme in human blood monocytes and macrophage subsets. Biochem Pharmacol. 1998;56:1105–1110. doi: 10.1016/s0006-2952(98)00105-1. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya KK, Brake PB, Eltom SE, Otto SA, Jefcoate CR. Identification of a rat adrenal cytochrome P450 active in polycyclic hydrocarbon metabolism as rat CYP1B1. Demonstration of a unique tissue-specific pattern of hormonal and aryl hydrocarbon receptor-linked regulation. J Biol Chem. 1995;270:11595–15602. doi: 10.1074/jbc.270.19.11595. [DOI] [PubMed] [Google Scholar]
- Buters JT, Sakai S, Richter T, Pineau T, Alexander DL, Savas U, Doehmer J, Ward JM, Jefcoate CR, Gonzalez FJ. Cytochrome P450 CYP1B1 determines susceptibility to 7, 12-dimethylbenz[a]anthracene-induced lymphomas. Proc Nat Acad Sci USA. 1999;96:1977–1982. doi: 10.1073/pnas.96.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buters J, Quintanilla-Martinez L, Schober W, Soballa VJ, Hintermair J, Wolff T, Gonzalez FJ, Greim H. CYP1B1 determines susceptibility to low doses of 7,12-dimethylbenz[a]anthracene-induced ovarian cancers in mice: correlation of CYP1B1-mediated DNA adducts with carcinogenicity. Carcinogenesis. 2003;24:327–334. doi: 10.1093/carcin/24.2.327. [DOI] [PubMed] [Google Scholar]
- Dean JH, Ward EC, Murray MJ, Lauer LD, House RV, Stillman W, Hamilton TA, Adams DO. Immunosuppression following 7,12-dimethylbenz[a]anthracene exposure in B6C3F1 mice--II. Altered cell-mediated immunity and tumor resistance. International journal of immunopharmacology. 1986;8:189–198. doi: 10.1016/0192-0561(86)90058-5. [DOI] [PubMed] [Google Scholar]
- Funk PE, Kincade PW, Witte PL. Native associations of early hematopoietic stem cells and stromal cells isolated in bone marrow cell aggregates. Blood. 1994;83:361–369. [PubMed] [Google Scholar]
- Galvan N, Jaskula-Sztul R, MacWilliams PS, Czuprynski CJ, Jefcoate CR. Bone marrow cytotoxicity of benzo[a]pyrene is dependent on CYP1B1 but is diminished by Ah receptor-mediated induction of CYP1A1 in liver. Toxicol Appl Pharmacol. 2003;193:84–96. doi: 10.1016/s0041-008x(03)00338-7. [DOI] [PubMed] [Google Scholar]
- Galvan N, Teske DE, Zhou G, Moorthy B, MacWilliams PS, Czuprynski CJ, Jefcoate CR. Induction of CYP1A1 and CYP1B1 in liver and lung by benzo(a)pyrene and 7,12-d imethylbenz(a)anthracene do not affect distribution of polycyclic hydrocarbons to target tissue: role of AhR and CYP1B1 in bone marrow cytotoxicity. Toxicol Appl Pharmacol. 2005;202:244–257. doi: 10.1016/j.taap.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Galvan N, Page TJ, Czuprynski CJ, Jefcoate CR. Benzo(a)pyrene and 7,12-dimethylbenz(a)anthrecene differentially affect bone marrow cells of the lymphoid and myeloid lineages. Toxicol Appl Pharmacol. 2006;213:105–116. doi: 10.1016/j.taap.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Gao J, Lauer FT, Dunaway S, Burchiel SW. Cytochrome P450 1B1 is required for 7,12-dimethylbenz(a)-anthracene (DMBA) induced spleen cell immunotoxicity. Toxicol Sci. 2005;86:68–74. doi: 10.1093/toxsci/kfi176. [DOI] [PubMed] [Google Scholar]
- Gao J, Lauer FT, Mitchell LA, Burchiel SW. Microsomal expoxide hydrolase is required for 7,12-dimethylbenz[a]anthracene (DMBA)-induced immunotoxicity in mice. Toxicol Sci. 2007;98:137–144. doi: 10.1093/toxsci/kfm089. [DOI] [PubMed] [Google Scholar]
- Gao J, Mitchell LA, Lauer FT, Burchiel SW. p53 and ATM/ATR regulate 7,12-dimethylbenz[a]anthracene-induced immunosuppression. Mol Pharmacol. 2008;73:137–46. doi: 10.1124/mol.107.039230. [DOI] [PubMed] [Google Scholar]
- Halberg RB, Larsen MC, Elmergreen TL, Ko AY, Irving AA, Clipson L, Jefcoate CR. Cyp1b1 exerts opposing effects on intestinal tumorigenesis via exogenous and endogenous substrates. Canc Res. 2008;68:7394–7402. doi: 10.1158/0008-5472.CAN-07-6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidel SM, MacWilliams PS, Baird WM, Dashwood WM, Buters JT, Gonzalez FJ, Larsen MC, Czuprynski CJ, Jefcoate CR. Cytochrome P4501B1 mediates induction of bone marrow cytotoxicity and preleukemia cells in mice treated with 7,12-dimethylbenz[a]anthracene. Canc Res. 2000;60:3454–3460. [PubMed] [Google Scholar]
- Igarashi H, Baba Y, Nagai Y, Jimi E, Ghosh S, Kincade PW. NF-kappaB is dispensable for normal lymphocyte development in bone marrow but required for protection of progenitors from TNFalpha. Int Immunol. 2006;18:653–659. doi: 10.1093/intimm/dxl002. [DOI] [PubMed] [Google Scholar]
- Kincade PW. B lymphopoiesis: global factors, local control. Proc Nat Acad Sci USA. 1994;91:2888–2889. doi: 10.1073/pnas.91.8.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecureur V, Ferrec EL, N'Diaye M, Vee ML, Gardyn C, Gilot D, Fardel O. ERK-dependent induction of TNFalpha expression by the environmental contaminant benzo(a)pyrene in primary human macrophages. FEBS Lett. 2005;579:1904–1910. doi: 10.1016/j.febslet.2005.01.081. [DOI] [PubMed] [Google Scholar]
- Lotem J, Sachs L. Cytokine control of developmental programs in normal hematopoiesis and leukemia. Oncogene. 2002;21:3284–3294. doi: 10.1038/sj.onc.1205319. [DOI] [PubMed] [Google Scholar]
- Mann KK, Matulka RA, Hahn ME, Trombino AF, Lawrence BP, Kerkvliet NI, Sherr DH. The role of polycyclic aromatic hydrocarbon metabolism in dimethylbenz[a]anthracene-induced pre-B lymphocyte apoptosis. Toxicol Appl Pharmacol. 1999;161:10–22. doi: 10.1006/taap.1999.8778. [DOI] [PubMed] [Google Scholar]
- Mann KK, Doerre S, Schlezinger JJ, Sherr DH, Quadri S. The role of NF-kappaB as a survival factor in environmental chemical-induced pre-B cell apoptosis. Molec Pharmacol. 2001;59:302–309. doi: 10.1124/mol.59.2.302. [DOI] [PubMed] [Google Scholar]
- Medina D, Stockman G, Griswold D. Significance of chemical carcinogen-induced immunosuppression in mammary tumorigenesis in BALB-c mice. Cancer research. 1974;34:2663–2668. [PubMed] [Google Scholar]
- Miyata M, Kudo G, Lee YH, Yang TJ, Gelboin HV, Fernandez-Salguero P, Kimura S, Gonzalez FJ. Targeted disruption of the microsomal epoxide hydrolase gene. Microsomal epoxide hydrolase is required for the carcinogenic activity of 7,12-dimethylbenz[a]anthracene. J Biol Chem. 1999;274:23963–23968. doi: 10.1074/jbc.274.34.23963. [DOI] [PubMed] [Google Scholar]
- Monteiro P, Gilot D, Le Ferrec E, Lecureur V, N'Diaye M, Le Vee M, Podechard N, Pouponnot C, Fardel O. AhR- and c-maf-dependent induction of beta7-integrin expression in human macrophages in response to environmental polycyclic aromatic hydrocarbons. Biochem Biophys Res Comm. 2007;358:442–448. doi: 10.1016/j.bbrc.2007.04.111. [DOI] [PubMed] [Google Scholar]
- Murante FG, Gasiewicz TA. Hemopoietic progenitor cells are sensitive targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J mice. Toxicol Sci. 2000;54:374–383. doi: 10.1093/toxsci/54.2.374. [DOI] [PubMed] [Google Scholar]
- N'diaye M, Le Ferrec E, Lagadic-Gossmann D, Corre S, Gilot D, Lecureur V, Monteiro P, Rauch C, Galibert MD, Fardel O. Aryl hydrocarbon receptor- and calcium-dependent induction of the chemokine CCL1 by the environmental contaminant benzo[a]pyrene. J Biol Chem. 2006;281:19906–19915. doi: 10.1074/jbc.M601192200. [DOI] [PubMed] [Google Scholar]
- Nagasawa T. Microenvironmental niches in the bone marrow required for B-cell development. Nature Rev. 2006;6:107–116. doi: 10.1038/nri1780. [DOI] [PubMed] [Google Scholar]
- Nagasawa T. The chemokine CXCL12 and regulation of HSC and B lymphocyte development in the bone marrow niche. Adv Exp Med Biol. 2007;602:69–75. doi: 10.1007/978-0-387-72009-8_9. [DOI] [PubMed] [Google Scholar]
- Nie L, Perry SS, Zhao Y, Huang J, Kincade PW, Farrar MA, Sun XH. Regulation of lymphocyte development by cell-type-specific interpretation of Notch signals. Mol Cell Biol. 2008;28:2078–2090. doi: 10.1128/MCB.00844-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page TJ, O'Brien S, Holston K, MacWilliams PS, Jefcoate CR, Czuprynski CJ. 7,12-Dimethylbenz[a]anthracene-induced bone marrow toxicity is p53-dependent. Toxicol Sci. 2003;74:85–92. doi: 10.1093/toxsci/kfg115. [DOI] [PubMed] [Google Scholar]
- Page TJ, MacWilliams PS, Suresh M, Jefcoate CR, Czuprynski CJ. 7-12 Dimethylbenz[a]anthracene-induced bone marrow hypocellularity is dependent on signaling through both the TNFR and PKR. Toxicol Appl Pharmacol. 2004;198:21–28. doi: 10.1016/j.taap.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Pelayo R, Welner R, Perry SS, Huang J, Baba Y, Yokota T, Kincade PW. Lymphoid progenitors and primary routes to becoming cells of the immune system. Curr Opin Immunol. 2005;17:100–107. doi: 10.1016/j.coi.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Pelayo R, Welner RS, Nagai Y, Kincade PW. Life before the pre-B cell receptor checkpoint: specification and commitment of primitive lymphoid progenitors in adult bone marrow. Sem Immunol. 2006;18:2–11. doi: 10.1016/j.smim.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annual Review of pharmacology and toxicology. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- Shimada T, Sugie A, Shindo M, Nakajima T, Azuma E, Hashimoto M, Inoue K. Tissue-specific induction of cytochromes P450 1A1 and 1B1 by polycyclic aromatic hydrocarbons and polychlorinated biphenyls in engineered C57BL/6J mice of arylhydrocarbon receptor gene. Toxicol Appl Pharmacol. 2003;187:1–10. doi: 10.1016/s0041-008x(02)00035-2. [DOI] [PubMed] [Google Scholar]
- Singh KP, Wyman A, Casado FL, Garrett R, Gasiewicz TA. Treatment of mice with the Ah receptor agonist and human carcinogen dioxin Results in altered numbers and function of hematopoietic stem cells. Carcinogenesis. 2009;30:11–19. doi: 10.1093/carcin/bgn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithgall TE, Harvey RG, Penning TM. Oxidation of the trans-3,4-dihydrodiol metabolites of the potent carcinogen 7,12-dimethylbenz(a)anthracene and other benz(a)anthracene derivatives by 3 alpha-hydroxysteroid-dihydrodiol dehydrogenase: effects of methyl substitution on velocity and stereochemical course of trans-dihydrodiol oxidation. Cancer research. 1988;48:1227–1232. [PubMed] [Google Scholar]
- Smithgall TE. Signal transduction pathways regulating hematopoietic differentiation. Pharmacol Rev. 1998;50:1–19. [PubMed] [Google Scholar]
- Tang Y, Scheef EA, Wang S, Sorenson CM, Marcus CB, Jefcoate CR, Sheibani N. CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood. 2009;113:744–754. doi: 10.1182/blood-2008-03-145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurmond TS, Gasiewicz TA. A single dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin produces a time- and dose-dependent alteration in the murine bone marrow B-lymphocyte maturation profile. Toxicol Sci. 2000;58:88–95. doi: 10.1093/toxsci/58.1.88. [DOI] [PubMed] [Google Scholar]
- Thurmond TS, Staples JE, Silverstone AE, Gasiewicz TA. The aryl hydrocarbon receptor has a role in the in vivo maturation of murine bone marrow B lymphocytes and their response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol. 2000;165:227–236. doi: 10.1006/taap.2000.8942. [DOI] [PubMed] [Google Scholar]
- Uno S, Dalton TP, Derkenne S, Curran CP, Miller ML, Shertzer HG, Nebert DW. Oral exposure to benzo[a]pyrene in the mouse: detoxification by inducible cytochrome P450 is more important than metabolic activation. Mol Pharmacol. 2004;65:1225–1237. doi: 10.1124/mol.65.5.1225. [DOI] [PubMed] [Google Scholar]
- Uno S, Dalton TP, Dragin N, Curran CP, Derkenne S, Miller ML, Shertzer HG, Gonzalez FJ, Nebert DW. Oral benzo[a]pyrene in Cyp1 knockout mouse lines: CYP1A1 important in detoxification, CYP1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol Pharmacol. 2006;69:1103–1114. doi: 10.1124/mol.105.021501. [DOI] [PubMed] [Google Scholar]
- van Grevenynghe J, Rion S, Le Ferrec E, Le Vee M, Amiot L, Fauchet R, Fardel O. Polycyclic aromatic hydrocarbons inhibit differentiation of human monocytes into macrophages. J Immunol. 2003;170:2374–2381. doi: 10.4049/jimmunol.170.5.2374. [DOI] [PubMed] [Google Scholar]
- van Grevenynghe J, Sparfel L, Le Vee M, Gilot D, Drenou B, Fauchet R, Fardel O. Cytochrome P450-dependent toxicity of environmental polycyclic aromatic hydrocarbons towards human macrophages. Biochem Biophys Res Comm. 2004;317:708–716. doi: 10.1016/j.bbrc.2004.03.104. [DOI] [PubMed] [Google Scholar]
- van Grevenynghe J, Monteiro P, Gilot D, Fest T, Fardel O. Human endothelial progenitors constitute targets for environmental atherogenic polycyclic aromatic hydrocarbons. Biochem Biophys Res Comm. 2006;341:763–769. doi: 10.1016/j.bbrc.2006.01.028. [DOI] [PubMed] [Google Scholar]
- Ward EC, Murray MJ, Lauer LD, House RV, Irons R, Dean JH. Immunosuppression following 7,12-dimethylbenz[a]anthracene exposure in B6C3F1 mice. I. Effects on humoral immunity and host resistance. Toxicol Appl Pharmacol. 1984;75:299–308. doi: 10.1016/0041-008x(84)90212-6. [DOI] [PubMed] [Google Scholar]
- Ward JM, Nikolov NP, Tschetter JR, Kopp JB, Gonzalez FJ, Kimura S, Siegel RM. Progressive glomerulonephritis and histiocytic sarcoma associated with macrophage functional defects in CYP1B1-deficient mice. Toxicol Pathol. 2004;32:710–8. doi: 10.1080/01926230490885706. [DOI] [PubMed] [Google Scholar]
- Wilson NM, Christou M, Turner CR, Wrighton SA, Jefcoate CR. Binding and metabolism of benzo[a]pyrene and 7,12-dimethylbenz[a]anthracene by seven purified forms of cytochrome P-450. Carcinogenesis. 1984;5:1475–1483. doi: 10.1093/carcin/5.11.1475. [DOI] [PubMed] [Google Scholar]