

Abstract
The solution structure of Ca2+-bound regulatory domain of cardiac troponin C (cNTnC) in complex with the switch region of troponin I (cTnI147-163) and the calmodulin antagonist, N-(6-aminohexyl)-5-chloro-1-naphthalenesulfinamide (W7), has been determined by NMR spectroscopy. The structure reveals that the W7 naphthalene ring interacts with the terminal methyl groups of M47, M60, and M81 as well as aliphatic and aromatic side-chains of several other residues in the hydrophobic pocket of cNTnC. The H3 ring proton of W7 also contacts the methyl groups of I148 and M153 of cTnI147-163. The N-(6-aminohexyl) tail interacts primarily with the methyl groups of V64 and M81, which are located on the C- and D-helices of cNTnC. Compared to the structure of the cNTnC•Ca2+•W7 complex (Hoffman, R. M. B. and Sykes, B. D. (2009) Biochemistry 48, 5541-5552), the tail of W7 reorients slightly towards the surface of cNTnC while the ring remains in the hydrophobic pocket. The positively charged -NH3+ group from the tail of W7 repels the positively charged R147 of cTnI147-163. As a result, the N-terminus of the peptide moves away from cNTnC and the helical content of cTnI147-163 is diminished, when compared to the structure of cNTnC•Ca2+•cTnI147-163 (Li, M. X., Spyracopoulos, L., and Sykes B. D. (1999) Biochemistry 38, 8289-8298). Thus the ternary structure cNTnC•Ca2+•W7•cTnI147-163 reported in this study offers an explanation for the ∼13-fold affinity reduction of cTnI147-163 for cNTnC•Ca2+ in the presence of W7, and provides a structural basis for the inhibitory effect of W7 in cardiac muscle contraction. This generates molecular insight into structural features that are useful for the design of cTnC-specific Ca2+-desensitizing drugs.
Keywords: troponin, structure, drugs, mechanism, inhibition
A healthy human heart generates ∼3 billion contractile cycles over an average life span. Each contractile cycle involves systolic activation and diastolic relaxation, regulated by Ca2+ association and dissociation from troponin in the cardiac myofilaments. Troponin is a heterotrimeric complex composed of the Ca2+-binding subunit, troponin C (cTnC), inhibitory subunit, troponin I (cTnI), and the tropomyosin binding subunit, troponin T (cTnT). During diastole, troponin holds tropomyosin in a conformational state that blocks the interaction between myosin and actin. When Ca2+ binds cTnC during systole, the switch region of cTnI associates with the N-domain of cTnC (cNTnC). This is coupled with the removal of the inhibitory region of cTnI from actin so that the troponin-tropomyosin complex no longer inhibits the actin-myosin interaction. Subsequently, tension producing actin-myosin cross bridges form, which potentiates actomyosin ATPase activity, and ultimately force generation (for reviews, see [1,2]).
In diseased heart, the contractile cycle is compromised by either Ca2+-desensitization with diminished systolic contractility (e.g. in cases of congestive heart failure) or Ca2+-oversensitization accompanied with insufficient diastolic relaxation (e.g. in cases of hypertrophic cardiomyopathy). The ability to sensitize or desensitize cardiac muscle to Ca2+ has therapeutic potential for the treatment of cardiac dysfunction. Ideally, this mechanism would avoid altering Ca2+ transients in myocardial cells, which would perturb the regulation of other Ca2+-based signaling pathways; but rather involve modulating the altered Ca2+ response of the myofilaments (for a review, see [3]). The essential role of troponin in the regulation of the contractile cycle makes it an attractive and logical target for the design of cardiotonic drugs. Toward this goal, a group of Ca2+-sensitizing drugs have been developed. One example is levosimendan, a novel Ca2+-sensitizer discovered by using cTnC as a target protein (for a recent review, see [4]). This drug has been proved to be a well-tolerated, effective treatment for patients with severe decompensated heart failure. A recent study has shown that a myosin inhibitor, blebbistatin (1-phenyl-1,2,3,4-tetrahydro-4-hydroxypyrrolo(2,3-b)-7-methylquinolin-4-one) functions as an effective Ca2+-desensitizer in cardiac muscle contraction without causing arrhythmia, suggesting that Ca2+-desensitization might be beneficial to individuals with hypertrophic cardiomyopathy [5].
Many hydrophobic compounds are known to bind to CaM and perturb CaM-target interactions. Because of the structural homology between cTnC and CaM, these agents may also interact with cTnC and be good candidates as cardiotonic drugs. An earlier study has shown that some CaM antagonists (calmidazolium, bepridil, trifluoperazine, chlorpromazine, pimozide) stimulate myofibrillar ATPase activity while others (W7, haloperiodol, mastoparan) inhibit ATPase activity [6]. This suggests that CaM antagonists differentially affect the properties of troponin, probably via different modes of action on the cTnC-cTnI interface. Structural studies have identified multiple binding sites of TFP and bepridil on cTnC [7]. In the X-ray structure of the cTnC•3Ca2+•3bepridil complex [8], two bepridil molecules pull the N- and C-domains close together to result in a compact structure for cTnC, while a third bepridil appears to stabilize an open regulatory domain conformation by binding to the hydrophobic pocket much like the switch region of cTnI (cTnI147-163) as shown in the structures of cNTnC•Ca2+•cTnI147-163[9] and cTnC•3Ca2+•cTnI31-210•cTnT183-288 [10] complexes. The NMR structure of the cNTnC•Ca2+•cTnI147-163•bepridil complex shows that bepridil and cTnI147-163 bind to the hydrophobic pocket of cNTnC•Ca2+ concurrently [11]. In the X-ray structure of cNTnC•Ca2+•2TFP (PDB: 1WRK and 1WRL), two TFP molecules fit in the hydrophobic pocket of cNTnC•Ca2+ with the −CF3 group of each TFP pointed towards the hydrophobic cleft. When compared to the structure of cNTnC•Ca2+•cTnI147-163•bepridil, it appears that one of the TFP molecules would be replaced by cTnI147-163 peptide. In the X-ray structure of the sTnC•4Ca2+•sTnI1-182•sTnT156-262 complex [12], a polyoxyethylene detergent molecule, anapoe, binds specifically to the Ca2+-saturated N-domain of sTnC together with the switch region of sTnI (sTnI115-131) and this binding is likely responsible for the increase of the contractile force of muscle fibers in the presence of anapoe. The Ca2+-sensitizing effect of levosimendan has been shown to be due to a network of interactions between the drug, cNTnC, and cTnI144-163 (a longer version of switch region of cTnI) that creates a binding site for levosimendan and the net result is the stabilization of the open conformation of cNTnC•Ca2+ [13].
W7 is a CaM antagonist that has been used to explore a wide range of physiological processes involving Ca2+-signaling in cardiomyocytes. Focusing on delineating the role of W7 in the interplay of troponin- and myosin-based pathways of Ca2+-activation in skeletal and cardiac muscle, Adhikari and Wang have shown that in both skeletal and cardiac muscle fibers, W7 inhibits the maximum ATPase activity and Ca2+ sensitivity [14]. The W7 inhibition is most likely mediated via specific interactions between W7 and TnC. This notion is supported by the observation that W7 binds specifically to TnC and not to tropomyosin, actin, or myosin [15]. We have shown that W7 binds specifically to cNTnC, and this binding can occur concurrently with the switch region of cTnI [16]. Moreover, we have shown that the affinity of W7 for cNTnC in the cTnC•3Ca2+•cTnI34-71•cTnI128-163 complex is ∼ 2-fold weaker than for the isolated cNTnC•Ca2+ [17]. This is in line with the functional data demonstrating that W7 is an effective inhibitor in cardiac muscle contraction. We have also determined the solution structure of cNTnC•Ca2+•W7 that located the binding site of W7 in the hydrophobic pocket of cNTnC•Ca2+ [18].
In order to elucidate the mechanism of W7 inhibition, we have determined the solution structure of a ternary cNTnC•Ca2+•cTnI147-163•W7 complex. In the structure cTnI147-163 binds at the A/B interhelical interface and W7 binds in the hydrophobic cavity formed between cNTnC•Ca2+ and cTnI147-163. In the structure, the N-terminal R147 of cTnI147-163 is electrostatically repelled from the positively charged tail of the W7, and thus moves away from cNTnC•Ca2+. This offers an explanation for the ∼13-fold affinity reduction of cTnI147-163 for cNTnC•Ca2+ in the presence of W7. The structure provides insights into the design of cardiotonic drugs that modulate the Ca2+-sensitivity of the myofilament via perturbation of the cTnC-cTnI interaction.
Materials and Methods
Sample preparation
The engineering of the cNTnC (1-89) construct and the expression of 15N- and 15N/13C-labeled cNTnC in E. coli were as described previously [11]. The synthetic peptide cTnI147-163, Acetyl-RISADAMMQALLGARAK-Amide, was obtained from GL Biochem Ltd. (Shanghai, China). The purity and mass were verified by HPLC and electrospray mass spectrometry, respectively. W7 was purchased from Sigma-Aldrich. Stock solutions of 100 mM W7 in DMSO-d6 (Cambridge Isotopes Inc.) were prepared and since W7 is sensitive to light, the vial was wrapped in aluminum foil. All NMR samples were 500 μL in volume. The buffer conditions were 100 mM KCl, 10 mM imidazole in 90% H2O/10% D2O or 5% H2O/95% D2O, 15 mM dithiothreitol (DTT), 0.5 mM 3-(trimethylsilyl)-1-propanesulfonic acid (DSS), and the pH adjusted to 6.8. For structure determination, NMR samples contain 0.7 mM cNTnC, 7 mM Ca2+, 3 mM W7, and 3.2 mM cTnI147-163.
cTnI147-163 titration of 15N-cNTnC•Ca2+•W7
Previously, cTnI147-163was titrated into 15N-cNTnC•Ca2+ in the absence of W7 [9]. The titration was performed by adding solid peptide and amino acid analysis was used to determine the peptide/protein ratio at every titration point. This is because cTnI147-163 peptide is only soluble in aqueous solution at low concentrations (∼ 1 mM) and tends to form a gel at higher concentrations. A similar procedure was employed in this work for the titration of cTnI147-163 to 15N-cNTnC•Ca2+ in the presence of W7. The sample was mixed thoroughly with each addition. Changes in pH associated with cTnI147-163 additions were compensated by adjusting to pH 6.8 at every titration point. 1D 1H and 2D 1H,15N-HSQC NMR spectra of 15N-cNTnC were acquired at every titration point.
NMR spectroscopy
NMR data used in this study were collected at 30°C using Varian Inova 800-MHz, Unity 600-MHz, and Varian Inova 500-MHz spectrometers. All spectrometers are equipped with triple resonance probes with Z-pulsed field gradients. The chemical shifts of backbone and side-chain atoms of cNTnC as well as the bound W7 and cTnI147-163 were assigned using 2D and 3D NMR experiments outlined in the Supplemental Table S1. 1D 1H and 2D DQF-COSY spectra of free W7 in DMSO-d6 were obtained (as described in Hoffman and Sykes [18]) to help in the assignment of the W7 in the ternary complex. 2D DQF-COSY NMR spectra of a NMR sample of cNTnC•Ca2+•cTnI147-163•W7 in D2O were acquired for the assignment of bound W7. Resonances for aromatic residues were assigned using 2D homonuclear DQF-COSY and NOESY experiments in D2O. 2D 13C/15N-filtered TOCSY and 2D 13C/15N-filtered NOESY [19-21] spectra of a sample of cNTnC•Ca2+•cTnI147-163•W7 in H2O were acquired for the assignment of bound W7 and bound cTnI147-163. The intermolecular NOEs between W7 and cTnI147-163 were assigned from 2D 13C/15N-filtered NOESY. The intermolecular NOEs between 13C/15N labeled cNTnC and unlabeled cTnI147-163 and W7 were obtained using 3D-13C-edited HMQC-NOESY [22,23] experiment with 150 ms mixing time.
Data processing and peak calibration
All 2D and 3D NMR data were processed using NMRPipe [24] and all 1D NMR data were processed using VNMRJ (Varian Inc.). Assignment of chemical shifts was carried out in NMRViewJ [25] and backbone assignments were aided with the software package SmartNotebook [26]. The chemical shifts that were assigned in NMRView were converted to CYANA nomenclature, for NOE calibration. Initial structures of cNTnC were generated using program CYANA 2.1. Unambiguous restraints were assigned manually and were forced to keep their assignments during the first four runs of CYANA [27] calculations, after which they were open for automatic assignment with the “noeassign” script of CYANA. Distance restraints were calibrated with CYANA standard procedure using upper limits of 6 Å. After the CYANA refinement, the final restraints were converted to XPLOR-NIH [28,29] nomenclature. Intramolecular NOEs of cNTnI147-163 and W7 and all intermolecular NOEs were assigned manually and categorized as weak (1.8-6.0 Å).
Coordinates and topology of W7
Representation of W7 was defined using HIC-Up and XPLO2D servers (http://xray.bmc.uu.se) and checked manually. Positive charge on W7 tail (-CH2-NH3+ group) was added manually in analogy to positive charge on -CH2-NH3+ group in LYS+.
Structural calculations
Dihedral angle restraints from TALOS [30,31] as well as 6 distance restraints from X-ray crystallographic data of chelating oxygen atoms to the Ca2+ ion in site II were used for structure calculation in addition to NOE distance restraints. The 20 conformers of cNTnC obtained from CYANA were averaged in XPLOR-NIH, and used as a template structure in the simulated annealing protocol, with 10,000 high temperature steps and 6000 cooling steps. After the structure of cNTnC was well defined, the ternary cNTnC•Ca2+•cTnI147-163•W7 structure was solved in a similar manner, starting with an extended conformation of cNTnC. The intramolecular and intermolecular NOE restraints used in the calculations are summarized in Table 1. 30 lowest energy conformers of the 300 calculated were refined in explicit solvent by XPLOR-NIH with a water box edge length of 18.8 Å. The final ensemble discussed in this article represent the 20 lowest energy conformers obtained after water refinement (see Table 1 for statistics). The final refined ensemble has been deposited in the protein data bank (www.rcsb.org) with the PDB accession code of 2KRD.
Table 1.
Structural statistics of the family of the 20 structures calculated.
| Intramolecular NOE restraints in cNTnC | ||
| Total | 1184 (∼13/residue) | |
| Short range (|i-j|≤1) | 817 | |
| Medium-range (2≤|i-j|≤4) | 211 | |
| Long-range (|i-j|≥5) | 156 | |
| Dihedral Restraints in cNTnC | ||
| φ | 59 | |
| ψ | 57 | |
| Intermolecular NOEs between cNTnC and W7 | 30 | |
| Intermolecular NOEs between cNTnC and the cTnI147-163 peptide | 24 | |
| Intermolecular NOEs between the cTnI147-163 peptide and W7 | 3 | |
| Intramolecular NOE restraints in W7 | 6 | |
| Intramolecular restraints in cTnI147-163 peptide | ||
| NOEs | 15 | |
| φangle restraints | 5 | |
| ψ angle restraints | 5 | |
| Artificial restraints to Ca2+ | 6 | |
| NOE violations/structure > 0.2 Å | ||
| Before water refinement | 0.0±0.0 | |
| After water refinement | 8.7±2.1 | |
| Dihedral violations/structure >1° | ||
| Before water refinement | 0.55±0.12 | |
| After water refinement | 1.00±0.22 | |
| φ, ψin core or allowed regionsc | ||
| Before water refinement | 99.5% | |
| After water refinement | 98.9% | |
| R.M.S.D. from the average structure (Å)a | ||
| Well-defined residues | Helices | |
| Before water refinement | 0.80±0.11 | 0.32±0.05 |
| After water refinement | 0.87±0.12 | 0.37±0.06 |
Well-defined backbone atoms (N, C, CA) with R.M.S.D smaller than 1Å. The R.M.S.D. for helices are calculated from the weighted mean average of the R.M.S.D. of individual helices.
The final KNOE and Kdihedral used were 50 kcalmol-1 and 200 kcalmol-1rad-2, respectively.
As determined using PROCHECK
Results and Discussion
Effect of W7 on the stability of cNTnC•Ca2+•cTnI147-163
We have previously characterized the interaction of cTnI147-163 and cNTnC•Ca2+ and determined the affinity of cTnI147-163 for cNTnC•Ca2+ (KD = 154 ± 30 μM) [9]. In the present study, the assigned 2D 1H, 15N-HSQC NMR spectrum of cNTnC•Ca2+•W7 was used to monitor the binding of cTnI147-163 to cNTnC•Ca2+•W7. Using four residues (L29, A31, K39, and E66) as examples, a comparison of the cTnI147-163-induced 2D 1H, 15N-HSQC NMR peak shifts of cNTnC•Ca2+ and of cNTnC•Ca2+•W7 is depicted in Figure 1A and 1B, respectively. In both titrations, the chemical shift changes fall into the fast exchange limit on the NMR scale, and the movement of the cross peaks indicates that the binding of cTnI147-163 to cNTnC•Ca2+ or cNTnC•Ca2+•W7 occurs with a 1:1 stoichiometry (Supplemental Figure S1). The cTnI147-163-induced chemical shift changes are larger in Figure 1A, corresponding to a closed to open conformational transition in cNTnC as characterized previously [9]. The smaller cTnI147-163-induced chemical shift changes in Figure 1B corresponds to the binding of cTnI147-163 to an already partially open cNTnC in the cNTnC•Ca2+•W7 complex [18]. Plotting the chemical shift changes in Figure 1B as a function of the peptide to protein ratio, a binding dissociation constant (KD) of 2000 ± 50 μM gives the best fit as shown in Supplemental Figure S1. A global fitting for the titration of cTnI147-163 to cNTnC•Ca2+ is also shown in Figure S1 for the purpose of a direct comparison. This suggests that the affinity of cTnI147-163 for cNTnC•Ca2+ is decreased ∼ 13-fold (from 154 ± 30 μM to 2000 ± 50 μM) by the presence of W7. This helps explain why W7 is effective in inhibiting the activity of actomyosin ATPase and force generation in muscle fibers [6,14].
Figure 1.
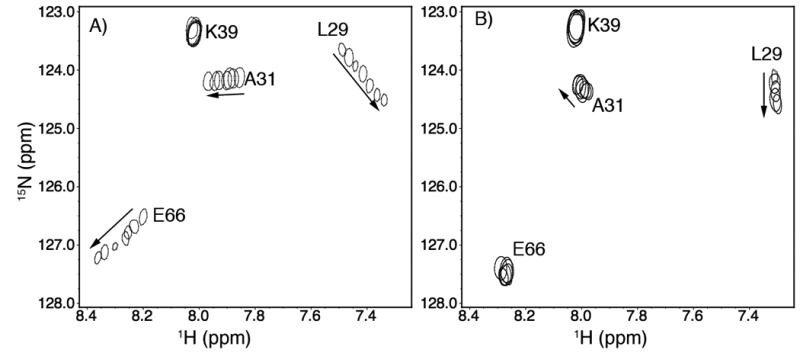
Titration of (A) cNTnC•Ca2+and (B) cNTnC•Ca2+•W7, respectively, with cTnI147-163. 2D 1H,15N-HSQC NMR spectra for residues L29, A31, K39, and E66 of cNTnC at various peptide additions are superimposed, showing the progressive shift of peaks with increasing cTnI147-163 concentrations.
Overall structure of the ternary complex
The 2D 1H, 15N-HSQC NMR spectrum of cNTnC in the ternary cNTnC•Ca2+•cTnI147-163•W7 complex is well resolved, allowing the chemical shifts of the backbone and the side chain atoms to be assigned using 15N- and/or 15N/13C-labeled protein. Intramolecular distance restraints for cNTnC in the ternary complex were obtained by analyzing the 3D 15N- and 13C-edited NOESY experiments. Dihedral angle restraints for cNTnC in the ternary complex were obtained from TALOS [30].
The proton NMR chemical shift assignments and intramolecular distance restraints for the bound cTnI147-163 and W7 required the collection of 15N/13C filtered 2D-TOCSY and 2D-NOESY experiments using unlabeled cTnI147-163 and W7 in complex with 15N/13C labeled cNTnC. These experiments removed all of the resonance peaks arising from 15N/13C labeled cNTnC. 2D DQF-COSY NMR spectra of free (see reference [18]) and bound W7 (see Supplemental Figure S2) were also acquired to aid in the assignment of bound W7. Intermolecular NOEs between cNTnC and W7 (Figure 2A) and between cNTnC and cTnI147-163 (Figure 2B) were determined from 3D-13C-edited/filtered HMQC-NOESY data. These experiments were optimized for detecting NOEs between protons attached to 12C and protons attached to aliphatic 13C, thereby editing out all the intramolecular NOEs. Three intermolecular NOEs between cTnI147-163 and W7 were obtained from the 15N/13C filtered 2D NOESY experiment (Figure 2C). A total of 1264 NOE distance restraints were used for structure calculations. These include 1184 intramolecular cNTnC distance restraints (∼ 13 restraints per residue), 15 intramolecular cTnI147-163 distance restraints, 6 intramolecular W7 distance restraints, 24 intermolecular contacts between cNTnC and cTnI147-163, 30 intermolecular contacts between cNTnC and W7, and 3 intermolecular contacts between cTnI147-163 and W7. 116 dihedral restraints for cNTnC, 10 dihedral restraints for cTnI147-163, and 6 artificial distance restraints to Ca2+ were used to calculate the structure of the cNTnC•Ca2+•cTnI147-163•W7 complex (see Table 1). Figure 3A depicts the ensemble of the 20 lowest energy structures of the ternary complex in stereo view, with overall structural statistics and conformational energies for the ensemble provided in Table 1. The ribbon representations of the structure of the ternary complex are shown in Figure 3B. The structure of cNTnC in the complex is better defined than those of cTnI147-163 or W7, as a result of more restraints for the protein than for the ligands.
Figure 2.


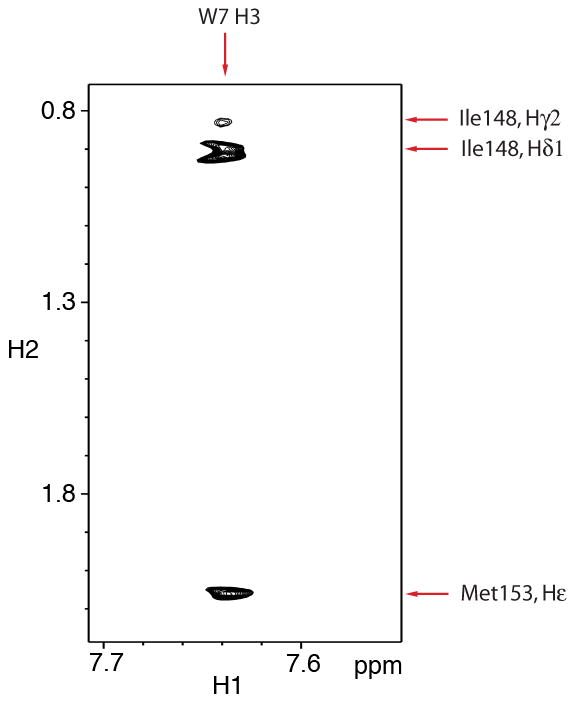
Intermolecular NOE contacts between cNTnC•Ca2+ and W7 (A), between cNTnC•Ca2+ and cTnI147-63 (B), and between cTnI147-163 and W7 (C). The NMR sample of cNTnC•Ca2+•cTnI147-163•W7 contains 0.7mM cNTnC, 7mM Ca2+, 3.4mM cTnI147-163, and 3mM W7: (A) The chemical structure of W7. The number designation and the chemical shift assignments of W7 are indicated. Strips from the 3D-15N/13C-edited HMQC-NOESY NMR experiments showing the intermolecular NOE contacts between the ring of W7 and cNTnC and between the tail of W7 and cNTnC. The circled peaks are intermolecular NOE contacts between the cTnI147-163 peptide and cNTnC (left panel) and are unassigned (right panel); (B) The sequence of cTnI147-163 peptide. Example strips from the 3D-15N/13C-edited HMQC-NOESY NMR experiments showing the intermolecular NOE contacts between cNTnC and cTnI147-163; (C) Intermolecular NOE contacts between the methyl groups of I148 and M153 of cTnI147-163 to H3 from the naphthalene ring of W7, obtained from 2D-15N/13C-filtered-NOESY NMR spectra.
Figure 3.
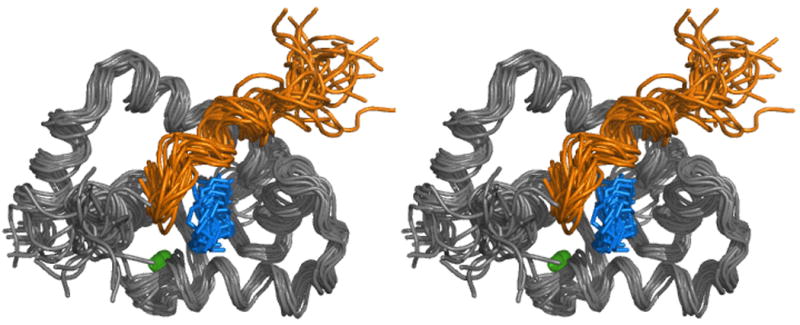

A) The solution structure of the cNTnC•Ca2+•cTnI147-163•W7 complex in stereo view. The backbones (N, Cα, C′) of a family of 20 structures are shown in gray. The assembly of the cTnI147-163 structures is shown in gold. The assembly of W7 is shown in blue. The Ca2+ ions are shown as green spheres.
B) The ribbon representation of the lowest energy structure in stereo view. The protein is shown in green and Ca2+ ions are shown as gray spheres. Heavy atom skeleton of W7 is shown in blue. The cTnI147-163 peptide is shown in purple.
Structure of cNTnC in the ternary complex
cNTnC in the present structure exhibits an overall fold resembling other Ca2+-bound domains in the EF-hand family. The secondary structural elements of cNTnC in the ternary cNTnC•Ca2+•cTnI147-163•W7 complex are similar to those in the cNTnC•apo (PDB: 1SPY), cNTnC•Ca2+ ((PDB: 1AP4), cNTnC•Ca2+•W7 (PDB: 2KFX), cNTnC•Ca2+•cTnI147-163 (PDB: 1MXL), and cNTnC•Ca2+•cTnI147-163•bepridil (PDB: 1LXF) complexes. The five helices, N, A, B, C, and D, are well-defined, superimposing with individual backbone rmsds of 0.31±0.12Å for helix N (residues 5-11), 0.54±0.14Å for helix A (residues 14-27), 0.20±0.13Å for helix B (residues 41-46), 0.39±0.13Å for helix C (residues 54-63), and 0.42±0.11Å for helix D (residues 74-86). The two EF hands Ca2+-binding sites are joined by a short anti-parallel β-sheet. The β-sheet (residues 35-37, 71-73) is well-defined with a backbone rmsd of 0.39±0.09Å, while the Ca2+-binding sites have a slightly more flexible structure (a backbone rmsd of 0.92±0.28Å). The N- and C-terminal residues (residues 1-4 and 86-89) are less well defined than the helices or the β-sheet. The global fold and the orientations of the five helices of cNTnC in the ternary cNTnC•Ca2+•cTnI147-163•W7 complex are similar to the open cNTnC•Ca2+•cTnI147-163, cNTnC•Ca2+•W7, and cNTnC•Ca2+•cTnI147-163•bepridil structures (see Figure 5). The effects of cTnI147-163 and W7 on the structural fold of cNTnC will be discussed below.
Figure 5.
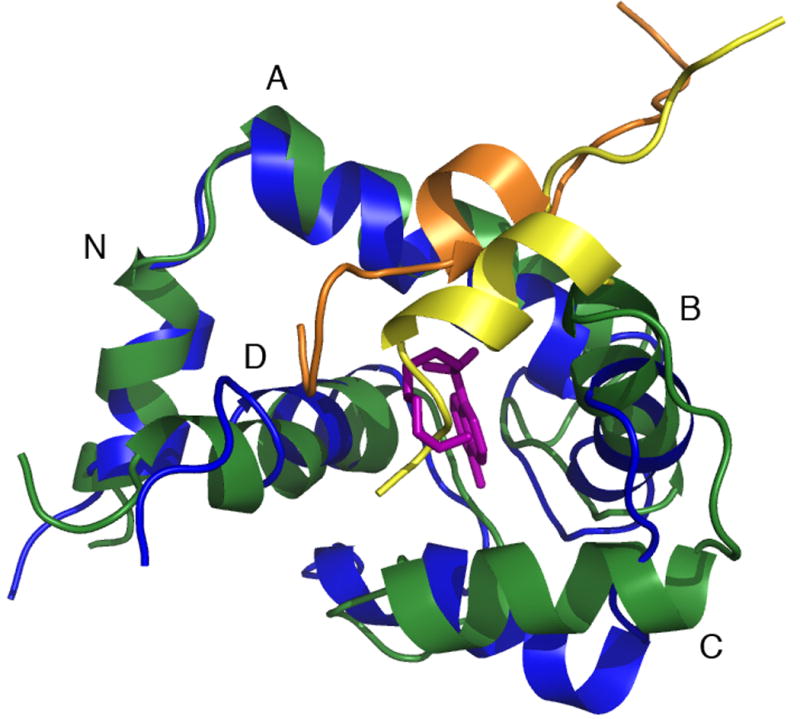
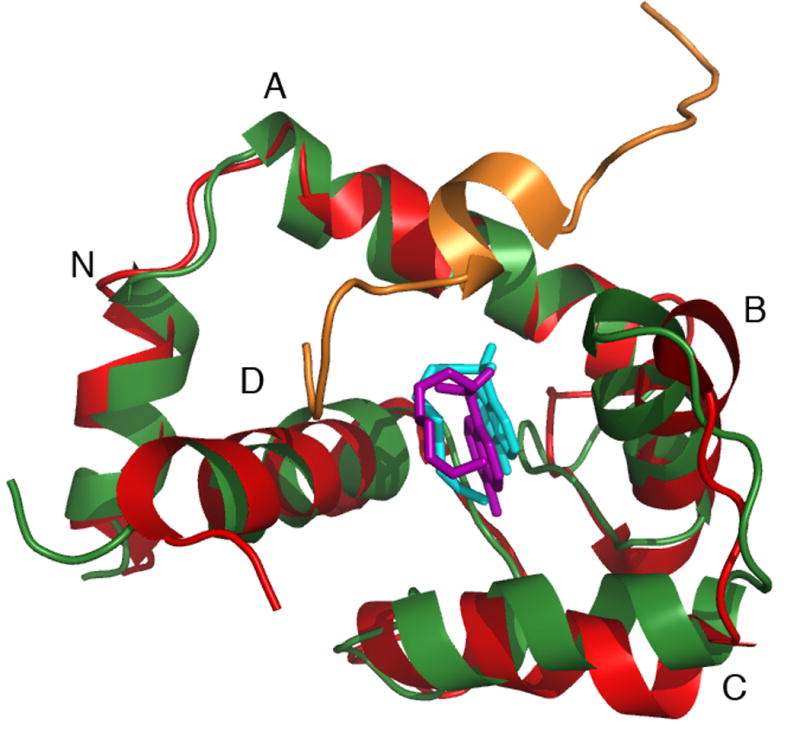

A) Backbone overlay of cNTnC in the cNTnC•Ca2+•cTnI147-163•W7 complex (green) and cNTnC in the cNTnC•Ca2+•cTnI147-163 complex (blue). Note the position shift and helical content changes of cTnI147-163 from yellow to gold in the presence of W7 (pink).
B) Backbone overlay of cNTnC in the cNTnC•Ca2+•cTnI147-163•W7 complex (green) and cNTnC in the cNTnC•Ca2+•W7 complex (red). Note the change of position of W7 from cyan to pink when cTnI147-163 is present.
C) Backbone overlay of cNTnC in the cNTnC•Ca2+•cTnI147-163•W7 complex (cNTnC in green, cTnI147-163 in gold, W7 in pink) and cNTnC in the cNTnC•Ca2+•cTnI147-163•bepridil complex (cNTnC in brown, cTnI147-163 in gray, bepridil in green).
Structure of W7 in the ternary complex
W7 consists of a naphthalene group and an N-(6-aminohexyl) tail (Figure 2A). The binding site and conformation of W7 in the ternary complex is similar as in the structure of cNTnC•Ca2+•W7. The orientation of W7 was determined by intermolecular NOEs between cNTnC and W7 and between cTnI147-163 and W7. Strip plots taken from the 3D 15N/13C F1-filtered, F3-edited spectra of cNTnC•Ca2+•cTnI147-163•W7 complex are shown in Figure 2A and 2B. In the spectra, only NOEs arising from the 12C-attached protons in cTnI147-163 and W7 and terminating on 13C-attached protons in cNTnC are observed. The naphthalene ring of W7 makes a number of contacts with residues that line the hydrophobic pocket of cNTnC, such as A23, L41, V44, M47, M60, I61, M81, I36, V64, and V72. As a result, the naphthalene ring of W7 is nestled deep in the hydrophobic cleft. The contacts between H3 of W7 and the methyl groups of I148 and M153 of cTnI147-163 fix the orientation of the bicyclic ring relative to cTnI147-163 (Figure 4). The N-(6-aminohexyl) tail of W7 makes contacts to the C/D helices of cNTnC (e.g. H16 to the methyl groups of V64 and M81). The positively charged −NH3+ moiety of W7 and the N-terminal R147 of cTnI147-163 repel each other and the two ligands reorient their positions relative to those in the binary cNTnC•Ca2+•cTnI147-163 or the cNTnC•Ca2+•W7 complex to accommodate each other in the hydrophobic pocket of cNTnC•Ca2+ (see below). When the heavy atoms of W7 superimpose onto the average structure in the ternary complex, the average rmsd for the naphthalene group is 1.1 Å, while that for the tail is 2.1 Å.
Figure 4.
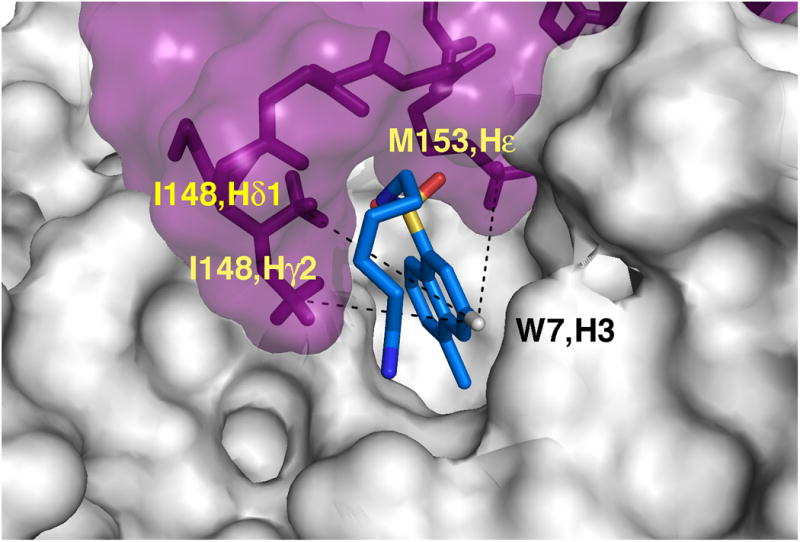
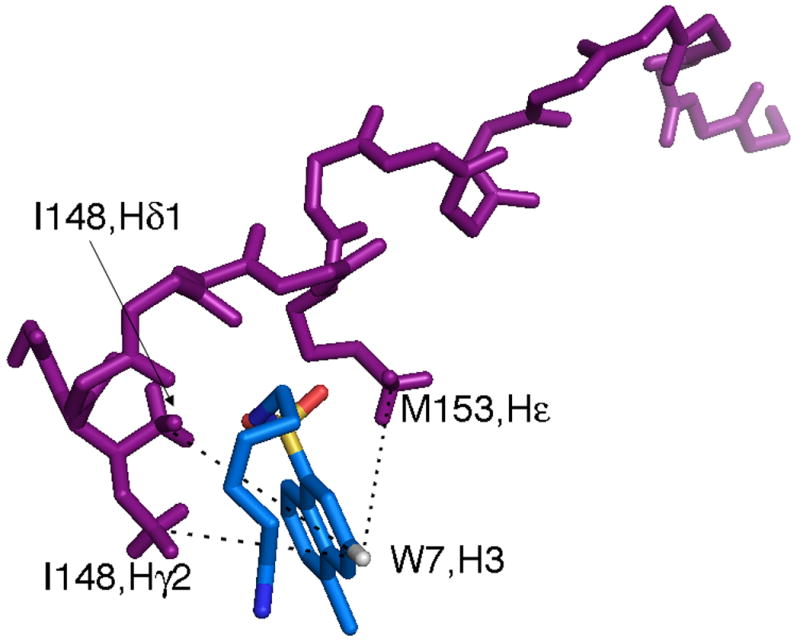
A close up view of the skeleton representation of the peptide backbone and W7, showing the interaction between cTnI147-163 and W7 with (A) and without (B) the protein surface present.
Structure of cTnI147-163 in the ternary complex
The structure of cTnI147-163 when complexed with cNTnC•Ca2+ has been determined in three separate structures. In the structure of the cNTnC•Ca2+•cTnI147-163, residues 150-157 form a short α-helix. This short helix was defined primarily by the use of chemical shift index and several intermolecular NOE restraints between cNTnC and the peptide (there were no intramolecular NOEs assigned). In the structure of cNTnC•Ca2+•cTnI147-163•bepridil, 24 intramolecular NOEs including 3 dNN and 6 dαN(i,i+3) (the hallmark of a helical structure) restraints for the 17 residue cTnI147-163 peptide were assigned. A combination of these NOEs and the dihedral angle restraints better defined the peptide. In the X-ray structure of the core domain cardiac troponin, cTnC•3Ca2+•cTnI34-210•cTnT182-288, this fragment of cTnI displays a near identical structure as in the two NMR structures (superimpose to 0.2 Å). In the present structure, cNTnC•Ca2+•cTnI147-163•W7, the cTnI147-163 conformation is calculated by using both intra- and inter-molecular NOEs and dihedral restraints. The overall conformation and orientation is similar to those in the other three structures but the helical component is diminished. In the ensemble of solution structures (Figure 3), the backbone atoms of the 17 residues superimpose on to the average peptide structure with an rmsd of 1.9±0.3 Å. The assigned intermolecular NOEs are summarized in Figure 2. These NOE contacts dictate a similar orientation for the cTnI147-163 in the ternary cNTnC•Ca2+•cTnI147-163•W7 complex as compared to that in the cNTnC•Ca2+•cTnI147-163 or cNTnC•Ca2+•cTnI147-163•bepridil complexes. The peptide inserts between the A/B helices in cNTnC with the N-terminus (residues 147-149) and the helical region interacts with the protein, while the C-terminus does not interact with the protein and remains disordered in the ensemble of the solution structures (Figure 3). Specifically, the helical portion of the peptide (residues 152-157) sits between the A/B helical interface, contacting the methyl group of residues A22, A23, and I26 in the A-helix and L41, K43, M47 and L48 in the B-helix. The N-terminus of the peptide extends into the hydrophobic pocket of cNTnC, making contacts to the methyl groups of M60, V82 and M85 in cNTnC. As discussed above, the N-terminal residue R147 of the peptide moves away from the tail of W7 because of the electrostatic repulsion.
Comparison of the cNTnC•Ca2+•cTnI147-163•W7 structure with the cNTnC•Ca2+•cTnI147-163, cNTnC•Ca2+•W7, and cNTnC•Ca2+•cTnI147-163•bepridil structures
The conformational change in cNTnC•Ca2+ that occurs upon binding cTnI147-163, bepridil, and W7 involves the straightening of the B-helix and the BC unit moving away from the NAD unit, consisting of a ‘closed’ to ‘open’ structural transition. This peptide or drug induced structural change in cNTnC•Ca2+ is similar to that observed for the apo to Ca2+-saturated transition in sNTnC [32]. An interesting finding from the current study is that while the cNTnC in this structure exhibits an open conformation, the A/B and C/D EF hands behave as separate units. When the NAD unit of cNTnC in cNTnC•Ca2+•cTnI147-163•W7 is superimposed onto the NAD unit of cNTnC in cNTnC•Ca2+•cTnI147-163, the B helix moved up along with cTnI147-163 (up 4±1 Å toward the A-helix) while the C-helix moved down (Figure 5A). As a result, the A/B pair is more closed but the C/D pair is more open. This change is also reflected in the A/B and C/D interhelical angle changes (see Table 2). It seems the C/D pair needs to be more open to accommodate W7 and the repulsion between W7 and cTnI147-163 shifts the peptide away from the hydrophobic pocket with a concomitant destabilization of the helix. As a result, the NOE contacts between cNTnC and cTnI147-163 from cNTnC•Ca2+•cTnI147-163 to cNTnC•Ca2+•cTnI147-163•W7 are altered. For instance, in cNTnC•Ca2+•cTnI147-163, A23 but not A22 on the A-helix makes a strong contact to L157 of cTnI147-163 peptide, while both A22 and A23 make strong contacts to L157 in cNTnC•Ca2+•cTnI147-163•W7. When the NAD unit of cNTnC in cNTnC•Ca2+•cTnI147-163•W7 superimpose onto the NAD unit of cNTnC in cNTnC•Ca2+•W7, the A/B unit did not change but the C-helix moved down (Figure 5B, see the A/B and C/D interhelical angle changes listed Table 2). In both the W7 bound structures, the strong NOE contacts between the hydrophobic residues in cNTnC and the naphthalene ring of W7 serve as the primary anchoring site for this molecule in the hydrophobic core of the protein. In the presence of cTnI147-163, W7 needs to rearrange its ring and makes more contacts with the C/D interhelical interface. As a result, the C/D pair is more open. When the NAD unit of cNTnC in cNTnC•Ca2+•cTnI147-163•W7 superimpose onto the NAD unit of cNTnC in cNTnC•Ca2+•cTnI147-163•bepridil, both the A/B and C/D pairs exhibit more open interhelical angels (Figure 5C). As a result, in cNTnC•Ca2+•cTnI147-163•W7, the BC unit is farther away from the NAD unit than that in cNTnC•Ca2+•cTnI147-163•bepridil. This could be attributed to the different effects of W7 and bepridil on the interaction of cNTnC and cTnI147-163.
Table 2.
Inter-helical angles of various EF hands.
| Calcium Binding Protein | Interhelical angles(°)a | Accession Code (PDB) | |
|---|---|---|---|
| A-B | C-D | ||
| cNTnC·apo (NMR) | 140±3 | 128±4 | 1SPY |
| cNTnC·Ca2+ (NMR) | 132±3 | 113±4 | 1AP4 |
| cNTnC·Ca2+·cTnI147-163 (NMR) | 102±5 | 96±5 | 1MXL |
| cTnC·3Ca2+·cTnI31-210·cTnT183-288 (X-RAY) | 103 | 96 | 1J1E |
| cNTnC·Ca2+·bepridil·cTnI147-163 (NMR) | 121±4 | 91±4 | 1LXF |
| cTnC·3Ca2+·bepridil (X-RAY) | 92 | 90 | 1DTL |
| cNTnC·Ca2+·W7 (NMR) | 114±3 | 92±2 | 2KFX |
| cNTnC·Ca2+·W7·cTnI147-163 (NMR) | 113±5 | 66±4 | 2KRD |
| CaM(N-domain) ·Ca2+·W7 (NMR) | 102±4 | 75±5 | 1MUX |
| CaM(C-domain) ·Ca2+·W7 (NMR) | 105±4 | 85±4 | 1MUX |
The 180° interhelical angle corresponding to a completely closed EF hand domain (antiparallel helices), whereas 0° angle defines a completely open conformation (parallel helices). The extent of openness is characterized by the A/B and C/D interhelical angles. cNTnC in 2KRD is open as compared to the closed 1SPY or 1AP4. The A/B helical pair is more closed than 1MXL, while the C/D pair is more open. As compared to 2KFX, the A/B pair is similar but the C/D pair is more open. Both the A/B and C/D pairs of 2KRD are more open than cNTnC in 1LXF. While the C/D helical pair in this complex is more open than that in all the open cNTnC structures, it is similar to the corresponding helical pair in CaM(N-domain)•2Ca2+•W7 and CaM(C-domain)•2Ca2+•W7.
The axis for an α-helix is defined by two points, taken as the average coordinates of the first and last 11 backbone atoms of the α-helix. Inter-helical angels were calculated using the program interhelix (K. Yap, University of Toronto).
The docking of W7 and cTnI147-163 on cNTnC is represented by comparing the molecular surfaces of cNTnC•Ca2+•cTnI147-163, cNTnC•Ca2+•cTnI147-163•W7, and cNTnC•Ca2+•W7 in Figure 6. In cNTnC•Ca2+•cTnI147-163, the helical region fit nicely between the A/B interhelical interface and the positive charged N-terminal R147 forms salt bridges with the acidic residues on the C/D helices of cNTnC (Figure 6A). To accommodate W7, the helical region of the peptide unwinds slightly and moves up toward the A helix while the N-terminal R147 moves away from the tail of W7 as a result of electrostatic repulsion (Figure 6B). In the absence of cTnI147-163, W7 finds its place in the hydrophobic pocket of cNTnC•Ca2+ (Figure 6C) and it reorients its position slightly to allow the concurrent binding of cTnI147-163 (see Figure 5B).
Figure 6.
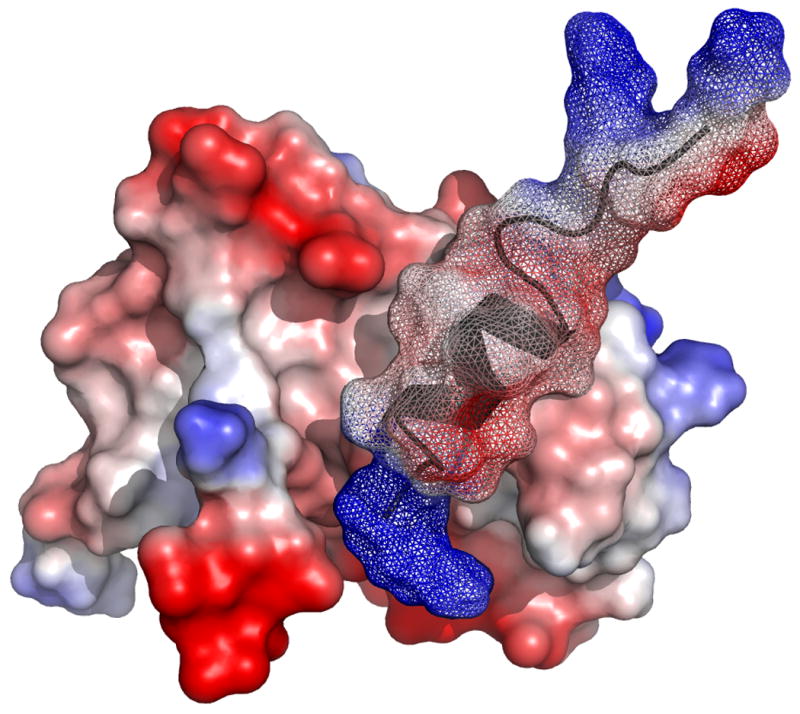
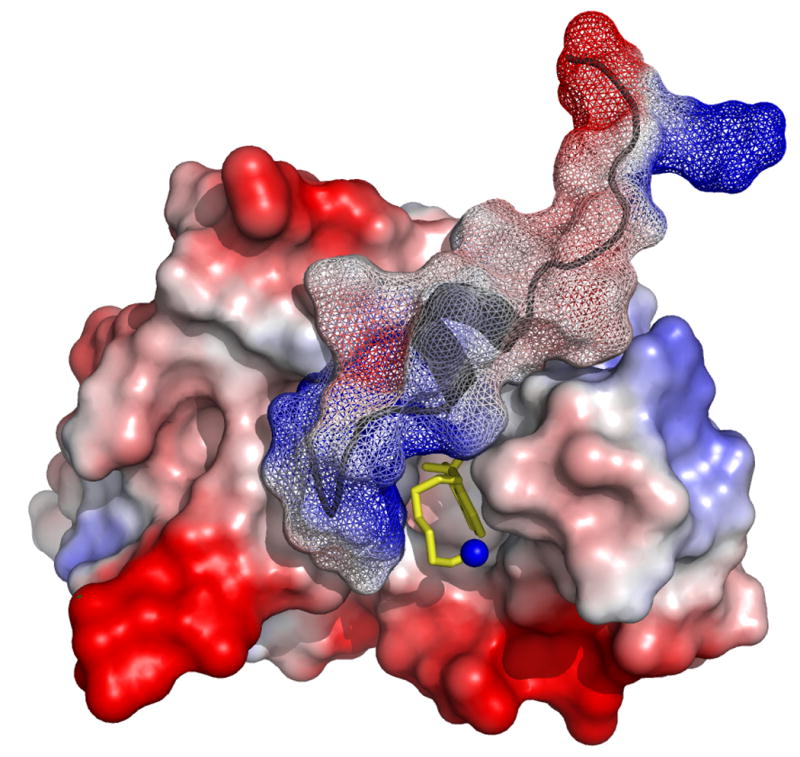
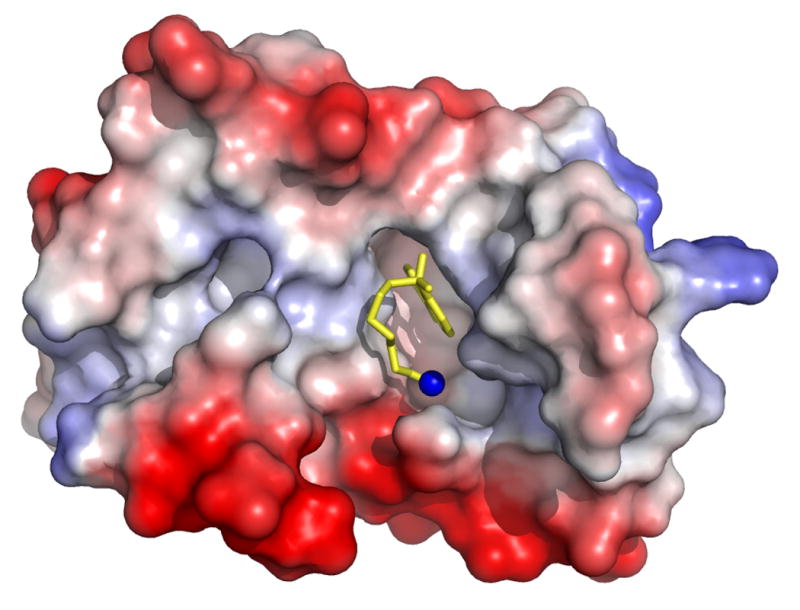
Molecular surface representations of (A) cNTnC•Ca2+•cTnI147-163 (PDB, 1MXL), (B) cNTnC•Ca2+•cTnI147-163•W7 (PDB, 2KRD), and (C) cNTnC•Ca2+•W7 (PDB, 2KFX). For cNTnC and cTnI147-163, positively charged residues are shown in blue, negatively charged residues are shown in red, and other residues are shown as charge gradient. Non-hydrogen atoms of W7 are shown as stick models in yellow and nitrogen is shown in blue.
In summary, the interaction between the cTnI147-163 peptide and W7 in the cNTnC•Ca2+•cTnI147-163•W7 ternary complex makes the two ligands reorient their positions relative to those in the cNTnC•Ca2+•cTnI147-163 or cNTnC•Ca2+•W7 complexes. The consequence is a rearrangement of interhelices of both AB and CD EF hands in the ternary complex than in the binary complexes. The interference between W7 and cTnI147-163 explains the weaker affinity of cTnI147-163 for cNTnC•Ca2+in the presence of W7 than in cNTnC•Ca2+ alone.
Comparison of the structures of cNTnC•Ca2+•cTnI147-163•W7 and cNTnC•Ca2+•W7 with the structure of CaM•4Ca2+•2W7
The NMR structure of CaM•4Ca2+ in complex with two W7 molecules was determined [33]. In this structure, each domain of CaM interacts with one W7 molecule. W7 binds CaM via many Van der Waals interactions between the naphthalene ring of the W7 and the hydrophobic residues of the CaM binding pocket. Osawa et al. compared the CaM binding mode between W7 and TFP and between W7 and tryptophan of CaM target peptide and concluded that the naphthalene ring is the largest that can fit into the hydrophobic pocket of CaM domain. As the N-terminal and C-terminal domains of CaM are similar, the orientation of W7 within the binding pocket is similar as well. The tail of W7 in the CaM domains is flexible but more defined in the structures of cNTnC•Ca2+•W7 and cNTnC•Ca2+•cTnI147-163•W7. Thus, W7 appears to inhibit the CaM-mediated activation of target proteins by blocking the hydrophobic pocket of each domain with its naphthalene ring that normally plays a role in anchoring targets. However, in cNTnC, the tail of W7 is playing a major role in interfering with cTnC's major target, cTnI.
Relevance to the future design of cTnC-specific pharmacological agents for the treatment of heart disease
There is a need for the development of novel approaches in the therapy of heart disease. The current therapeutic paradigm for decreased systolic function is the employment of drugs that increase cystolic Ca2+-levels; however, prolonged use leads to increased heart rate, hypertension, arrhythmias, and mortality. Presently, there is no drug available for the treatment of familial hypertrophic cardiomyopathy that is associated with hypertrophy and fibrosis contributing to the risk of ventricular arrhythmias and sudden cardiac death. Mutations in myofilament proteins or altered myofilament structures lead to changes in the myofilament response to Ca2+ and thus represent good targets for the design of novel inotropic agents. A group of Ca2+-sensitizers has been developed, which have been shown to increase the myocardial contractility by generating more force for a given amount of cytosolic Ca2+. Levosimendan and EMD 57033 are typical examples. Recently, a new class of pharmacological agents, cardiac myosin activators (e.g. CK-1827452), have been shown to directly target the kinetics of the myosin head (for a review see [34]. In vitro studies have demonstrated that these agents increase the rate of effective myosin-actin cross-bridge formation and improve myocyte energy utilization with no effect on intracellular Ca2+ or cAMP. Animal model and in human studies have demonstrated significant increases in systolic ejection time and cardiac output, suggesting that myosin activators offer the promise of a safe and effective treatment for heart failure.
The aforementioned compounds all belong to a class of compounds known as Ca2+-sensitizers, and are effective in treating symptoms of decreased cardiac output; however, there are less treatment options for patients suffering from impaired diastolic relaxation or increased contractility. The therapeutic advantage of compounds that target the contractile machinery was illustrated in a recent study that showed arrhythmias susceptibility caused by the Ca2+-sensitizing agent EMD 57033 in animal models is prevented by the myosin inhibitor, blebbistatin [5,35]. The protective effect of blebbistatin provides the first direct evidence that Ca2+-desensitization in myofilaments is antiarrhythmic and might be beneficial to individuals with hypertrophic cardiomyopathy.
The data presented in this work provide the structural basis of W7 inhibition of the regulatory cTnC-cTnI interaction. W7 functions as a Ca2+-desensitier by decreasing the binding affinity of cTnI147-163 for cNTnC•Ca2+. The NMR solution structure of the cNTnC•Ca2+•cTnI147-163•W7 ternary complex furthers understanding of the molecular mechanism underlying the effects of cardiotonic drugs on the cTnC-cTnI interface. Together with a series of structures determined previously in our laboratory, it provides a molecular model important for the design of cTnC-specific cardiotonic drugs.
Supplementary Material
Acknowledgments
We thank Ryan Hoffman for insightful discussions. We thank David Corson and Melissa Crane for the help with the cNTnC protein expression. We would like to thank the Canadian National High Field NMR Centre (NANUC) for their assistance and use of the facilities. Operation of NANUC is funded by the Canadian Institutes of Health Research, the Natural Science and Engineering Research Council of Canada and the University of Alberta. We also would like to acknowledge Jeff DeVries, and Nicolas Shaw for spectrometer maintenance.
Supported by the Canadian Institutes of Health Research (Grant MOP 37760), the National Institutes of Health Grant (R01 HL-085234), and the Heart and Stroke Foundation of Canada. Marta Oleszczuk is supported by an Alberta Heritage Foundation for Medical Research postdoctoral fellowship and Ian Robertson is supported by an Alberta Heritage Foundation for Medical Research studentship.
Abbreviations
- TnC
troponin C
- cTnC
cardiac troponin C
- cNTnC
N-domain (residues 1-89) of cTnC
- cCTnC
C-domain (residues 91-161) of cTnC
- sTnC
skeletal troponin C
- CaM
calmodulin
- TnI
troponin I
- cTnI
cardiac troponin I
- cTnI147-163
cTnI peptide Acetyl-RISADAMMQALLGARAK-Amide
- sTnI
skeletal troponin I
- sTnI115-131
sTnI residues 115-131
- TFP
trifluoperazine
Footnotes
Data Deposition: The atomic coordinates have been deposited in the RCSB Protein Data Bank (PDB accession code: 2KRD)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- 2.Li MX, Wang X, Sykes BD. Structural based insights into the role of troponin in cardiac muscle pathophysiology. J Muscle Res Cell Motil. 2004;25:559–579. doi: 10.1007/s10974-004-5879-2. [DOI] [PubMed] [Google Scholar]
- 3.Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006;113:305–315. doi: 10.1161/CIRCULATIONAHA.105.542407. [DOI] [PubMed] [Google Scholar]
- 4.Nieminen MS, Pollesello P, Vajda G, Papp Z. Effects of levosimendan on the energy balance: preclinical and clinical evidence. J Cardiovasc Pharmacol. 2009;53:302–310. doi: 10.1097/FJC.0b013e31819c9a17. [DOI] [PubMed] [Google Scholar]
- 5.Baudenbacher F, et al. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–3903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silver PJ, Pinto PB, Dachiw J. Modulation of vascular and cardiac contractile protein regulatory mechanisms by calmodulin inhibitors and related compounds. Biochem Pharmacol. 1986;35:2545–2551. doi: 10.1016/0006-2952(86)90052-3. [DOI] [PubMed] [Google Scholar]
- 7.Kleerekoper Q, Liu W, Choi D, Putkey JA. Identification of binding sites for bepridil and trifluoperazine on cardiac troponin C. J Biol Chem. 1998;273:8153–8160. doi: 10.1074/jbc.273.14.8153. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Love ML, Putkey JA, Cohen C. Bepridil opens the regulatory N-terminal lobe of cardiac troponin C. Proc Natl Acad Sci U S A. 2000;97:5140–5145. doi: 10.1073/pnas.090098997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li MX, Spyracopoulos L, Sykes BD. Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 10.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Li MX, Sykes BD. Structure of the regulatory N-domain of human cardiac troponin C in complex with human cardiac troponin I147-163 and bepridil. J Biol Chem. 2002;277:31124–31133. doi: 10.1074/jbc.M203896200. [DOI] [PubMed] [Google Scholar]
- 12.Vinogradova MV, et al. Ca(2+)-regulated structural changes in troponin. Proc Natl Acad Sci U S A. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robertson IM, Baryshnikova OK, Li MX, Sykes BD. Defining the binding site of levosimendan and its analogues in a regulatory cardiac troponin C-troponin I complex. Biochemistry. 2008;47:7485–7495. doi: 10.1021/bi800438k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adhikari BB, Wang K. Interplay of troponin- and Myosin-based pathways of calcium activation in skeletal and cardiac muscle: the use of W7 as an inhibitor of thin filament activation. Biophys J. 2004;86:359–370. doi: 10.1016/S0006-3495(04)74112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hidaka H, Yamaki T, Naka M, Tanaka T, Hayashi H, Kobayashi R. Calcium-regulated modulator protein interacting agents inhibit smooth muscle calcium-stimulated protein kinase and ATPase. Mol Pharmacol. 1980;17:66–72. [PubMed] [Google Scholar]
- 16.Li MX, Hoffman RM, Sykes BD. Interaction of cardiac troponin C with calmodulin antagonist [corrected] W7 in the presence of three functional regions of cardiac troponin I. Biochemistry. 2006;45:9833–9840. doi: 10.1021/bi060779a. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman RM, Li MX, Sykes BD. The binding of W7, an inhibitor of striated muscle contraction, to cardiac troponin C. Biochemistry. 2005;44:15750–15759. doi: 10.1021/bi051583y. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman RM, Sykes BD. Structure of the inhibitor W7 bound to the regulatory domain of cardiac troponin C. Biochemistry. 2009;48:5541–5552. doi: 10.1021/bi9001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gemmecker G, Olejniczak ET, Fesik SW. An Improved Method for Selectively Observing Protons Attached to C-12 in the Presence of H-1-C-13 Spin Pairs. J Magn Reson. 1992;96:199–204. [Google Scholar]
- 20.Ikura M, Bax A. Isotope-Filtered 2d NMR of a Protein Peptide Complex - Study of a Sceletal-Mucsle <yosin Light Chain Kinase Fragment Bound to Calmodulin. J Am Chem Soc. 1992;114:2433–2440. [Google Scholar]
- 21.Ogura K, Terasawa H, Inagaki F. An Improved Double-Tuned and Isotope-Filtered Pulse Scheme Based on a Pulse Field Gradient and a Wide-Band Inversion Shaped Pulse. J Biomol NMR. 1996;8:492–498. doi: 10.1007/BF00228150. [DOI] [PubMed] [Google Scholar]
- 22.Lee W, Revington MJ, Arrowsmith C, Kay LE. A pulsed field gradient isotope-filtered 3D 13C HMQC-NOESY experiment for extracting intermolecular NOE contacts in molecular complexes. FEBS Lett. 1994;350:87–90. doi: 10.1016/0014-5793(94)00740-3. [DOI] [PubMed] [Google Scholar]
- 23.Roberston IM, Spyracopoulos L, Sykes BD. The evaluation of Isotope editing and filtering for protein-ligand interaction elucidation by NMR. NATO Science for Peace and Security Series - B: Physics and Biophysics 2009;Biophysics and the Challenges of Emerging Threats. :101–119. [Google Scholar]
- 24.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 25.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 26.Slupsky CM, Boyko RF, Booth VK, Sykes BD. Smartnotebook: a semi-automated approach to protein sequential NMR resonance assignments. J Biomol NMR. 2003;27:313–321. doi: 10.1023/a:1025870122182. [DOI] [PubMed] [Google Scholar]
- 27.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 28.Linge JP, Williams MA, Spronk CA, Bonvin AM, Nilges M. Refinement of protein structures in explicit solvent. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 29.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 30.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 31.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009 doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gagne SM, Tsuda S, Li MX, Smillie LB, Sykes BD. Structures of the troponin C regulatory domains in the apo and calcium-saturated states. Nat Struct Biol. 1995;2:784–789. doi: 10.1038/nsb0995-784. [DOI] [PubMed] [Google Scholar]
- 33.Osawa M, et al. Solution structure of calmodulin-W-7 complex: the basis of diversity in molecular recognition. J Mol Biol. 1998;276:165–176. doi: 10.1006/jmbi.1997.1524. [DOI] [PubMed] [Google Scholar]
- 34.Teerlink JR. A novel approach to improve cardiac performance: cardiac myosin activators. Heart Fail Rev. 2009;14:289–298. doi: 10.1007/s10741-009-9135-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allingham JS, Smith R, Rayment I. The structural basis of blebbistatin inhibition and specificity for myosin II. Nat Struct Mol Biol. 2005;12:378–379. doi: 10.1038/nsmb908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.