

Abstract
Background
Ghrelin is a potent orexigenic hormone that likely impacts eating via several mechanisms. Here, we hypothesized that ghrelin can regulate extra-homeostatic, hedonic aspects of eating behavior.
Methods
In the current study, we assessed the effects of different pharmacological, physiological and genetic models of increased ghrelin and/or ghrelin signaling blockade on two classic behavioral tests of reward behavior: conditioned place preference (CPP) and operant conditioning.
Results
Using both CPP and operant conditioning, we found that ghrelin enhanced the rewarding value of high-fat diet (HFD) when administered to ad lib-fed mice. Conversely, wild-type mice treated with ghrelin receptor antagonist and ghrelin receptor-null mice both failed to show CPP to HFD normally observed under calorie restriction. Interestingly, neither pharmacologic nor genetic blockade of ghrelin signaling inhibited the body weight homeostasis-related, compensatory hyperphagia associated with chronic calorie restriction. Also, ghrelin's effects on HFD reward were blocked in orexin-deficient mice and wild-type mice treated with an orexin 1 receptor antagonist.
Conclusions
Our results demonstrate an obligatory role for ghrelin in certain rewarding aspects of eating that is separate from eating associated with body weight homeostasis and that requires the presence of intact orexin signaling.
Keywords: ghrelin, orexin, food reward, food intake
Introduction
Food intake involves a well-integrated regulatory system in which hormones that sense changes in the body's energy stores interact with homeostatic brain circuits to maintain body weight and also interact with reward circuits to drive the consumption of rewarding foods (1). However, little is known about those processes mediating food reward or the relative contributions of feeding-related hormones to homeostatic vs. hedonic controls of feeding behavior.
One likely candidate to mediate both body weight homeostasis and food reward processing is the orexigenic hormone ghrelin (2). Preprandial and calorie restriction-associated increases in plasma ghrelin and hypothalamic expression of ghrelin receptors (GHSRs) suggest a role for ghrelin in body weight homeostasis-related eating (3-6). GHSRs also are expressed in and ghrelin interacts with several brain regions involved with reward processing (7-9). For instance, ghrelin increases action potential frequency in ventral tegmental area (VTA) neurons and induces dopamine release into the nucleus accumbens (10-12). VTA microinjection of ghrelin increases food intake while VTA microinjection of a GHSR antagonist decreases food intake in response to i.p.-injected ghrelin (10, 13). Ghrelin also activates orexin neurons and increases food intake when microinjected into the lateral hypothalamic area (LHA) (14-17). Furthermore, ghrelin is taken up by and increases spine synapse density within the hippocampus (18). Thus, it has been hypothesized that ghrelin can affect various reward behaviors (7, 10). Such is supported by work showing that ghrelin augments cocaine hyperactivity, lowers the threshold dose of cocaine required to establish a CPP, is required for alcohol reward and itself can elicit CPP (19-22). Ghrelin induces anti-depressant-like properties, which are known to involve regulation of brain reward circuits including those in which orexin participates (23). Importantly, ghrelin also increases neuronal activity in brain reward centers in humans shown images of appealing foods (24). However, whether ghrelin affects specific behaviors associated with rewarding aspects of eating remains unknown. The experiments within this report were designed to further characterize ghrelin's effects on food reward.
Methods and Materials
Animals and housing
Male mice were housed in a 12-h light/dark cycle with regular chow (RC; 4 g% fat, diet #7001, Harlan-Teklad, Madison, WI), which provides 2.9 kcal/g of energy, and water available ad lib, except when indicated. All animal procedures were carried out in accordance with NIH guidelines and UTSW Institutional Animal Care and Use Committee guidelines. Adult (8-10 weeks old) C57BL6/J mice were from Jackson Laboratory. Orexin-deficient mice and GHSR-null mice with their respective wild-type littermates were generated as reported previously (25, 26). Study animals of both genetic models were derived from crosses between heterozygous animals back-crossed > 10 generations onto a C57BL6/J genetic background.
Conditioned place preference task
We used a balanced paradigm in a three-chamber apparatus (Med Associates Inc., St Albans, VT), which consisted of a central, brightly-lit shuttle chamber (4.5 × 6 inches) between two dimly-lit, larger conditioning chambers (9.5 × 6 inches each), which differed from each other in wall pattern and floor texture. The animal's location within the chamber was monitored by photobeams (Med PC IV software, Med Associates Inc.). On Day 0, animals were exposed in their home cage to a HFD pellet weighing ∼1.0 g to avoid neophobia during the task. A pretest session was performed on Day 1, during which a single mouse was placed in the shuttle chamber of the CPP apparatus and allowed free access to the adjacent conditioning chambers in the absence of food, for 20 min. The amount of time the mouse spent in each chamber was used to calculate a pretest CPP score upon subsequent assignment of the conditioning chambers to either RC or HFD (CPP score = time spent in chamber paired to HFD minus time spent in chamber paired to RC). We assigned the HFD-paired side in a balanced manner such that the mean pretest CPP score was close to zero. Conditioning sessions were performed on Days 2-13. On even days, mice were confined to one conditioning chamber in the presence of a HFD pellet for 30 min. On odd days, mice were confined to the other conditioning chamber in the presence of an RC pellet for 30 min. The RC and HFD pellets used each contained 2.4 kcal. The RC pellets are described above. The HFD pellets were from Research Diets [Rodent Chow #D12331, New Brunswick, NJ, which provides 5.56 kcal/g of energy (35.8 g% of fat or 58 kcal% of fat)]. The test session was performed on Day 14, during which the mice were placed in the shuttle chamber and allowed free access to the adjacent conditioning chambers, in the absence of food, for 20 min. Test CPP scores were determined, as above.
Operant Responding Task
Mice were trained to poke their nose into a lit portal to obtain a 20 mg custom-prepared HFD pellet reward (see Supplementary Materials) in standard operant conditioning chambers (Model ENV307A, Med Associates Inc.) equipped with three nose poke portals. Mice were rewarded for nose poking in the middle portal only; the side portals were inactive but monitored. During the training period, mice were allowed access to an amount of RC corresponding to 60% of their average daily food intake. For the training sessions, mice initially received the HFD pellet rewards under a fixed ratio schedule (from a fixed ratio 1 schedule up to a fixed ratio 5 schedule). Then, mice were moved to a stepped progressive ratio reinforcement schedule, whereby the response requirement for each successive pellet was raised by progressive increments according to the following series: 5, 10, 20, 30, 50, 70, 100, 130, etc. Breakpoint was defined as the last progressive ratio which an animal successfully completed to receive a reinforcement within a 10 min period. Once mice demonstrated a stable 10 min breakpoint for 3 successive days, they were allowed ad lib access to RC, and continued to be challenged in the operant chambers. When they again showed a stable 10 min breakpoint in the ad lib-fed condition for 3 successive days, testing began.
Immunohistochemistry
Free-floating coronal sections of mouse brains (25 μm thickness) were processed sequentially by dual-label immunohistochemistry for c-Fos expression and then orexin expression, as previously described, with minor modifications (29). Antisera used included anti-c-fos antibody (Calbiochem/Oncogene, Temecula, CA, cat# PC38, 1:30,000) and anti-orexin antibody (Phoenix Pharmaceuticals, cat# H-003-30, 1:10,000). Quantitative analysis was performed in four animals per condition (ghrelin vs. saline administration during conditioning). All orexin neurons on both the left and right sides of every fifth section of the brain were counted (486 ± 39 and 529 ± 24 neurons, per mouse in saline- and ghrelin-treated groups, respectively).
Statistics
Data are expressed as mean ± s.e.m. The paired t-test was used to compare pretest vs. test CPP scores within each CPP group. For the operant conditioning experiments, paired t-test was used to compare the ghrelin vs. saline breakpoint in each group of mice (vehicle-pretreated wild-type mice, SB-334867-pretreated wild-type mice, and vehicle-pretreated orexin-deficient mice). ANOVA was performed when assessing the effects of genotype or treatment on ghrelin-induced food intake. p < 0.05 was considered statistically significant for all the experiments. 0.05 ≤ p < 0.09 was considered evident of a statistical trend. No significant differences (p > 0.09) were observed between the three groups of wild-type animals (those from Jackson Laboratory vs. wild-type littermates of the orexin-deficient mice vs. wild-type littermates of the GHSR-null mice) in any measures taken, and thus their data were pooled. See Supplementary materials for further details about the experimental procedures.
Results
To investigate a role for ghrelin in the rewarding properties of HFD, we adapted a version of the CPP task which is typically used in drug abuse studies. In this task, mice were conditioned to associate one chamber of the CPP apparatus with HFD and a 2nd chamber with an equal calorie amount of regular chow (RC). After a 12 day conditioning period (6 days on the HFD-paired side alternating every-other-day with 6 days on the RC-paired side), mice were permitted free access to both chambers in the absence of food. CPP scores were calculated by subtracting the time spent in the RC-paired chamber from the time spent in the HFD-paired chamber. Place preference for HFD was demonstrated by a positive CPP score, meaning that the mice preferred to spend time in the chamber that they associate with the more rewarding food. To ascertain whether ghrelin modulates CPP for HFD, ad lib-fed wild-type mice were injected with ghrelin (2 μg/g BW) or saline 20 min prior to placement in the CPP apparatus on the test day. Saline-treated mice spent a similar amount of time in each chamber after the conditioning period (on the test day) as before the conditioning period (on the pretest day; Fig. 1A, n=29). In contrast, mice treated with ghrelin prior to testing spent significantly more time in the HFD-paired chamber after the conditioning period (Fig. 1A, n=15). This finding suggests that rises in ghrelin are sufficient to induce expression of CPP for HFD or may act like a cue used in the expression of CPP for HFD. Next, mice were treated with ghrelin (2 μg/g BW) prior to each conditioning session, but not on the test day. Mice treated this way displayed a clear preference for the HFD-paired chamber (Fig. 1A, n=22), indicating that rises in ghrelin also enable acquisition of CPP for HFD. To confirm the specificity of our findings, we repeated the experiment using GHSR-null mice (Fig. 1B). Saline-treated GHSR-null mice spent a similar amount of time in each chamber after and before the conditioning period (n=20). In contrast to wild-type mice, GHSR-null mice did not elicit CPP for HFD upon administration of ghrelin (2 μg/g BW) either prior to testing (n=16) or prior to each conditioning session (n=16).
Figure 1.

Ghrelin affects the rewarding value of HFD in a food CPP task. A. Administration of ghrelin (2 μg/g BW, s.c.) to ad lib-fed wild-type mice either acutely before the test or before each conditioning session increases the preference for the HFD-paired chamber. B. Administration of ghrelin (2 μg/g BW, s.c.) to ad lib-fed GHSR-null mice either acutely before the test or before each conditioning session fails to affect the preference for the HFD-paired chamber. Data represent the mean ± s.e.m. ** p<0.01, *** p<0.0001.
It is well known that ghrelin is physiologically increased upon calorie restriction, and that in this situation there is an enhancement in food CPP performance (30, 31). To determine whether the enhanced food CPP performance associated with calorie restriction is due to the elevated levels of endogenous ghrelin associated with calorie restriction, we performed CPP experiments using a recently reported GHSR antagonist, Compound 26 (27, 28). Calorie-restricted mice were provided ad lib access to RC in their home cages between 12:00 p.m. and 4:00 p.m. Mice required one week to adjust their food intake amount to this 4-h calorie restriction experimental paradigm. Thus, during the first week of calorie restriction, mice gradually increased and then stabilized their daily intake of RC during the 4 h that food was available, as follows: 1.36±0.10, 1.55±0.18, 1.84±0.13, 2.13±0.07, 2.11±0.09, 2.27±0.12, and 2.42±0.13 g of RC. Behavioral studies on calorie restricted mice were started following this initial adjustment week. Throughout the remaining days of the experiment, the mice ate an average of 2.87±0.15 g of RC (nearly 8.0 kcal/day), which represents approximately 70% of the daily food intake of ad lib-fed wild type mice in our experimental condition (4.09±0.14 g or nearly 11.9 kcal of RC). After the initial adjustment week, calorie restricted mice maintained body weights that were about 85% the average body weights of the ad lib-fed wild- type mice. Food CPP tasks were started after the initial adjustment week and were performed just prior to the 4 h period of food availability, when plasma acylated ghrelin levels were found to be increased two-fold (13.2±1.0 vs. 5.8±0.6 pg/mL in calorie-restricted vs. ad lib-fed mice, p<0.01). Compound 26 (30 μg/g BW) was administered either 1 h before the test or 1 h before each conditioning session to calorie-restricted mice. As expected, calorie-restricted mice treated with vehicle spent more time in the HFD-paired chamber on the test day as compared to the pretest day (Fig. 2A, n=34). Calorie-restricted mice treated with Compound 26 only on the test day also displayed a higher mean test CPP score than pretest CPP score (Fig. 2A, n=10). In contrast, Compound 26 given during the conditioning period completely blocked the acquisition of food CPP in calorie-restricted mice (Fig. 2A, n=14). Thus, the endogenous rise in ghrelin that occur in the setting of calorie restriction is required for acquisition but not expression of CPP for HFD.
Figure 2.

Ghrelin signaling is required for the enhancement in food CPP performance induced by calorie restriction. A. The GHSR antagonist Compound 26 (30 μg/g BW, via gavage) blocks the acquisition of food CPP in calorie-restricted mice when used before each conditioning session, whereas it is ineffective when administrated only on the test day. B. In contrast to wild-type mice, GHSR-null mice fail to show food CPP under a mild calorie-restriction protocol. Data represent the mean ± s.e.m. * p<0.05, **** p<0.000001.
To corroborate our findings with Compound 26, we also performed CPP experiments using GHSR-null mice. For these experiments, GHSR-null and wild-type littermates were calorie restricted during the week preceding conditioning and throughout the pretest, conditioning and test days. Instead of the 4-h calorie restriction paradigm used above, here animals were provided access to an amount of RC corresponding to 80% of their average daily food intake [an average of 3.31±0.21 g of RC (nearly 9.5 kcal/day)]. Both groups of 80% calorie restricted mice maintained body weights that were about 95% the average body weights of ad lib-fed mice. Food CPP tasks were performed just prior to the feeding period. Using this 80% calorie restriction protocol, wild-type mice spent more time in the HFD-paired chamber on the test day as compared to the pretest day (Fig. 2B, n=22). In contrast, GHSR-null mice failed to show a calorie restriction-induced CPP (Fig. 2B, n=22). Thus, both genetic and pharmacologic interference with endogenous ghrelin signaling have the ability to block CPP to HFD. Of note, different results were observed with the GHSR-null mice when the more food-limiting 4-h calorie restriction paradigm was used instead of the 80% calorie restriction. Under the 4-h protocol, both wild-type mice and GHSR-null littermates demonstrated CPP for HFD (Fig. S3).
It is well established that following periods of calorie restriction, mice respond with a homeostatic drive to replenish energy stores through hyperphagia. Interestingly, in our experiments described above, Compound 26 administration to calorie-restricted mice only on the test day or only during the conditioning period both failed to block the compensatory hyperphagia observed at the end of the test when the mice were given free access to RC (Fig. 3A). Similarly, 80% calorie-restricted wild-type mice and GHSR-null littermates both experienced compensatory hyperphagia at the end of the test when the mice were given free access to RC (Fig. 3B). As such, while calorie restriction-associated increases in endogenous ghrelin are required to demonstrate CPP for HFD (a measure of food incentive value), ghrelin signaling does not appear to be required for the compensatory hyperphagia (a measure of homeostatic drive) that normally follows chronic calorie restriction.
Figure 3.

Ghrelin signaling is not required for the compensatory hyperphagia that follows chronic calorie restriction. A. Compound 26 administered before either the test session or each conditioning session fails to block calorie restriction-associated overeating of freely available food. B. Wild-type and GHSR-null mice have similar levels of calorie restriction-associated overeating of freely available food. Data represent the mean ± s.e.m.
As previous work indicates that orexin neurons become activated by cues associated with different rewards, including addictive drugs and food (32), we next investigated whether orexin neurons contribute to ghrelin's enhancement of CPP for HFD. We assessed c-fos expression, a marker of neuronal stimulation, within orexin neurons in mice given either saline or ghrelin prior to each conditioning session but not prior to testing. For this procedure, brains were collected from mice 2 h following their test day exposure to the CPP apparatus. Mice that exhibited ghrelin-induced acquisition of food CPP showed increased c-fos expression within LHA orexin neurons. Quantitative analysis indicated that 29.0 ± 5.2% of orexin-positive neurons were positive for c-fos in the LHAs of saline-treated mice, while in the LHAs of ghrelin-treated mice, 43.2 ± 2.3% of orexin-positive neurons were positive for c-fos (p<0.05 vs. saline-treated mice, Fig. 4A-B). The distribution of the orexin-positive neurons positive for c-fos in the LHA did not show any particular topography within the LHA.
Figure 4.
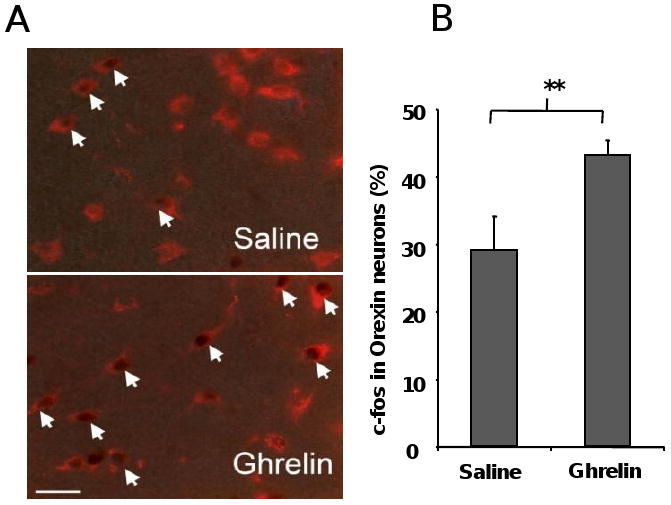
Ghrelin-induced acquisition of food CPP activates orexin neurons in the lateral hypothalamus. Representative photomicrographs of brains processed by dual-label immunohistochemistry for orexin (red) and c-fos (brown) (A) and quantification of dual-labeled neurons (B) demonstrating that mice that exhibited ghrelin-induced preference for the HFD-paired chamber show increased c-fos induction within LHA orexin cells. Dual-labeled neurons are identified by arrows. Data represent the mean ± s.e.m. ** p<0.01.
Next, we determined if orexin signaling is required for the ghrelin-induced acquisition of food CPP. Administration of the orexin receptor 1 selective antagonist SB-334867 (10 μg/g BW) to mice subsequently receiving ghrelin before each conditioning session blocked the acquisition of food CPP (Fig. 5A) while pretreatment with SB-334867 alone (without subsequent ghrelin) did not affect the performance in the food CPP protocol (Fig. S4). This dose of SB-334867 was shown to have no anorexigenic (or orexigenic) effect on intake of freely-available food (data not shown). In addition, orexin-deficient mice (25) failed to show ghrelin-induced acquisition of food CPP (Fig. 5A). SB-334867-pretreated mice and orexin-deficient mice both displayed full orexigenic responses to ghrelin (for both HFD and RC) as measured during the conditioning phase of the CPP trial (Fig. 5B). Thus, orexin pathways facilitate the ghrelin-induced acquisition of food CPP, however, ghrelin-induced stimulation of intake of freely-available food seems to be independent of orexin signaling under these particular experimental conditions.
Figure 5.

Ghrelin's actions on food CPP require intact orexin signaling. A. Orexin 1 receptor antagonist (SB-334867, 10 μg/g BW, i.p.)-pretreated mice and orexin-deficient mice fail to demonstrate ghrelin-induced acquisition of food CPP. B. SB-334867-pretreated and orexin-deficient mice show similar ghrelin-induced increases in food intake for both HFD and RC after 30 min of treatment. Data represent the mean ± s.e.m. ****, p<0.00001.
Finally, we tested mice using an operant conditioning protocol under a progressive ratio schedule (33). In this measure of food motivation, the number of nose pokes required to obtain each successive reinforcing HFD pellet increases by progressive increments. The session continues until a 10 min period elapses during which the mouse does not obtain a HFD pellet. The last progressive ratio successfully completed before the end of the session is defined as the breakpoint. Ad lib-fed mice receiving ghrelin (2 μg/g BW) 10 min prior to placement into the operant chamber showed a higher breakpoint as compared to saline-treated mice (Fig. 6, n=19). Pretreatment of ad lib-fed mice with SB-334867 (10 μg/g BW) blocked the ghrelin-induced increase in breakpoint (Fig. 6, n=16), without altering the performance of mice injected with saline. In addition, orexin-deficient mice treated with ghrelin also failed to show a significant increase of the breakpoint (Fig. 6, n=5). These results confirm that ghrelin increases the motivation to obtain HFD pellets, and that this behavior requires intact orexin signaling.
Figure 6.

Ghrelin increases the motivation to obtain HFD pellets in an orexin-dependent manner. Ghrelin increases the breakpoint for HFD pellets in an operant conditioning trial using a progressive ratio schedule. In contrast, SB-334867-pretreated (10 μg/g BW, i.p.) and orexin-deficient mice fail to show a ghrelin-induced increase of breakpoint for HFD pellets. Data represent the mean ± s.e.m. ** p<0.01
Discussion
The present study identifies a required role for ghrelin in mediating certain hedonic and motivational components of eating. Our results, together with the previous fMRI findings of ghrelin-induced increases in the neural response to food pictures within human brain regions implicated in encoding the incentive value of food cues, allow us to add food reward to a growing list of other reward behaviors, including cocaine-seeking and alcohol-reward, influenced by ghrelin (21, 22, 24). Furthermore, the ghrelin-orexin pathway investigated here appears to be distinct from those circuitries controlling homeostatic drives that promote food intake in response to reduced energy stores, and therefore is highly relevant to the current obesity crisis where food intake occurs in the context of adequate adipose reserves. These findings raise several interesting considerations.
Mechanism of Action
Our food CPP studies reveal several intricacies regarding ghrelin's roles in modulation of the rewarding value of HFD and food intake. In particular, pharmacologic increases in ghrelin are sufficient to enhance both the associative learning of HFD's reward value (or rather, the acquisition of CPP for HFD) and its retrieval (expression of CPP for HFD). However, our studies with Compound 26 suggested that the physiological increases in ghrelin associated with calorie restriction are only important for acquisition of CPP for HFD, but are not required for expression of the learned association. In this regard, ghrelin signaling pathways seem to behave similarly to leptin and insulin. Just as does ghrelin, leptin and insulin act at the central level to regulate energy homeostasis and food reward behaviors [as reviewed in (31)]. In fact, differential effects on acquisition and expression of food CPP also have been demonstrated for both leptin and insulin, although in contrast to ghrelin, both hormones specifically block the expression of food CPP, but not the acquisition of food CPP (34). It is highly likely that ghrelin works in concert with other satiety and body weight related peripheral hormones, such as leptin and insulin, to fine tune the body's responses to rewarding substances such as HFD.
In the present study, we also were able to dissociate the obligatory role for ghrelin in food reward-related eating behaviors from eating that occurs as part of the process to maintain body weight homeostasis. In particular, our restricted access to food protocol highlights previous studies showing that caloric restriction induces not only compensatory hyperphagia when free access to food is again permitted, but also various food reward behaviors. However, we now show that while pharmacologic interference with ghrelin action does block HFD reward behavior associated with chronic caloric restriction, it does not block the rebound overeating that occurs when animals are switched from restricted to ad lib access to food. Similar results were observed using a genetic method of interfering with endogenous ghrelin action: mice lacking GHSR failed to show CPP for HFD upon testing under a moderate calorie restriction protocol (exposure to 80% of average food intake). Similar to the effects of Compound 26 in wild-type mice, genetic deletion of GHSR did not affect the compensatory hyperphagia normally observed in wild-type mice upon cessation of the moderate calorie restriction protocol. This observed dissociation may be due to inherent redundancies in pathways responsible for body weight homeostasis-related eating that are not present in the machinery controlling food reward behavior.
Our CPP results using 80% calorie restricted GHSR-null vs. wild-type mice support our CPP results using Compound 26-treated vs. vehicle-treated calorie-restricted wild-type mice (namely, that genetic and pharmacologoic interference with endogenous ghrelin signaling both block CPP for HFD associated with calorie restriction). However, the same was not true when the more limiting 4-h calorie restriction paradigm was used for the genetic model. The exact reason for this discrepancy is unclear. Certainly, mice do experience a more limiting exposure to food with the 4-h protocol than with the 80% protocol, but it is uncertain how such a change would offset the effects of GHSR deletion observed with the more moderate 80% restriction protocol. We speculate but cannot confirm that developmental compensations within the GHSR-null model, which may not be as obvious under the less food restrictive conditions, may be at fault. Developmental compensations were proposed previously to explain the finding of normal rebound hyperphagia in GHSR-knockout mice following an acute fast but blunted rebound feeding in acutely fasted wild-type mice given a GHSR antagonist (10).
Neural Circuitry
Our data reveal that ghrelin's action on food reward requires orexin signaling as evidenced by the finding that ghrelin's effects on CPP and operant conditioning were blocked in orexin-deficient mice and wild-type mice given an orexin 1 receptor antagonist. Yet, the exact neuronal pathway (or pathways) that link ghrelin and orexin to food reward remains unknown. The most obvious pathway would involve direct binding of ghrelin to GHSRs present on orexin neurons. Such would be supported by previous studies demonstrating GHSRs within the LHA of rat (8) as well as those showing that ghrelin can induce action potentials and depolarization in isolated orexin neurons (17). In turn, these ghrelin-engaged orexin-containing LHA neurons presumably would then project to the VTA, where activation of mesolimbic dopaminergic circuitry would ensue. In fact, previous work has demonstrated that orexin neurons in the LHA are critical in reward seeking behaviors (32), and that orexin action in the VTA is involved in activation of mesolimbic dopaminergic reward circuitry (35). Alternatively, ghrelin might indirectly engage the orexin system by targeting neurons at other locations which, in turn, project to the LHA. For instance, several studies suggest direct ghrelin action on AgRP/NPY neurons of the hypothalamic arcuate nucleus as playing a key role in stimulating food intake [as reviewed in (6, 36)]. Not only do AgRP/NPY neurons project to LHA orexin neurons (37), but also NPY has been shown to have the capacity to induce CPP (38). Indirect activation of orexin neurons by ghrelin also could occur via other pathways. For instance, neurons of the nucleus accumbens project to and activate LHA orexin neurons to drive the intake of palatable foods (39). GHSR-expressing neurons in both the VTA and hippocampus exist, and neurons in both of these sites are known to project to the nucleus accumbens (40-42). Future studies will be required to establish the exact neuronal circuits by which ghrelin increases the rewarding value of HFD.
Conclusions
The findings of a role for ghrelin in food reward will likely have important implications in our overall understanding of energy balance regulation in both lean and obese individuals. Furthermore, the findings of a dissociation between ghrelin's effects on food reward from its effects on homeostatic eating behaviors suggests the plausibility of the design of pharmacologic agents that can target specific reward behaviors associated with eating while leaving other aspects of eating untouched.
Supplementary Material
Acknowledgments
The authors thank Joel Elmquist, Debbie Clegg, Stephen Benoit, and David Self for many helpful discussions. We also thank Douglas Frantz of the UTSW Medical Center Synthetic Chemistry Core for assistance in synthesizing Compound 26. This work was supported by 1R01DA024680-01, K08DK068069-01A2, R01DK71320, RL1DK081185-01, a Foundation for Prader-Willi Research Grant, two NARSAD Young Investigator Awards, and two UTSW Disease-Oriented Clinical Scholar Awards.
Footnotes
Financial disclosures: All authors reported no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36:199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 2.Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 3.Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714–1719. doi: 10.2337/diabetes.50.8.1714. [DOI] [PubMed] [Google Scholar]
- 4.Nagaya N, Uematsu M, Kojima M, Date Y, Nakazato M, Okumura H, et al. Elevated circulating level of ghrelin in cachexia associated with chronic heart failure: relationships between ghrelin and anabolic/catabolic factors. Circulation. 2001;104:2034–2038. doi: 10.1161/hc4201.097836. [DOI] [PubMed] [Google Scholar]
- 5.Otto B, Cuntz U, Fruehauf E, Wawarta R, Folwaczny C, Riepl RL, et al. Weight gain decreases elevated plasma ghrelin concentrations of patients with anorexia nervosa. Eur J Endocrinol. 2001;145:669–673. [PubMed] [Google Scholar]
- 6.Zigman JM, Elmquist JK. Minireview: From anorexia to obesity--the yin and yang of body weight control. Endocrinology. 2003;144:3749–3756. doi: 10.1210/en.2003-0241. [DOI] [PubMed] [Google Scholar]
- 7.Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494:528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell V, Bouret S, Beauvillain JC, Schilling A, Perret M, Kordon C, et al. Comparative distribution of mRNA encoding the growth hormone secretagogue-receptor (GHS-R) in Microcebus murinus (Primate, lemurian) and rat forebrain and pituitary. J Comp Neurol. 2001;429:469–489. doi: 10.1002/1096-9861(20010115)429:3<469::aid-cne8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 9.Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res. 1997;48:23–29. doi: 10.1016/s0169-328x(97)00071-5. [DOI] [PubMed] [Google Scholar]
- 10.Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006 doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jerlhag E, Egecioglu E, Dickson SL, Douhan A, Svensson L, Engel JA. Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict Biol. 2007;12:6–16. doi: 10.1111/j.1369-1600.2006.00041.x. [DOI] [PubMed] [Google Scholar]
- 12.Jerlhag E, Egecioglu E, Dickson SL, Andersson M, Svensson L, Engel JA. Ghrelin stimulates locomotor activity and accumbal dopamine-overflow via central cholinergic systems in mice: implications for its involvement in brain reward. Addict Biol. 2006;11:45–54. doi: 10.1111/j.1369-1600.2006.00002.x. [DOI] [PubMed] [Google Scholar]
- 13.Naleid AM, Grace MK, Cummings DE, Levine AS. Ghrelin induces feeding in the mesolimbic reward pathway between the ventral tegmental area and the nucleus accumbens. Peptides. 2005;26:2274–2279. doi: 10.1016/j.peptides.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence CB, Snape AC, Baudoin FM, Luckman SM. Acute central ghrelin and GH secretagogues induce feeding and activate brain appetite centers. Endocrinology. 2002;143:155–162. doi: 10.1210/endo.143.1.8561. [DOI] [PubMed] [Google Scholar]
- 15.Olszewski PK, Li D, Grace MK, Billington CJ, Kotz CM, Levine AS. Neural basis of orexigenic effects of ghrelin acting within lateral hypothalamus. Peptides. 2003;24:597–602. doi: 10.1016/s0196-9781(03)00105-0. [DOI] [PubMed] [Google Scholar]
- 16.Kohno D, Gao HZ, Muroya S, Kikuyama S, Yada T. Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2+ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes. 2003;52:948–956. doi: 10.2337/diabetes.52.4.948. [DOI] [PubMed] [Google Scholar]
- 17.Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, et al. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- 18.Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9:381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- 19.Jerlhag E. Systemic administration of ghrelin induces conditioned place preference and stimulates accumbal dopamine. Addict Biol. 2008;13:358–363. doi: 10.1111/j.1369-1600.2008.00125.x. [DOI] [PubMed] [Google Scholar]
- 20.Wellman PJ, Davis KW, Nation JR. Augmentation of cocaine hyperactivity in rats by systemic ghrelin. Regul Pept. 2005;125:151–154. doi: 10.1016/j.regpep.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 21.Davis KW, Wellman PJ, Clifford PS. Augmented cocaine conditioned place preference in rats pretreated with systemic ghrelin. Regul Pept. 2007;140:148–152. doi: 10.1016/j.regpep.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jerlhag E, Egecioglu E, Landgren S, Salome N, Heilig M, Moechars D, et al. Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci U S A. 2009;106:11318–11323. doi: 10.1073/pnas.0812809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, et al. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci. 2008;11:752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malik S, McGlone F, Bedrossian D, Dagher A. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab. 2008;7:400–409. doi: 10.1016/j.cmet.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 26.Zigman JM, Nakano Y, Coppari R, Balthasar N, Marcus JN, Lee CE, et al. Mice lacking ghrelin receptors resist the development of diet-induced obesity. J Clin Invest. 2005;115:3564–3572. doi: 10.1172/JCI26002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudolph J, Esler WP, O'Connor S, Coish PD, Wickens PL, Brands M, et al. Quinazolinone derivatives as orally available ghrelin receptor antagonists for the treatment of diabetes and obesity. Journal of medicinal chemistry. 2007;50:5202–5216. doi: 10.1021/jm070071+. [DOI] [PubMed] [Google Scholar]
- 28.Esler WP, Rudolph J, Claus TH, Tang W, Barucci N, Brown SE, et al. Small-molecule ghrelin receptor antagonists improve glucose tolerance, suppress appetite, and promote weight loss. Endocrinology. 2007;148:5175–5185. doi: 10.1210/en.2007-0239. [DOI] [PubMed] [Google Scholar]
- 29.Lutter M, Krishnan V, Russo SJ, Jung S, McClung CA, Nestler EJ. Orexin signaling mediates the antidepressant-like effect of calorie restriction. J Neurosci. 2008;28:3071–3075. doi: 10.1523/JNEUROSCI.5584-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Figlewicz DP, Higgins MS, Ng-Evans SB, Havel PJ. Leptin reverses sucrose-conditioned place preference in food-restricted rats. Physiol Behav. 2001;73:229–234. doi: 10.1016/s0031-9384(01)00486-3. [DOI] [PubMed] [Google Scholar]
- 31.Figlewicz DP, Benoit SC. Insulin, leptin, and food reward: update 2008. Am J Physiol Regul Integr Comp Physiol. 2009;296:R9–R19. doi: 10.1152/ajpregu.90725.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437:556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- 33.Colby CR, Whisler K, Steffen C, Nestler EJ, Self DW. Striatal cell type-specific overexpression of DeltaFosB enhances incentive for cocaine. J Neurosci. 2003;23:2488–2493. doi: 10.1523/JNEUROSCI.23-06-02488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Figlewicz DP, Bennett J, Evans SB, Kaiyala K, Sipols AJ, Benoit SC. Intraventricular insulin and leptin reverse place preference conditioned with high-fat diet in rats. Behav Neurosci. 2004;118:479–487. doi: 10.1037/0735-7044.118.3.479. [DOI] [PubMed] [Google Scholar]
- 35.Narita M, Nagumo Y, Hashimoto S, Khotib J, Miyatake M, Sakurai T, et al. Direct involvement of orexinergic systems in the activation of the mesolimbic dopamine pathway and related behaviors induced by morphine. J Neurosci. 2006;26:398–405. doi: 10.1523/JNEUROSCI.2761-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kojima M, Kangawa K. Drug insight: The functions of ghrelin and its potential as a multitherapeutic hormone. Nat Clin Pract Endocrinol Metab. 2006;2:80–88. doi: 10.1038/ncpendmet0080. [DOI] [PubMed] [Google Scholar]
- 37.Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, et al. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol. 1998;402:442–459. [PubMed] [Google Scholar]
- 38.Josselyn SA, Beninger RJ. Neuropeptide Y: intraaccumbens injections produce a place preference that is blocked by cis-flupenthixol. Pharmacol Biochem Behav. 1993;46:543–552. doi: 10.1016/0091-3057(93)90542-2. [DOI] [PubMed] [Google Scholar]
- 39.Zheng H, Patterson LM, Berthoud HR. Orexin signaling in the ventral tegmental area is required for high-fat appetite induced by opioid stimulation of the nucleus accumbens. J Neurosci. 2007;27:11075–11082. doi: 10.1523/JNEUROSCI.3542-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fallon JH, Moore RY. Catecholamine innervation of the basal forebrain. IV. Topography of the dopamine projection to the basal forebrain and neostriatum. J Comp Neurol. 1978;180:545–580. doi: 10.1002/cne.901800310. [DOI] [PubMed] [Google Scholar]
- 41.Mogenson GJ, Nielsen M. A study of the contribution of hippocampal-accumbens-subpallidal projections to locomotor activity. Behav Neural Biol. 1984;42:38–51. doi: 10.1016/s0163-1047(84)90412-6. [DOI] [PubMed] [Google Scholar]
- 42.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.