

Abstract
To date, more than 230 disease causing mutations have been linked to the slow/cardiac muscle myosin gene, β-MyHC (MYH7). The majority of these mutations are located in the globular head region of the protein and result in cardiomyopathies. Recently, however, a number of novel disease causing mutations have been described in the long, α-helical, coiled-coil tail region of the β-MyHC protein. Mutations in this region are of particular interest because they are associated with a multitude of human diseases, including both cardiac and skeletal myopathies. Here, we attempt to dissect the mechanism(s) by which mutations in the rod region of β-MyHC can cause a variety of diseases by analyzing two mutations at a single amino acid (R1500P and R1500W) which cause two distinct diseases (Laing-type early onset distal myopathy and dilated cardiomyopathy, respectively). For diseases linked to the R1500 residue, we find that each mutation displays distinct structural, thermodynamic, and functional properties. Both R1500P and R1500W cause a decrease in thermodynamic stability, though the R1500W phenotype is more severe. Both mutations also affect filament assembly, with R1500P causing an additional decrease in filament stability. In addition to furthering our understanding of the mechanism of pathogenesis for each of these diseases, these data also suggest how the variance in molecular phenotype may be associated with the variance in clinical phenotype present with mutations in the β-MyHC rod.
Keywords: Dilated Cardiomyopathy, Distal Myopathy, Myosin, MPD1, LMM
1. Introduction
Myosin is a major component of heart and skeletal muscle, and plays a key role in muscle contractility. In vivo, myosin exists as a dimer of globular heads linked to an α-helical coiled-coil rod domain. Each head contains ATP hydrolysis and actin binding sites, while the rod is involved in thick filament formation[1–3]. Seven sarcomeric myosin isoforms are present in humans, with expression varying by muscle type. β-myosin (β-MyHC), expressed in the adult heart and slow skeletal muscle fibers, is important clinically as it is associated with at least 230 disease causing mutations[4]. The majority of these mutations are in the head region and many have been discussed previously. Mutations in the β-MyHC rod, however, have only recently begun to be characterized. To date a single subset of mutations, responsible for the disease myosin storage myopathy (MSM), have been investigated. Results from these studies suggest that there are several mechanisms, including thermodynamic instability and diminished self-assembly, by which mutations in the β-MyHC rod can cause disease [5].
Mutations in the myosin rod are particularly interesting because of their clinical heterogeneity. While mutations in the head are typically associated with cardiomyopathies, rod mutations are associated with several disparate diseases including both cardiac and skeletal myopathies[4]. We sought to investigate the phenotypic variability of mutations in the β-MyHC rod by characterizing two different mutations found at a single amino acid that, interestingly, are able to cause distinct diseases. Both diseases result from the mutation of amino acid R1500. When mutated to proline (R1500P), patients develop a skeletal myopathy known Laing-type early-onset distal myopathy or MPD1, whereas when this residue is mutated to tryptophan (R1500W) patients develop dilated cardiomyopathy[6, 7]. MPD1 is a slowly progressive congenital myopathy with variable pathology, but typically includes distal weakness of ankle, finger, and neck flexion, atrophic type I muscle fibers, and the occasional presence of rimmed vacuoles[8]. Dilated cardiomyopathy (DCM) is also clinically and etiologically heterogeneous, but is primarily associated with left ventricular dilation and reduced ejection fraction[9]. In both cases, the mutation is seen in heterozygous patients and is hypothesized to cause disease by disrupting interactions between MyHC rods during thick filament assembly.
Assembly of MyHC into thick filaments relies upon charge-based interactions between adjacent α-helical coiled-coil rods. The α-helices are composed of a repeating heptad of residues (denoted a-g), and residues in outer positions are typically charged and meditate thick filament formation through electrostatic interactions between coiled-coils. The R1500 residue of β-MyHC is located in the outermost f position and, as such, mutations in this residue are predicted to alter the proper assembly of the thick filament (Fig 1A). To test this, we characterized the effect of each mutation when expressed in the C-terminal fragment of the MyHC rod known as light meromyosin (LMM) (Fig. 1B). Using a variety of assays we previously established for investigating mutations in this region of the protein, we show each R1500 mutation displays unique thermodynamic, structural, and functional differences[5]. These results intimate at how varying molecular phenotypes may be responsible for the variety of diseases associated with mutations in the MyHC rod, and in conjunction with previously published data provide further characterization of mutations in this region of the protein which will be necessary to understand their pathogenicity.
Fig 1.
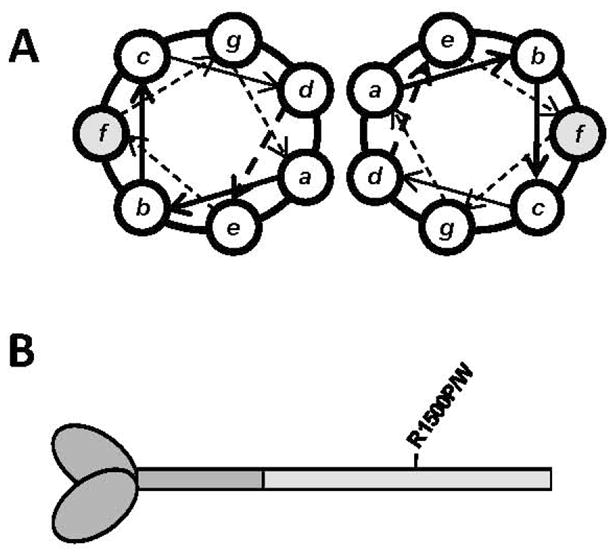
Location of R1500P and R1500W mutations in the coiled-coil rod of β-MyHC. (A) Relative amino acid positions within the heptad repeat of a coiled-coil, denoted by a-g. R1500P and R1500W are located in the shaded f position. (B) Diagram of β-MyHC structure. Heavy meromyosin is comprised of the globular head and N-terminal portion of the myosin rod, and is shown in dark gray. LMM is the C-terminal coiled-coil tail region and is shaded light gray, with the relative position of the R1500P and R1500W mutations marked.
2. Materials and Methods
2.1 Protein expression and purification
Wild-type β-MyHC LMM (aa residues 1231-1938) was cloned into the pUC18 expression vector and R1500P and R1500W mutations were introduced via inverse PCR, verified by sequencing, and subcloned into the pET3a expression vector as described previously (modified pET vector provided by R. Thompson). Constructs were transformed into BL-21 cells and protein extraction and purification were performed as previously described[5]. Briefly, proteins were purified on Ni-NTA agarose, eluted with imidazole, and further purified by anion exchange chromatography. LMM containing fractions were analyzed via SDS-PAGE for purity, pooled, and concentrated using Amicon Ultracel 50K centrifuge columns.
2.2 Circular dichroism measurements
Wild-type and mutant LMM proteins were dialyzed into high salt buffer (HSB; 10 mM TES, 300 mM NaCl, and 3.5 mM EDTA, pH 7.3), diluted to approximately 0.3 mg/mL, and reduced with 1 mM TCEP. Circular dichroism (CD) analysis was performed using a J-815 spectropolarimeter (Jasco Inc, Easton, MD) with constant N2 flushing. All measurements were performed at 4 °C with a 1-mm path length optical cell. Spectra were determined from 240 nm to 200 nm with a 0.1 nm data pitch, a continuous scanning speed of 100 nm/min, and averaged over 5 accumulations. Baseline spectra for buffer were collected in the same manner and subtracted from protein spectra. The spectra were normalized by mean residue molar ellipticity and percent α-helix was calculated as previously described[10].
2.3 Thermal denaturation monitored by CD222
The thermal stability of protein in HSB was measured by CD at 222 nm during temperature-induced protein denaturation from 4 °C to 90 °C. Data were collected at 1 °C intervals at a scan rate of 60 °C per hour. To determine transition thermodynamics, the change in mean residue ellipticity as a function of temperature was modeled using a non-linear least squares algorithm that assumes the two-state transition of monomer from a folded to an unfolded state with no change in heat capacity between the folded and unfolded forms[11]. The fraction folded at any given temperature was calculated using a model similar to one which we have previously described, but because the heat capacities of the folded and unfolded states are assumed to be equal, ΔCp was fixed at zero in all models described here[5].
2.4 Differential scanning calorimetry (DSC)
Wild-type and mutant proteins were prepared at concentrations of approximately 2 mg/ml in HSB and reduced with 5 mM TCEP. Exact concentrations for each protein were determined by μBCA assay. DSC was performed using a VP-DSC differential scanning microcalorimeter (MicroCal, Northampton, MA) at a heating rate of 90 °C/hour and under a constant pressure of 2.07·105 Pa. Data were analyzed and plotted using Origin (OriginLab, Northampton, MA) after baseline subtraction and normalization for concentration.
2.5 Static Light Scattering
The self-assembly of wild-type and mutant protein was measured in real-time by 90° light scattering using a PTI QM-2000-6SE fluorescence spectrometer (Photon Technology International, Birmingham, NJ) essentially as described previously[5]. Briefly, measurements were taken for buffer without salt (10 mM TES, 3.5 mM EDTA, 1mM TCEP, pH 7.3) for two minutes to obtain a baseline for scattering, at which point an equal volume of 400 nM protein in HSB was added, diluting the buffer to 150 mM NaCl and allowing for self-assembly at a final protein concentration of 200 nM. Reactions were allowed to proceed to completion (40 minutes) before 5M NaCl was added to return the NaCl concentration to 300 mM. Data were analyzed using Kaleidagraph (Synergy Software, Reading, PA) with background buffer scattering subtracted from each reading.
2.6 Paracrystal Formation and Visualization
Paracrystals of wild-type and mutant protein were formed by dialyzing 100 ug of protein from HSB into 10 mM bis-Tris propane, 100 mM NaCl, 3.5 mM EDTA, pH 7.3, and imaged as previously described[5].
2.7 Limited Trypsinization
Paracrystals of wild type and mutant protein were formed as described above, reduced with 1 mM TCEP, and trypsinization was performed as previously described[5, 12]. Briefly, paracrystals were trypsinized with 0.002 mg/mL porcine trypsin and incubated at 30 °C. Reactions were quenched at various times by mixing in 40X excess soybean trypsin inhibitor. Samples were run on 4% – 20% SDS-PAGE gels, stained with Imperial Protein Stain (Thermo Scientific, Rockford, IL) and imaged with a Li-COR Odyssey infrared imager (Li-COR Inc., Lincoln, NE).
3. Results
3.1 R1500 mutations do not affect protein secondary structure
To determine if R1500P and R1500W mutations affect the ability of the MyHC rod to properly form coiled-coils, we obtained far-UV circular dichroism (CD) spectra for wild type and both mutant LMM proteins to determine secondary structure. All three proteins display canonical α-helical spectra, with characteristic minima at 208 nm and 222 nm (Fig. 2). The calculated α-helical content of each is also similar, with both R1500P and R1500W LMM having values similar to the 91.5 % α-helix seen for wild type LMM, suggesting that neither mutation affects the gross secondary structure of the protein (Table 1).
Fig 2.
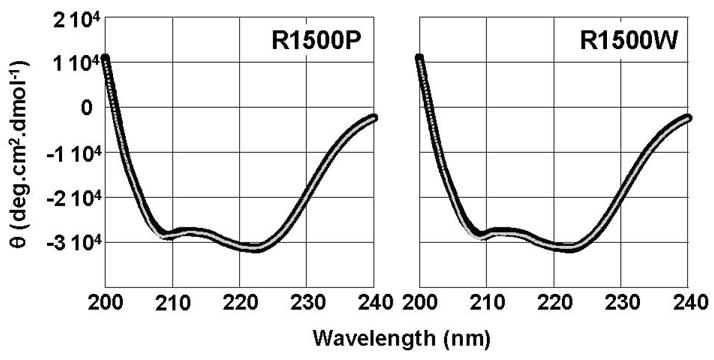
R1500P and R1500W mutations do not detectibly alter the secondary structure of LMM. Far-UV CD spectra of WT (black) and mutant (gray) LMM were obtained from 250 nm – 200 nm at 4 °C. WT LMM has nearly identical secondary structure profiles to R1500P and R1500W LMM. All proteins display canonical α-helical spectra with characteristic minima at 208 nm and 222 nm, and the calculated percentage of α-helix is similar for all three proteins.
Table 1.
Biophysical data for myosin tail constructs
| Construct | [θ]222 | α-Helix | Tm | ΔGapp,37 | ΔH | ΔS |
|---|---|---|---|---|---|---|
| deg.cm2.dmol−1 | % | °C | kcal.mol−1 | kcal.mol−1 | kcal.mol−1.K−1 | |
| Wild type | −31,422 | 91.5 | 50.9 | −3.04 | −71.1 | −0.220 |
| R1500P | −31,280 | 91.0 | 50.9 | −2.46 | −57.5 | −0.177 |
| R1500W | −31,103 | 91.8 | 48.6 | −1.70 | −47.3 | −0.146 |
3.2 Both R1500P and R1500W Show Unique Thermodynamic Profiles
The thermodynamic stability of wild type and mutant LMM was assayed by monitoring α-helical content, determined by CD at 222 nm (CD222), as the temperature was increased from 4 °C to 90 °C, and thermodynamic data were obtained by modeling the thermal denaturation of each protein (Fig. 3). The thermal midpoint of unfolding (calculated at K = 1) is unchanged between WT and R1500P LMM, whereas R1500W shows a relatively large 2.3 °C decrease in Tm. Thermodynamic parameters for R1500W also indicate a decrease in protein stability. R1500W LMM has a ΔΔG at 37 °C of −1.34 kcal·mol−1, and ΔH and ΔS are both decreased by 34%. Interestingly, although the Tm of R1500P LMM is similar to WT LMM, the ΔΔG at 37 °C is −0.58 kcal/mol, and ΔH and ΔS are both decreased by 19% (Table 1). These results suggest that under physiological conditions both R1500P and R1500W proteins are less thermodynamically stable than WT, and that the thermodynamic phenotype associated with R1500W appears to be more severe.
Fig 3.

Thermal denaturation of LMM reveals variation in R1500P and R1500W thermodynamic profiles. (A) The α-helical secondary structure of LMM was monitored by θ222 during thermally denaturation. Measured θ222 data (black) were fit to a theoretical model (gray) to derive thermodynamic parameters. (B) Residuals for the fit of the theoretical model to the measured data are calculated as the difference between the two at each point. Residuals are all around zero, indicating that our model fits well and shows no systematic deviation. (C) Fit θ222 data are plotted over the central segment of denaturation (40 °C to 60 °C) to directly illustrate differences in thermal stability. R1500W LMM (green) melts at a lower temperature and exhibits a wider transition than WT (red) and R1500P (blue) LMM, indicating a decreased cooperativity of melting for R1500W.
3.3 Differential scanning calorimetry reveals R1500W LMM instability
To further understand how R1500 mutations are affecting protein stability, we utilized differential scanning calorimetry (DSC) to directly measure thermodynamic parameters of protein unfolding. DSC results are in agreement with the CD222 results and are closely approximated by a two-state transition model (Fig. 4). WT LMM displays a single endotherm having a Tm of 50.5 °C, with a calorimetric enthalpy of unfolding (ΔHcal) of 134 kcal·mol−1 and a van’t Hoff enthalpy of unfolding (ΔHvH) of 181 kcal·mol−1. R1500P LMM also displays a single endotherm and has a similar Tm of 50.5 °C, although both ΔHcal and ΔHvH of R1500P LMM are lower than WT LMM (Table 2). Interestingly, DSC of R1500W LMM reveals two prominent endotherms. Deconvolution analysis of the scans show each endotherm to have an associated Tm of 44.9 °C and 50.0 °C, with ΔHcal of 87.9 kcal·mol−1 and 260 kcal·mol−1, and ΔHvH of 178 kcal·mol−1 and 269 kcal·mol−1, respectively (Table 2). While one of the endotherms seen in R1500W LMM has a Tm similar to WT, the presence of a second, less stable endotherm helps to explain the large decrease in Tm observed by CD222 melts.
Fig 4.

DSC thermograms of WT, R1500P, and R1500W LMM. Thermograms show theoretical fits of the data modeled using a two-state transition. Two peaks were deconvoluted for R1500W and thermodynamic parameters calculated separately for each.
Table 2.
Differential scanning calorimetry data for myosin tail constructs
| Construct | Tm | ΔHcal | ΔHvH | ΔHvH/ΔHcal |
|---|---|---|---|---|
| °C | kcal.mol−1 | kcal.mol−1 | ||
| Wild type | 50.5 | 134 | 181 | 1.35 |
| R1500P | 50.5 | 69.9 | 162 | 2.32 |
| R1500W, 1 | 44.9 | 87.9 | 178 | 2.02 |
| R1500W, 2 | 50.0 | 260 | 269 | 10.35 |
Also interesting are the ratios of ΔHvH/ΔHcal, which reflect the apparent number of associated molecules in a cooperative unit during thermal transition. Ratios for WT and R1500P LMM are around 2, which may be indicative of a dimeric assembly such as the coiled-coil. The less stable endotherm seen in DSC analysis of R1500W has a similar ratio, but the endotherm which appears similar to WT LMM with a Tm of 50.0 °C has a much larger ΔHvH/ΔHcal ratio, and may be indicative of protein misfolding (Table 2).
3.4 R1500 mutations alter the ability of LMM to form filaments
The location of R1500 in the f position of the heptad repeat suggests both mutations may be interfering with the ability of the MyHC rod to assemble into the thick filament. To test this, we utilized static light scattering to determine if the self-assembly of LMM is affected. WT and mutant LMM protein was diluted from high salt buffer where it exists stably as a coiled-coil into a physiologically relevant buffer where it will self-assemble into ordered structures. Formation of assemblies was followed in real time by monitoring 90° light scattering, as described previously [5, 13, 14]. WT LMM assembled as expected, with the majority of the reaction completed within the first 12 minutes (Fig. 5). Both R1500P LMM and R1500W LMM, however, showed defects in the ability to properly assemble. Each mutant appears to assemble at a rate similar to WT LMM, but the extent of the assembly is truncated to approximately 70% of that for WT LMM, in agreement with previous results for assembly-defective mutations in LMM [5].
Fig 5.
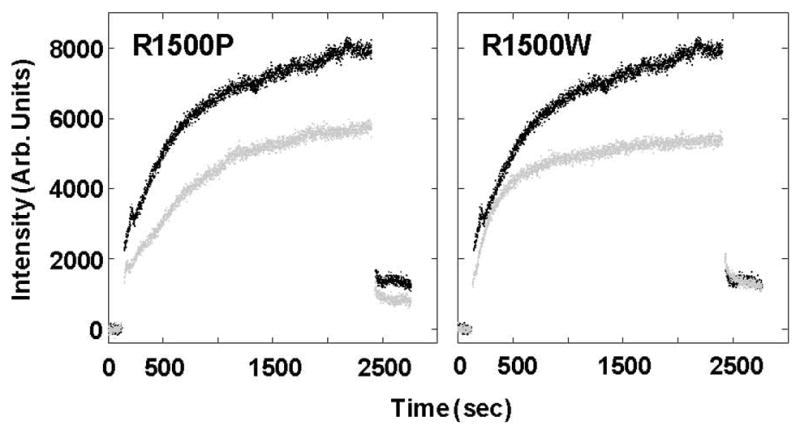
R1500P and R1500W LMM display similar defects in self-assembly. Baseline 90° light scattering in no salt buffer was obtained for 120 sec before an equal volume of either WT (Black) or mutant (gray) LMM was added, diluting the sample to a final concentration of 150 mM NaCl and 200 nM protein to initiate self-assembly. Data were collected at a rate of one point/sec, and the reaction was allowed to proceed for 40 min before the addition of 5M salt to return the buffer to 300 mM NaCl and demonstrate the reversibility of the reaction. The intensity of 90° light scattering is plotted in arbitrary units with respect to time.
3.5 R1500 mutations do not alter filament ultrastructure
We next determined the effect of these mutations on the ultrastructure of the filaments by visualizing paracrystals directly. Paracrystals are ordered assemblies of LMM that provide a well-established model for thick filament formation, and are formed in a manner similar to static light scattering experiments, stained, and analyzed by electron microscopy (Fig. 6) [15–21]. As expected, WT LMM formed paracrystals with 14.0 nm periodicity, similar to the 14.3 nm axial spacing seen for muscle fibers in vivo[15, 16, 19, 21]. Neither R1500P nor R1500W alters the periodicity of the paracrystals, with each being similar to WT and indicating that the gross morphology of filaments formed from these mutants is unaffected (Table 3).
Fig 6.

Ultrastructures of mutant and WT paracrystals are indistinguishable. (A) WT and mutant paracrystals have similar periodicities. WT and mutant LMM were dialyzed overnight into crystallization buffer to induce paracrystal formation. Paracrystals were adsorbed onto carbon mesh grids and electron micrographs were recorded at a magnification of X92000. Paracrystal periodicity measurements were taken over the range of several striations and divided by the number of striations to obtain an average periodicity per paracrystal.
Table 3.
LMM paracrystal parameter
| Striations(nm) | +/− | n= | |
|---|---|---|---|
| Wild type | 14.0 | 0.44 | 20 |
| R1500P | 14.3 | 0.67 | 12 |
| R1500W | 14.4 | 0.52 | 20 |
3.6 R1500 proteins alter filament properties
To further characterize the effect of each mutation on filament formation we examined the stability of paracrystal assemblies by limited proteolysis. Paracrystals were formed as described, and limited proteolysis was performed by digestion with a low concentration of porcine trypsin. Reactions were quenched at several time points up to 90 minutes and proteolytic digestion was analyzed via SDS-PAGE (Fig. 7A). Band intensity of full length protein was measured and plotted over time, and the data were fit to an exponential decay curve (Fig. 7B). WT and R1500W LMM formed paracrystals of similar proteolytic stability, with approximately 80% of the protein remaining undigested after 90 minutes. R1500P LMM, however, is more readily proteolyzed. Although the extent of proteolysis after 90 minutes is similar between WT LMM and both mutant proteins, R1500P LMM degradation occurs four times more rapidly than WT LMM, with 20% percent of the full length protein digested within the first 20 minutes.
Fig 7.

R1500 mutations have differential effects on the proteolytic stability of LMM. (A) Time course for limited proteolytic digestion of LMM. Lane 12 shows molecular weight markers of 125 kDa and 82 kDa. (B) Proteolysis data are plotted as relative band intensity of full length LMM, and fit to an exponential decay curve versus time. The intensity of full length LMM at time zero is defined as 1, and the intensity of the full length LMM band at each time point is plotted with respect to that value.
4. Discussion
Mutations in the rod region of β-MyHC are intriguing because they can result in a wide variety of clinical pathologies though expressed in a region of myosin assumed to be somewhat functionally homogeneous in its role of incorporating the protein into the thick filament. To further our understanding of how this may occur, we utilized an array of assays to investigate the mechanism by which two mutations at a single amino acid residue in the β-MyHC rod (R1500P and R1500W), which give rise to different diseases, affect the structural, thermodynamic, and functional properties of the protein. Similar to the variability in molecular phenotypes seen for MSM mutations, our data suggest that each mutation studied here, though located at the same amino acid, affects the protein in different ways. This variance may be responsible for the different phenotypes associated with these mutations in human patients, and may provides clues about the role of this region of the protein in incorporating the molecule into the thick filament.
4.1 R1500P
Proline is known to be detrimental to the structure of α-helices, leading to the hypothesis that R1500P alters the secondary structure of the MyHC rod and prevents proper incorporation into or formation of thick filaments[6]. Surprisingly, our CD data demonstrate that R1500P does not detectably alter the secondary structure or Tm of the protein, although it does appear to destabilize it. Thermodynamic measurements show WT and R1500P LMM display single endotherms with an identical Tm, but R1500P displays ΔHcal and ΔHvH values which are lower than those obtained for WT supporting the notion that R1500P is less thermodynamically stable.
Although no gross change in secondary structure was detected, the instability of the mutation does appear to have a functional impact. Results from 90° light scattering show R1500P LMM is able to self-assemble at a rate similar to WT but these assemblies are only approximately 70% of the size of those formed from WT. It is possible, however, that the reduced signal from R1500P assemblies may result from the presence of fewer assemblies similar in size to those formed from WT LMM. Paracrystals formed from R1500P LMM do have similar periodicity to WT, however, limited proteolysis experiments suggest that R1500P paracrystals are less stable than those formed with WT protein. R1500P paracrystals are degraded approximately four times faster than WT, suggesting that the proline may inhibit the ability of the coiled-coils to properly pack together, and that this less rigid packing structure allows proteolysis to occur more readily.
Taken together, these data are consistent with the hypothesis that mutating a charged amino acid in the f position of the α-helical coiled-coil alters the ability of the MyHC rod to properly assemble. Residues in the outer f position of the α-helix mediate critical charge based interactions between adjacent coiled-coils in the MyHC rod, and replacement of a single charged residue at this position with an aliphatic proline residue appears to interfere with this process.
4.2 R1500W
As expected, mutating R1500 to tryptophan did not cause any detectible shift in protein secondary structure, but does result in thermodynamic differences compared to WT. CD222 thermal melts show R1500W LMM has a lower Tm than WT and thermodynamic parameters suggest that at physiological temperatures R1500W LMM is significantly less stable. Calorimetric measurements help to explain these results. DSC experiments show two distinct denaturation events, represented by the two observed endotherms. The first endotherm is much less stable than the WT endotherm (ΔTm = −5.6 °C), but with a ΔHvH/ΔHcal of approximately 2 appears to represent the properly associated, dimeric cooperative unit. In contrast, the second endotherm has a Tm similar to WT (ΔTm = −0.5 °C), but has a ΔHvH/ΔHcal ratio of more than 10 which may represent an increase in protein misfolding. As noted elsewhere, high ΔHvH/ΔHcal ratios can occur in several ways[22]. One attractive scenario to explain our data is that the local region surrounding the R1500W mutation is destabilized and melts at a lower Tm than WT. This denatured region may then result in non-native intramolecular interactions which give rise to a high ΔHvH/ΔHcal ratio upon the thermal denaturation of the remaining protein at temperatures similar to WT denaturation.
The destabilization from R1500W has a functional impact as well. Similar to R1500P, 90° light scattering shows R1500W LMM assembles at a rate similar to WT but to a lesser extent. This may be due to the formation of smaller assemblies or to fewer WT-sized assemblies being formed. Surprisingly, we do not observe any change R1500W paracrystal ultrastructure, and, unlike R1500P, R1500W paracrystals of R1500W LMM display the same level of proteolytic susceptibility.
Here we present work characterizing the molecular mechanisms underlying two diseases caused by mutations at the same amino acid residue in the β-MyHC rod. For the two mutations at R1500, our data demonstrate variance in the molecular phenotype associated with each.
One possible model explaining the tissue specificity associated with each mutation involves varying demands placed upon each type of muscle. Due to increased demand cardiac sarcomeres may provide a sensitized background in which the variance in molecular phenotype becomes more apparent. The decreased stability of R1500P may mean that within the increased stringency of the heart, these proteins are not stably incorporated or are more readily degraded, leaving the majority of fibers in the heart as WT. Because skeletal muscle is not under the same demands, proteins harboring the R1500P mutation may be stably incorporated into the thick filament, but these filaments may not be able to function as well as wholly WT filaments, leading to a skeletal myopathy. Filaments formed from R1500W, however, do not display a decrease in stability by our assays. Protein containing this mutation, then, may be stably incorporated in the thick filaments of cardiac sarcomeres, but because of decreased protein stability may not be able to function as well in the heart. R1500W mutant myosin should also be incorporated into the sarcomeres of skeletal muscles, but if the molecular phenotype associated with this mutation is less severe it may not be detectible in a muscle where the sarcomeres are not placed under as much stress as those found in the heart.
A second possibility is that each mutation may differentially interfere with the association of a tissue specific isoform of a myosin rod-binding protein. Although proteins such as MyBP-C, MyBP-H, Myomesin, and Titan are not presently thought to interact with the R1500 residue of the myosin rod, much is unknown about sarcomere ultrastructure and this provides an intriguing alternative hypothesis[23–25]. While much remains to be learned about the pathogenicity of mutations in the MyHC rod, the work presented here helps to dissect the variance in pathology seen with mutations in this region of the protein and adds to a more complete characterization of mutations in this region of the molecule. Further work with cell and animal based models of these mutations will be necessary to correlate the link between molecular and physiological phenotypes to understand how molecular phenotypes contribute to disease development, as well as understanding how each mutation affects the function of heart and skeletal muscle in vivo.
Acknowledgments
We thank M. Buvoli and R. Thompson for discussion and suggestions, S. Bevers for technical advice and support, T. Giddings for EM technical assistance, and R. Thompson for bringing the SLS assay to our lab and for assistance with protein purification. This work was supported by National Institutes of Health Grants 5R01 HL085573-01 (to L.A.L.) and T32GM065103 (to T.Z.A.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McLachlan AD, Karn J. Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature. 1982 Sep 16;299(5880):226–31. doi: 10.1038/299226a0. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson SJ, Stewart M. Molecular interactions in myosin assembly. Role of the 28-residue charge repeat in the rod. J Mol Biol. 1992 Jul 5;226(1):7–13. doi: 10.1016/0022-2836(92)90118-4. [DOI] [PubMed] [Google Scholar]
- 3.Atkinson SJ, Stewart M. Molecular basis of myosin assembly: coiled-coil interactions and the role of charge periodicities. J Cell Sci Suppl. 1991;14:7–10. doi: 10.1242/jcs.1991.supplement_14.2. [DOI] [PubMed] [Google Scholar]
- 4.Buvoli M, Hamady M, Leinwand LA, Knight R. Bioinformatics assessment of beta-myosin mutations reveals myosin’s high sensitivity to mutations. Trends Cardiovasc Med. 2008 May;18(4):141–9. doi: 10.1016/j.tcm.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armel TZ, Leinwand LA. Mutations in the beta-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc Natl Acad Sci U S A. 2009 Apr 14;106(15):6291–6. doi: 10.1073/pnas.0900107106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1) Am J Hum Genet. 2004 Oct;75(4):703–8. doi: 10.1086/424760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karkkainen S, Helio T, Jaaskelainen P, Miettinen R, Tuomainen P, Ylitalo K, et al. Two novel mutations in the beta-myosin heavy chain gene associated with dilated cardiomyopathy. Eur J Heart Fail. 2004 Dec;6(7):861–8. doi: 10.1016/j.ejheart.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 8.Lamont PJ, Udd B, Mastaglia FL, de Visser M, Hedera P, Voit T, et al. Laing early onset distal myopathy: slow myosin defect with variable abnormalities on muscle biopsy. J Neurol Neurosurg Psychiatry. 2006 Feb;77(2):208–15. doi: 10.1136/jnnp.2005.073825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karkkainen S, Peuhkurinen K. Genetics of dilated cardiomyopathy. Ann Med. 2007;39(2):91–107. doi: 10.1080/07853890601145821. [DOI] [PubMed] [Google Scholar]
- 10.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1(6):2876–90. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenfield NJ. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat Protoc. 2006;1(6):2527–35. doi: 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sumida JP, Wu E, Lehrer SS. Conserved Asp-137 imparts flexibility to tropomyosin and affects function. J Biol Chem. 2008 Mar 14;283(11):6728–34. doi: 10.1074/jbc.M707485200. [DOI] [PubMed] [Google Scholar]
- 13.Sinard JH, Stafford WF, Pollard TD. The mechanism of assembly of Acanthamoeba myosin-II minifilaments: minifilaments assemble by three successive dimerization steps. J Cell Biol. 1989 Oct;109(4 Pt 1):1537–47. doi: 10.1083/jcb.109.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinard JH, Pollard TD. The effect of heavy chain phosphorylation and solution conditions on the assembly of Acanthamoeba myosin-II. J Cell Biol. 1989 Oct;109(4 Pt 1):1529–35. doi: 10.1083/jcb.109.4.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King MV, Young M. Selective non-enzymic cleavage of the myosin rod. Electron-microscopic studies on crystals and paracrystals of light meromyosin-C. J Mol Biol. 1970 Jun 14;50(2):491–507. doi: 10.1016/0022-2836(70)90207-x. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura A, Sreter F, Gergely J. Comparative studies of light meromyosin paracrystals derived from red, white, and cardiac muscle myosins. J Cell Biol. 1971 Jun;49(3):883–98. doi: 10.1083/jcb.49.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Safer D, Pepe FA. Axial packing in light meromyosin paracrystals. J Mol Biol. 1980 Feb 5;136(4):343–58. doi: 10.1016/0022-2836(80)90394-0. [DOI] [PubMed] [Google Scholar]
- 18.Bennett PM. The structure of spindle-shaped paracrystals of light meromyosin. J Mol Biol. 1981 Feb 25;146(2):201–21. doi: 10.1016/0022-2836(81)90432-0. [DOI] [PubMed] [Google Scholar]
- 19.Ward R, Murray JM. Three-dimensional structure of frozen-hydrated paracrystals of myosin rod. J Muscle Res Cell Motil. 1990 Oct;11(5):403–18. doi: 10.1007/BF01739761. [DOI] [PubMed] [Google Scholar]
- 20.Ward R, Bennett PM. Paracrystals of myosin rod. J Muscle Res Cell Motil. 1989 Feb;10(1):34–52. doi: 10.1007/BF01739855. [DOI] [PubMed] [Google Scholar]
- 21.Atkinson SJ, Stewart M. Expression in Escherichia coli of fragments of the coiled-coil rod domain of rabbit myosin: influence of different regions of the molecule on aggregation and paracrystal formation. J Cell Sci. 1991 Aug;99( Pt 4):823–36. doi: 10.1242/jcs.99.4.823. [DOI] [PubMed] [Google Scholar]
- 22.Stathopulos PB, Rumfeldt JA, Karbassi F, Siddall CA, Lepock JR, Meiering EM. Calorimetric analysis of thermodynamic stability and aggregation for apo and holo amyotrophic lateral sclerosis-associated Gly-93 mutants of superoxide dismutase. J Biol Chem. 2006 Mar 10;281(10):6184–93. doi: 10.1074/jbc.M509496200. [DOI] [PubMed] [Google Scholar]
- 23.Obermann WM, van der Ven PF, Steiner F, Weber K, Furst DO. Mapping of a myosin-binding domain and a regulatory phosphorylation site in M-protein, a structural protein of the sarcomeric M band. Mol Biol Cell. 1998 Apr;9(4):829–40. doi: 10.1091/mbc.9.4.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flashman E, Watkins H, Redwood C. Localization of the binding site of the C-terminal domain of cardiac myosin-binding protein-C on the myosin rod. Biochem J. 2007 Jan 1;401(1):97–102. doi: 10.1042/BJ20060500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welikson RE, Fischman DA. The C-terminal IgI domains of myosin-binding proteins C and H (MyBP-C and MyBP-H) are both necessary and sufficient for the intracellular crosslinking of sarcomeric myosin in transfected non-muscle cells. J Cell Sci. 2002 Sep 1;115(Pt 17):3517–26. doi: 10.1242/jcs.115.17.3517. [DOI] [PubMed] [Google Scholar]