

Summary
The mammalian target of rapamycin (mTOR) is a master regulator of cell growth and division that responds to a variety of stimuli, including nutrient, energy, and growth factors. In the last years, a significant number of pieces have been added to the puzzle of how mTOR coordinates and executes its functions. Extensive research on mTOR has also uncovered a complex network of regulatory loops that impact the therapeutic approaches aimed at targeting mTOR.
Introduction
Adequate cellular levels of energy and nutrients are a prerequisite for successful cell growth and division. Therefore, cells have acquired mechanisms to sense energy and nutrients before committing to grow and divide. In all eukaryotes, the target of rapamycin (TOR) protein is a master regulator that integrates the signals from nutrient and energy sensors with cell growth and proliferation, so as to ensure that they are triggered only during favorable conditions. The mammalian TOR (mTOR) also integrates growth factor signaling together with nutrients and energy as a mechanism to coordinate cell growth and proliferation in large numbers of cells within different organs. In addition to this, a network of regulatory loops affects the function of mTOR and impact on therapeutic approaches aimed at targeting mTOR, which will be discussed here.
The mTOR signaling pathway
mTOR forms two different protein complexes defined by the proteins to which it is bound, exerting different but related functions. The mTOR complex 1 (mTORC1) is defined by the presence of Raptor, mLST8/GβL, Deptor and Pras40 [1-7], whereas Rictor, GβL, Protor, Deptor and mSin form mTORC2 [8-12] (see Figure 1). In addition to their differential sensitivity to rapamycin, mTORC1 and 2 are activated in different ways and have distinct substrate specificity. mTORC1, which is sensitive to rapamycin, responds to energy, amino acids, growth factors and oxygen levels, whereas mTORC2 activation is ill-defined, but seems to be only by growth factors. Active mTORC1 phosphorylates, among other targets, S6K1 and 4EBP1, proteins involved in the regulation of translation initiation, protein synthesis and cell mass gain (see [13] for a comprehensive review). An overview of the mTOR complexes and signaling pathway is provided in Figure 1.
Figure 1. mTOR signaling pathway.
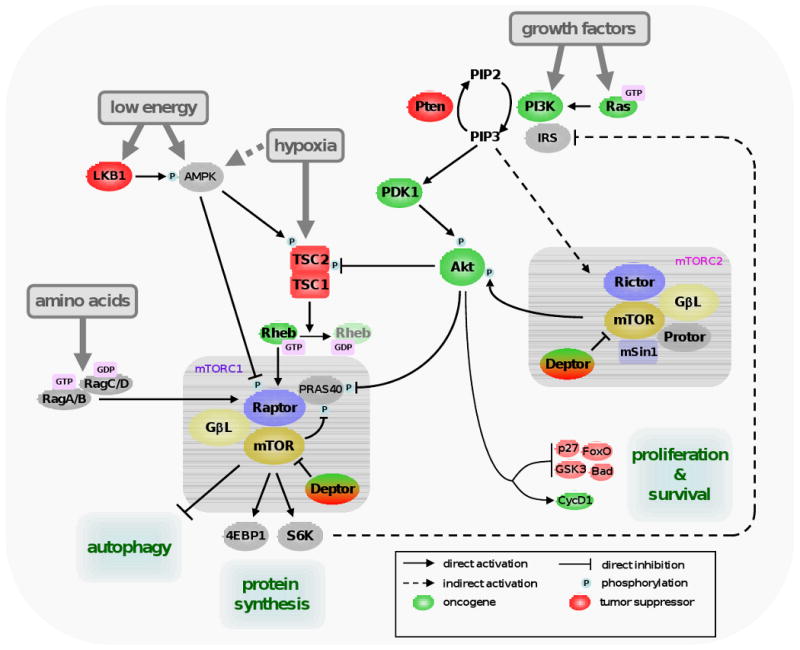
One branch of mTORC1 activation is mediated by the class I phosphoinositide-3 kinase (PI3K), Akt (also known as Pkb) and the tuberous sclerosis complex (TSC). TSC is formed by TSC1 and TSC2, and inhibits a direct activator of mTORC1, the GTPase Ras-homolog enriched in brain (Rheb) by hydrolyzing its GTP into GDP [52]. TSC2 is activated by phosphorylation by AMP-activated protein kinase (AMPK) [53], which is directly activated by a high AMP vs. ATP ratio. AMPK also directly phosphorylates and inactivates Raptor, so it inhibits mTORC1 by TSC-dependent and -independent manners [54]. The activity of AMPK is regulated by phosphorylation by the tumor suppressor LKB1. This protein, like TSC1/2, was found mutated in the germline of patients with different hamartomatous syndromes [55,56]. Akt is a serine/threonine kinase and an important player in regulating mTORC1 activity. Akt positively regulates mTORC1 by acting at different levels. First, Akt inactivates TSC1/2 by phosphorylating TSC2 [57]. Second, Akt inhibits Pras40, negative regulator of mTORC1 that counteracts Rheb function [6,7]. Akt is activated by PI3K, which responds to a variety of growth factors. When activated by insulin or insulin-like growth factors (IGFs), as well as other growth factors, class I PI3K catalyzes the formation of the lipidic second messenger phosphoinositide 3,4,5 tri-phosphate (PIP3) from the bi-phosphate form PIP2. PIP3 triggers the relocation of Akt to the inner surface of the plasma membrane, where it is activated by phosphoinositide-dependent kinase 1 (PDK1) and transduces the signal as described above. Opposing Akt function is the tumor suppressor Phosphatase and Tensin homolog deleted on chromosome ten (Pten), a lipid phosphatase that converts of PIP3 to PIP2, thus shutting off signaling from PI3K. Pten deficiency causes a series of hamartomatous syndromes collectively classified as PTEN hamartoma tumour syndrome (reviewed in [58]). Amino acids activate mTORC1 by an independent route mediated by the Rag family of proteins. The activation of mTORC2 is not well-understood, but this complex directly activates Akt (and Akt-related kinases) by phosphorylation. Akt, in addition, regulates many proteins involved in cell survival and cell cycle progression.
The Rag - amino acid link
The fact that amino acid-dependent activation of mTORC1 occurs independently of the Akt-TSC axis signaling has been known for years, but the identification of the molecular link between amino acids and mTORC1 has remained elusive. Recently, we and others showed that the Rag family of GTPases is required for mTORC1 activation by amino acids [14,15]. Rag proteins directly interact with Raptor in an amino acid-dependent manner, and this interaction leads to mTORC1 activation. Interestingly, the nucleotide-bound status of the Rag GTPases dictates its affinity to Raptor and the activation of mTORC1. This discovery shows a previously unknown regulatory mechanism of mTOR function that involves mTOR shuttling. In the presence of amino acids, Rag proteins mediate mTORC1 shuttle from disperse punctuate pattern in the cytoplasm to the endomembrane system of the cell, where signaling occurs [15]. According to this, appropriate localization and active PI3K-Akt-Rheb axis must coexist, explaining why both amino acids and growth factors are needed for activation of mTORC1. It is likely that Rag GTPases are not responsible for directly sensing amino acid levels; instead this information is probably communicated to the Rag proteins by additional mediators.
Complex loops of regulation - all roads lead to mTOR
mTOR, as a master regulator of a variety of inputs, is subjected to different mechanisms that tightly and coordinately regulate its activation. Many positive and feedback loops have been described lately and most likely other regulatory loops will be deciphered in the near future.
The S6K1-PI3K inhibition
A very important negative feedback involves the inhibition of PI3K by mTORC1, which occurs at many levels (see Figure 2A). Active S6K inhibits insulin receptor substrate 1 (IRS-1) by phosphorylating it at multiple sites, by inducing its degradation and by altering its localization, all of which ultimately dampen PI3K/Akt activation [16-19]. This loop is relevant in vivo and influences therapeutic responses based on mTOR inhibition (see below), and also plays a role in type 2 diabetes [20]. A functionally similar loop mediated by S6K1 is the inhibition of platelet-derived growth factor receptor (PDGFR), that also signals through PI3K [21]. Of note, the direct target of S6K1 that inhibits PDGFR activity is currently unknown. In addition, it has been recently shown that S6K1 is also responsible for inhibiting the activity of the ERK/MAPK pathway. This loop was empirically unmasked by therapeutic inhibition of mTORC1 in cancer patients, which correlated with an increase in ERK phosphorylation in vivo [22]. In spite of the fact that most research on mTORC1 is centered on S6K1, it is also conceivable that the effects on mTORC1 inhibition may not be solely through this target. These partially overlapping inhibitory loops have probably evolved as a mechanism to negatively control PI3K pathway after its activation, reflecting the necessity of putting a brake to excessive (acute or chronic) activation of the pathway.
Figure 2. Feedback loops modulating mTOR signaling pathway.
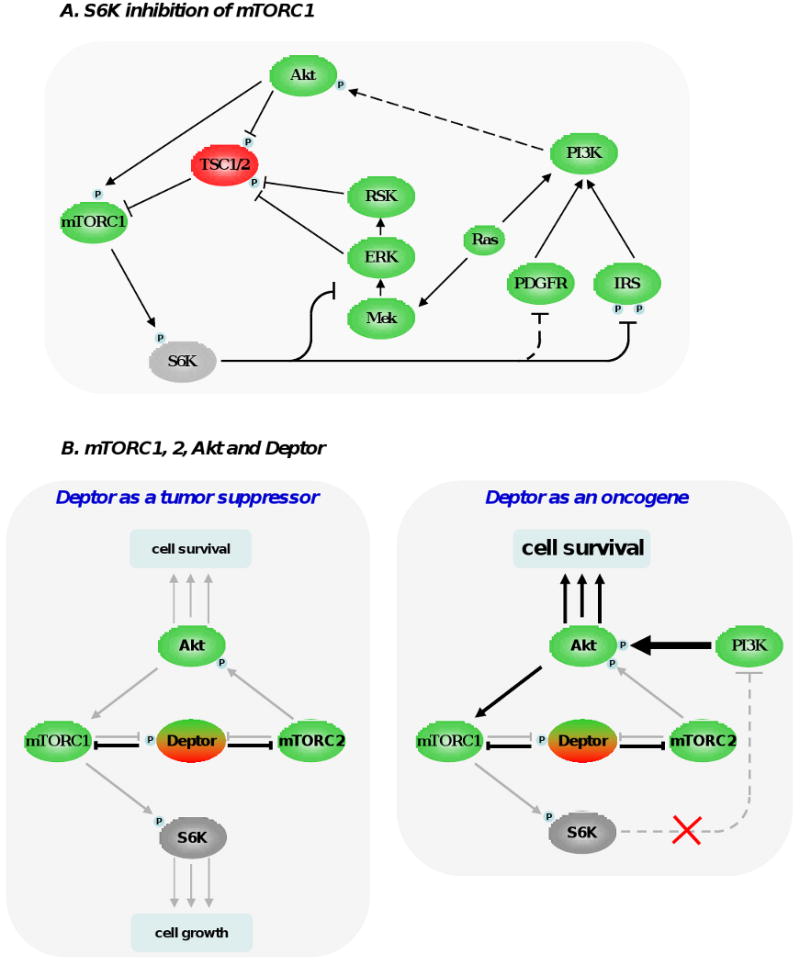
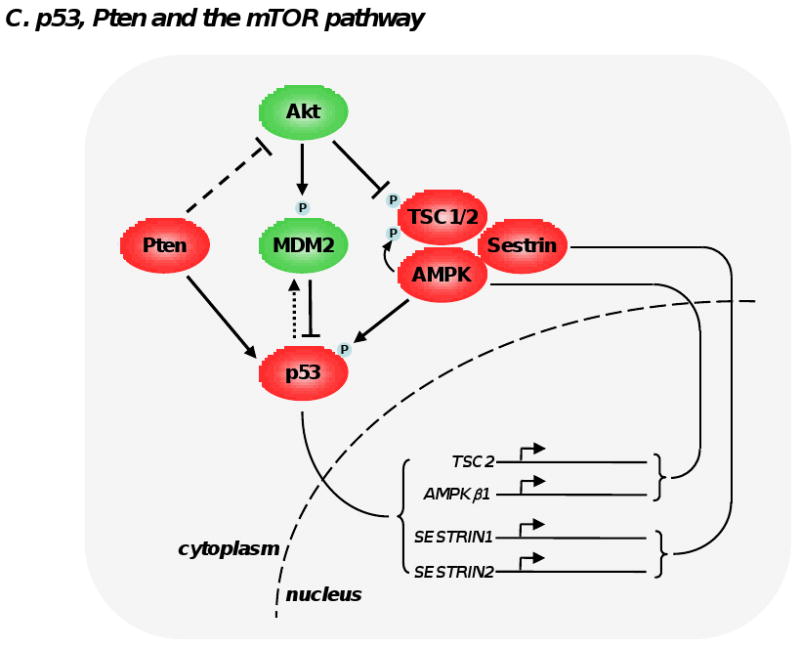
(A) S6K1 phosphorylation by mTORC1 triggers a number of feedback loops that negatively impact on PI3K signaling. S6K1 directly phosphorylates insulin receptor substrate 1 (IRS-1) at multiple sites, which leads a significant attenuation of insulin and and insulin-like growth factor effect at the cell membrane. S6K1 also inhibits PDGFR and the MEK/ERK signaling pathway through a not fully-understood mechanism. These 3 inhibitory loops ultimately dampen PI3K, Akt and mTORC1 activation and have likely evolved as a cellular response to buffer aberrant or excessive PI3K activity. (B) Deptor can have oncogenic and tumor suppressive properties. By blocking mTORC1 and mTORC2, Deptor inhibits protein synthesis, cell size increase and the proliferative and survival effects of Akt. However, under certain conditions, inhibition of mTORC1 by Deptor relieves the feedback inhibition from S6K1 to PI3K, boosting Akt activity. (C) The 2 most important tumor suppressors, namely Pten and p53, cooperate in the inhibition of mTORC1. p53 transactivates many negative regulators of mTORC1 (TSC2, AMPKβ1, Sestrin 1 and 2), which oppose Akt activity. Akt favors degradation of p53 by phosphorylating and stabilizing MDM2. Importantly, Pten and AMPK directly stabilize p53. Collectively, these interactions imply the existence of a coordinate regulation of mTOR signaling pathway by energy, nutrients, growth factors, oncogenic stress and DNA integrity.
Intra-complex loops
mTOR regulation involves a series of feedback loops triggered by mTORC1 and 2 components. Perhaps the most interesting loop where both mTOR complexes participate is the one involving mTOR regulation of Akt. It has been shown that mTORC2 is the kinase of Akt at the serine in position 473 [23]. This phosphorylation, together with the one at 308 performed by PDK1 upon PI3K activation, is needed for maximal Akt activity and conceptually locates mTOR both upstream and downstream Akt (Figure 2B).
Our group has recently described the role of Deptor in the regulation of both mTORC1 and 2 [5]. The functional relationship of Deptor and the mTORCs is not simple (Figure 2B). Deptor protein and mRNA levels are suppressed by mTORC1 and mTORC2 activation, and Deptor inhibits mTORC1 and 2 kinase activities in vitro and in cell-based assays. Consequently, activation of mTORC1 and 2 leads to Deptor inhibition, leading to further mTOR activity and in contrast, high levels of Deptor inhibit mTOR activity. Importantly, inhibition of mTORC1 by Deptor indirectly activates Akt, by relieving the inhibitory loop on PI3K triggered by S6K1. Considering this, theoretically Deptor could have both tumor suppressive (by inhibition of mTORC1 and 2) and oncogenic roles (by the activation of Akt) (Figure 2B). Although more work and in vivo data will help clarify this issue, transcriptional profiling of tumor cell lines showed that Deptor was overexpressed in multiple myeloma cells lines that show high Akt activity. In these cell lines, silencing of Deptor lead to inhibition of Akt and cell death, altogether suggesting that deregulated Deptor function may have oncogenic properties [5].
Recently, TSC, in addition to its role in the inhibition of mTORC1, was linked to mTORC2 function. TSC1/2 complex interacts with mTORC2 and positively regulates its kinase activity by an as yet undefined mechanism that seems to be independent of the inhibition of mTORC1 [24]. Although this finding requires further investigation, it locates TSC downstream and upstream of Akt. It also helps to explain the benign nature of tuberous sclerosis syndromes, because in TSC-deficient cells, in addition to the S6K1-PI3K-Akt feedback that inhibits Akt, its activity would be further diminished by reduced activation mTORC2. In addition, S6K1 was shown to regulate mTORC2 activity by direct phosphorylation of the mTORC2 component Rictor. This phosphorylation event does not regulate mTORC2 kinase activity in vitro nor alters mTORC2 effects on most of its targets in cell-based experiments, but it negatively regulates phosphorylation of Akt at serine 473 [25]. These two new regulatory loops point towards a cellular mechanism that prevents simultaneous activation of mTORC1 and mTORC2, in addition to the already described role of S6K1 in putting a brake on excessive PI3K activation.
p53 and mTORC1
The tumor suppressor p53 is activated by different types of cellular stress, including DNA damage and oncogene activation. p53 mediates transactivation of a large number of genes involved in cell-cycle arrest and apoptosis. p53 also transactivates negative regulators of mTORC1, namely TSC2 and AMPKβ1 [26]. Besides, AMPK phosphorylates p53 at the serine in position 15, which leads to stabilization and favors the transactivation capacity of p53 [27]. Hence, p53 and AMPK positively regulate each other. Moreover, p53 upregulates the transcription of Sestrins 1 and 2, which bind to the ternary complex TSC1/2-AMPK, inducing phosphorylation and activation of TSC2 by AMPK [28]. In addition Pten was shown to interact with p53, leading to p53 stabilization [29]. Interestingly, Akt is a negative regulator of p53 activity by phosphorylating an E3 ubiquitin ligase, namely MDM2, which drives its translocation to the nucleus where it destabilizes p53 [30]. These cross regulations of AMPK, Pten and p53 oppose Akt activity (see Figure 2C) and constitute a network coordination of division and growth after sensing nutrient availability, DNA integrity and oncogenic activity.
The impact of mTOR feedback loops in cancer
Rapamycin-based therapy
Extensive basic research with rapamycin showed high specificity towards mTORC1 inhibition. This has encouraged a number of clinical trials using this compound as an anticancer drug, but in fact, rapamycin and its first generation analogs (temsirolimus and everolimus) have proven modest success in clinical trials, reflecting an incomplete understanding of mTOR functions. These small molecules are allosteric inhibitors of mTOR that block S6K1 phosphorylation, but it has become evident that not all mTORC1 functions are blocked by rapamycin [31-35]. Rapamycin also stops the negative feedback loops emanating from S6K1 to PI3K signaling pathway, thus activating proliferative and prosurvival effectors, as Akt. This was shown to be relevant in many cancer cell lines and also in clinical samples [36], and may explain why rapamycin, although slowing proliferation in many cell lines, is a poor inducer of apoptosis. Altogether, this raises an important concern: if mTORC1 has rapamycin-resistant functions and rapamycin indirectly activates the PI3K signaling pathway, which is responsible for mTORC1 activation, then rapamycin could cause hyperactivation of those mTORC1-dependent functions not inhibited by rapamycin.
Akt is not the only target downstream PI3K activation but there is in vivo evidence that point towards Akt as perhaps the most important target affected by the feedback loop from S6K1, and contributing to the modest therapeutic success of rapamycin. TSC2-heterozygous mice have constitutive mTORC1 activity and, consequently, overactivation of the feedback loop that dampens Akt activity. Interestingly, double heterozygous mice for TSC2 and Pten have active Akt and accelerated tumorigenesis in some organs in comparison with single heterozygous mice [37,38]. Hay and colleagues [39] have shown that mTORC1 function downstream of PI3K-Akt is necessary for transformation and tumorigenesis in an experimental model of mammary and salivary tumors, favoring the hypothesis that rapamycin may be a good therapeutic strategy under conditions of Akt hyperactivation. However, Akt may drive tumorigenesis by regulating effectors different from mTORC1. In fact, Akt can regulate the Foxo proteins, cyclin D1, p27 and GSK3, all involved in cell cycle progression, and MDM2, caspase-9, IKKα and Bad, controlling the execution of apoptosis [30]. Only in those tumors where mTORC1 is the important downstream effector of the oncogenic activity of Akt, rapamycin would be a reasonable therapeutic strategy.
PI3K also has Akt-independent effectors, with potential clinical relevance, that would also be indirectly activated by rapamycin. For instance, high levels of PDK1 can sustain tumor growth in an Akt-independent manner, by hyperactivation of SGK3 [40]. SGK3 is part of the AGC family of proteins, that also includes Akt, and that share target specificity. Interestingly, SGK proteins are, as Akt, substrates of mTORC2 [41].
Although rapamycin is a highly selective mTORC1 inhibitor, this has been challenged by the fact that it can also block mTORC2 activity in a subset of cancer cell lines after prolonged treatment through a mechanism that may involve inhibition of mTORC2 assembly [42]. Moreover, high concentration of rapamycin was also shown to block mTORC2 activity [43]. However, whether such toxic high levels of rapamycin have a clinical applicability is unclear.
mTOR catalytic inhibitors
The development of a different class of mTOR inhibitors that blocks mTOR catalytic site (Torin [34], PP242 and PP30 [32], Ku-0063794 [33], and WAY-600, WYE-687 and WYE-354 [35]) was reported almost simultaneously by several groups. This approach presents an obvious advantage for therapeutics: by blocking mTOR, both mTORC1 and mTORC2 become inhibited. Consequently, all mTOR target activities are blocked, including Akt phosphorylation at position 473 [32-35]. The fact that mTORC2 is the kinase for Akt at Ser473 is now widely accepted and supported by genetic studies in mice, but it is important to determine to what extent the inhibition of this phosphorylation impairs Akt activity towards all of its targets, which may be differentially inhibited by abrogating Ser473 phosphorylation [9,44]. How phosphorylation at positions 308 and 473 cross-regulate Akt activity is particularly important in situations where S6K1 inhibition will boost PI3K activation (and, consequently, Akt phosphorylation at Thr308). Noteworthy, acute inhibition of mTORC2 by using these mTOR catalytic inhibitors suggests that phosphorylation at Ser473 is required for phosphorylation at Thr308 and, consequently, for Akt activation. Further research should address how these compensatory loops would affect tumor responsiveness.
Toxicity is a possible caveat to consider if all mTOR functions are inhibited. However, work in mice suggests that blocking mTORC2 could be tolerated in vivo. In a murine model of Pten deficiency-driven prostate tumorigenesis, deletion of Rictor (i.e. mTORC2) partially impairs prostate adenocarcinomas development, but does not affect normal prostate gland function and architecture [45]. This indicates that mTORC2-specific inhibitors would be a good therapeutic strategy to be further investigated. In a similar experimental approach, Pandolfi and colleagues also showed that mTORC2 enhances prostate tumorigenesis, but ablation of mTOR in adult prostate does not affect normal prostate function [46], providing strong evidence that mTOR catalytic inhibitors could be well-tolerated in post-mitotic cells. Supporting this notion, WYE-354 inhibited human cancer cells growing as xenografts in nude mice with no detectable toxicity in the short term [35].
Dual PI3K-mTOR inhibitors
An alternative way to circumvent the activation of feedback loops that may preclude tumor responsiveness is the combined inhibition of PI3K together with mTOR. There are several small molecules that block PI3K and mTORC1 and 2, currently under intense investigation (reviewed in [47]). In cell lines and experimental tumors with hyperactive PI3K signaling, dual inhibition of PI3K and mTOR was effective [48], but failed in the presence of K-Ras hyperactivation. In Ras-driven tumorigenesis, concomitant inhibition of PI3K and mTOR, and also blocking downstream mediators of Ras seems to be required [49-51]. Whether such a combinatorial signaling perturbation will be toxic also for normal cells is a reasonable caveat to be considered.
Conclusions
In the last years, substantial progression in the understanding of how mTOR signaling pathway occurs has taught us that there are still significant aspects of mTOR regulation that need further clarification. The mechanism underlying the induction of cell death versus cell-cycle arrest in different cell lines treated with mTOR inhibitors, the existence of additional feedback loops triggered by S6K1 and how these loops affect therapeutic responses should be addressed in the future. Moreover, a clearer picture of the relevance of Akt-dependent versus -independent signaling of PI3K, how do phosphorylation of Akt at Thr308 and at Ser473 influence each other and Akt activity is also needed. Gaining insight in these issues will certainly settle the bases for better therapeutic strategies against cancer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References with notes
- 1.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 2.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 3.Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, Tempst P, Sabatini DM. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 4.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 5.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. (") In this report, Deptor, a negative regulator of mTORC1 and 2, is identified. mTOR and Deptor inhibit each other, and interestingly, Deptor overexpression indirectly leads to Akt hyperactivation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 8.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16:1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 10.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 11.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 12.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. (") Comprehensive review of mTORC1 function, particularly in the regulation of translational control. [DOI] [PubMed] [Google Scholar]
- 14.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–945. doi: 10.1038/ncb1753. ("") [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. ("") References 14 and 15 identify the Rag proteins as the direct link of amino acid-dependent activation of mTORC1. In addition, in reference 14 the authors also show that lack of amino acid sensing confers a negative fitness to Drosophila melanogaster in vivo. Reference 15 also demonstrates direct binding of Rag protein to mTORC1 and Rag-dependent mTORC1 shuttling as a novel regulatory mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartley D, Cooper GM. Role of mTOR in the degradation of IRS-1: regulation of PP2A activity. J Cell Biochem. 2002;85:304–314. doi: 10.1002/jcb.10135. [DOI] [PubMed] [Google Scholar]
- 18.Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000;14:783–794. doi: 10.1210/mend.14.6.0446. [DOI] [PubMed] [Google Scholar]
- 19.Takano A, Usui I, Haruta T, Kawahara J, Uno T, Iwata M, Kobayashi M. Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol Cell Biol. 2001;21:5050–5062. doi: 10.1128/MCB.21.15.5050-5062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, Griffin JD, Kwiatkowski DJ. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–738. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28:4104–4115. doi: 10.1128/MCB.00289-08. (") A regulatory role of TSC in mTORC2 activity is shown here. The authors describe that TSC positively regulates mTORC2 activity, hence, also indirectly activating Akt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009 doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 27.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 28.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–130. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 30.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. (") [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, Alessi DR. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009;421:29–42. doi: 10.1042/BJ20090489. (") [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. (") [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009;69:6232–6240. doi: 10.1158/0008-5472.CAN-09-0299. (") This reference, together with 32, 33 and 34, describe the generation of clinically relevant small molecules that inhibit mTOR catalytic site. Reference 35 also shows an antitumoral effect of mTOR inhibitors in vivo, with apparently no toxicity in short-term assays. [DOI] [PubMed] [Google Scholar]
- 36.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma L, Teruya-Feldstein J, Behrendt N, Chen Z, Noda T, Hino O, Cordon-Cardo C, Pandolfi PP. Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev. 2005;19:1779–1786. doi: 10.1101/gad.1314405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19:1773–1778. doi: 10.1101/gad.1314605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, Hahn-Windgassen A, Kiyokawa H, Hay N. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10:269–280. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 40.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. (") Herein, support for the relevance of Akt-independent signaling downstream of PI3K in tumor cell lines and clinical samples is reported. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 42.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 43.Shor B, Zhang WG, Toral-Barza L, Lucas J, Abraham RT, Gibbons JJ, Yu K. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res. 2008;68:2934–2943. doi: 10.1158/0008-5472.CAN-07-6487. [DOI] [PubMed] [Google Scholar]
- 44.Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell. 2006;11:583–589. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 45.Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H, Sabatini DM. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–159. doi: 10.1016/j.ccr.2008.12.017. (") In this paper, ablation of mTORC2 in mice, although impacting on tumor development, shows no obvious alterations in normal prostate gland, suggesting therapeutic mTORC2 inhibition could be well-tolerated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nardella C, Carracedo A, Alimonti A, Hobbs RM, Clohessy JG, Chen Z, Egia A, Fornari A, Fiorentino M, Loda M, et al. Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Sci Signal. 2009;2:ra2. doi: 10.1126/scisignal.2000189. (") This paper provides evidence for lack of toxicity by mTOR inhibition in normal adult cells in vivo, encouraging therapies based on mTOR inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 48.Brachmann S, Fritsch C, Maira SM, Garcia-Echeverria C. PI3K and mTOR inhibitors: a new generation of targeted anticancer agents. Curr Opin Cell Biol. 2009;21:194–198. doi: 10.1016/j.ceb.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 49.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ihle NT, Lemos R, Jr, Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL, Powis G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69:143–150. doi: 10.1158/0008-5472.CAN-07-6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Torbett NE, Luna-Moran A, Knight ZA, Houk A, Moasser M, Weiss W, Shokat KM, Stokoe D. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J. 2008;415:97–110. doi: 10.1042/BJ20080639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 54.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 56.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 57.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 58.Orloff MS, Eng C. Genetic and phenotypic heterogeneity in the PTEN hamartoma tumour syndrome. Oncogene. 2008;27:5387–5397. doi: 10.1038/onc.2008.237. [DOI] [PubMed] [Google Scholar]