

Abstract
The role of changes in the extracellular potassium concentration [K+]o in epilepsy has remained unclear. Historically, it was hypothesized that [K+]o is the causal factor for epileptic seizures. This so-called potassium accumulation hypothesis led to substantial debate but subsequently failed to find wide acceptance. However, recent studies on the pathophysiology of tissue from epileptic human patients and animal epilepsy models revealed aberrations in [K+]o regulation. Computational models of cortical circuits that include ion concentration dynamics have catalyzed a renewed interest in the role of [K+]o in epilepsy. The authors here connect classical and more recent insights on [K+]o dynamics in the cortex with the goal of providing starting points for a next generation of [K+]o research. Such research may ultimately lead to an entirely new class of antiepileptic drugs that act on the [K+]o regulation system.
Keywords: Extracellular potassium concentration, Epilepsy, Neocortex, Hippocampus, Glia, Astrocytes, Dynamics, Computational model, Tonic-clonic seizures
Gradients between intracellular and extracellular ion concentrations are the basis for electrical signaling in the nervous system by means of transmembrane ion currents (Hille 2001; Somjen 2002). Because ion currents reflect net ion flux across the cell membrane, these concentration gradients would rapidly degrade in the absence of mechanisms to maintain intra- and extracellular ion concentrations. Although the central nervous system is endowed with powerful ion concentration homeostasis mechanisms, ion concentrations do not assume constant values in the living brain. Rather, they change over time in an activity-dependent manner and therefore are dynamic variables. Aberrant ion concentration homeostasis has been linked to a variety of severe neurological conditions that include epilepsy, stroke, hypoxic encephalopathy, and migraines (Hille 2001; Somjen 2002).
As even minor fluctuations result in measurable changes in the K+ equilibrium potential and therefore K+ currents (Box 1), we focus on extracellular potassium concentration [K+]o. Because K+ currents play an essential role in controlling neuronal excitability, it was initially hypothesized that [K+]o elevations are the cause of epileptiform activity (Green 1964; Fertziger and Ranck 1970). Measurements of [K+]o with K+ sensitive microelectrodes (KSMs, see Box 2) indeed showed increases in [K+]o of several millimolars during experimental seizures. These recordings, however, did not meet criteria for a causal role of [K+]o in seizure initiation and termination because [K+]o increases were delayed relative to the seizure onset and [K+]o did not reach levels that depolarized neurons sufficiently to prevent them from firing action potentials (see below; also, e.g., Somjen 1979). As a consequence, interest in [K+]o dynamics and their role in epilepsy waned.
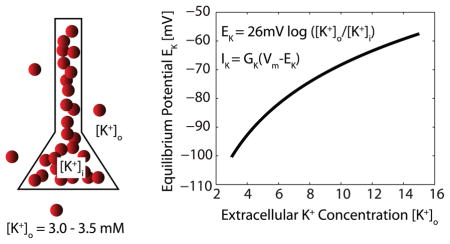
Box 1. Extracellular Potassium Concentration [K+]o.
Intracellular potassium concentration [K+]i is high (ca. 130 mM) in comparison to the extracellular concentration [K+]o, which is typically around 3 mM in the cortex. This concentration gradient defines the equilibrium potential EK (Nernst equation, top) for all potassium currents IK (bottom equation, GK is the conductance, Vm is the membrane potential). Changes in [K+]i have limited effect on the equilibrium potential (not shown), whereas changes in [K+]o can substantially depolarize the K+ reversal potential (right panel).
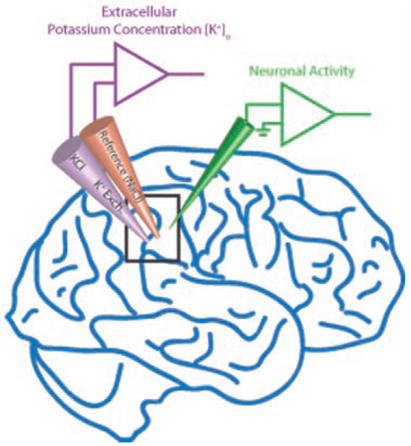
Box 2. Measuring [K+]o.
Typically, [K+]o is measured with potassium-ion selective microelectrodes (KSMs) (Walker 1971; Vyskocil and Kriz 1972; Neher and Lux 1973), often in combination with measurements of neural activity (e.g., with an extracellular recording electrode). KSMs are usually double-barreled glass electrodes. One barrel is filled with a column of potassium-selective ion exchanger and backfilled with KCl. The other barrel is filled with NaCl. The K+-dependent potential is determined by differential amplification of the signals from the two barrels. Half-max rise-time constants were measured to be smaller than 20 msec for a K+ source 10 μm away from the KSM (Lux and Neher 1973). The tip of the KSM creates an unnatural deadspace in neural tissue, and therefore the measured [K+]o values represent underestimates of the true values that would occur in the unperturbed case. Also, typically used K+ ion exchangers are sensitive to various neurotransmitters even in very low concentrations (Kuramoto and Haber 1981). Recently, K+-selective fluorescent probes have been developed and applied to measure [K+]o dynamics during experimental spreading depression (Padmawar and others 2005). Optical imaging represents an exciting new opportunity for relatively noninvasive measurements of [K+]o signals.
Recently, however, an increasing number of studies on the pathophysiology of tissue from both animal epilepsy models and human epileptic patients have strongly implicated impairment of [K+]o homeostasis apparatus in a variety of epilepsies with different etiologies. These more recent results thus are in apparent conflict with the previous conclusion that denied [K+]o a significant role in cortical seizures. Although there are many different explanations for these discrepancies, we argue here that the interaction between [K+]o and neural activity is a subtle one that is crucial in understanding dynamics. Computational models of cortical circuits that include ion concentration dynamics have provided novel insights in the complex interaction between neural activity and [K+]o.
We structured the remainder of this review as follows. First, we briefly highlight some of the classical findings on [K+]o in the cortex. We then review recent experimental and computational modeling findings on the role of [K+]o dynamics in epilepsy. The scope of this article is purposefully limited to hippocampal and neocortical networks because [K+]o dynamics in other preparations appear sufficiently distinct to deserve separate consideration. We conclude by proposing an integrated research approach to further clarify the role of [K+]o dynamics in epilepsy.
[K+]o Measurements in Vivo
Initial studies on [K+]o were mostly performed in the anesthetized in vivo preparation (Lux and Neher 1973; Prince and others 1973; Moody and others 1974), where [K+]o increased in the cortex in response to physiological stimuli (e.g., bars of light, see Fig. 1A). These findings indicated that [K+]o is not a parameter with a fixed value but rather a dynamic variable (Singer and Lux 1975; Connors and others 1979). Substantial [K+]o fluctuations were also found in the cat suprasylvian cortex during slow oscillations under ketamine-xylazine anesthesia (Fig. 1B). Few [K+]o recordings in the waking animal are reported in the literature. One group found surprisingly strong [K+]o transients in response to behaviorally relevant stimuli (Skinner and Molnar 1983). The interpretation of these results, however, is hampered by the fact that KSMs are sensitive to even very low concentrations of neurotransmitters and neuromodulators (Kuramoto and Haber 1981). Neurochemical changes in the awake animal in response to the stimuli used in that study (e.g., presentation of food or foot shocks) may have tainted the [K+]o recordings. During electrically or pharmacologically induced paroxysmal activity, [K+]o changed more substantially but never rose above a “ceiling value” of about 12 mM in the absence of spreading depression in the adult animal (Heinemann and Lux 1977).
Fig. 1.

In vivo recordings of [K+]o. A, Simultaneous measurement of [K+]o and single unit activity in response to visual stimulation with a moving bar in the anesthetized cat primary visual cortex. Arrows indicate directions of stimulus. 1 mV corresponds to approximately 0.17 mM (Kofuji and Newman 2004). Stimuli with orientations different from the preferred orientation elicited neither spiking nor increases in [K+]o (not shown). Figure adapted with permission from Singer and Lux (1975) © Elsevier. B, Simultaneous [K+]o and depth EEG recording during slow oscillation in ketamine-xylazine anesthetized cat in suprasylvian gyrus. Figure adapted with permission from Amzica and Steriade (2000) © Society for Neuroscience. C, Simultaneous [K+]o and field recording during tonic-clonic seizure in response to electrical stimulation in cat pericruciate cortex. Figure adapted with permission from Sypert and Ward (1974) © Elsevier.
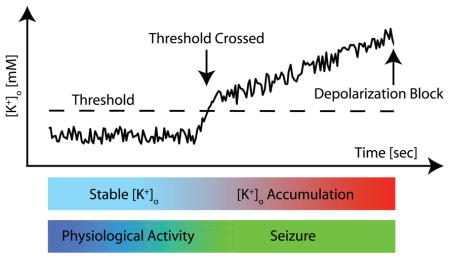
According to the so-called potassium accumulation hypothesis (Green 1964; Fertziger and Ranck 1970), an initial [K+]o increase above a certain threshold triggers a positive feedback cycle that mutually boosts [K+]o and neural activity until [K+]o reaches a value for which neurons are too depolarized to fire (Box 3). However, the initial predictions derived from this potassium accumulation hypothesis eluded experimental verification. In fact, little evidence was found for the expected 1) [K+]o threshold for seizure initiation (but see Sypert and Ward 1974), 2) monotonic increase in [K+]o during seizures, and 3) depolarization block of neurons at seizure cessation (Moody and others 1974; Sypert and Ward 1974; Heinemann and Lux 1977). Rather, dynamic changes in [K+]o appeared to be delayed in comparison to changes in neural activity. This delayed rise in [K+]o was interpreted as evidence that increased [K+]o is a result and not a cause of cortical seizures. Also, [K+]o increased during tonic firing phases and decreased during clonic bursting phases of the electrographic seizure (Moody and others 1974; Sypert and Ward 1974), as shown in Figure 1C. Recently, the interpretation of these findings and the rejection of [K+]o as an important factor in seizure generation have been reconsidered in the light of methodological concerns and novel insights from computational models (Somjen 2004; Frohlich and others 2007).
Box 3. Potassium Accumulation Hypothesis.
According to the potassium accumulation hypothesis, [K+]o fluctuates around a stable baseline (typically 3 mM) during physiological activity levels. A transient increase triggers the occurrence of a seizure during which [K+]o further accumulates. As a result, neurons become even more depolarized, fire more action potentials, and release even more K+ ions into the extracellular space. Eventually, these run-away dynamics (positive feedback) come to an end when the neurons are so depolarized that they can no longer spike due to sodium channel inactivation. At this point, the seizure terminates. Although the potassium accumulation hypothesis had originally been rejected, more recent computational studies of ion concentrations during seizures have provided more refined models that are partially based on such positive feedback dynamics. Also see text.
Ionic Models of Epilepsy in Vitro
With the advent of the in vitro preparation, brain slices perfused with artificial cerebrospinal fluid (ACSF) mimicking the ionic composition as measured in vivo during seizures have become an important model system for the study of hypersynchronous activation (“ionic models”). Although observations of in vitro “interictal” and “ictal” activity in ionic models do not represent a proof for a causal role of ionic disbalance in epileptogenesis, they show that changes of the extracellular ionic microenvironment are sufficient for network hyperactivation. Specifically, elevation of K+ concentration in the ACSF to 7.5 or 8.5 mM was sufficient to trigger both periodic network activation (“interictal spikes”) and in some cases events resembling electrographic seizures with a “tonic” firing and “clonic” bursting phase in the hippocampus (Korn and others 1987; Traynelis and Dingledine 1988; Jensen and Yaari 1997). Furthermore, seizure-like events occurred in an all-or-none fashion depending on the degree of [K+]o increase evoked by extracellular electrical stimulation or focal potassium injection in a high [K+]o and low [Ca2+]o model (Konnerth and others 1986; Yaari and others 1986). These findings contrast with the presumed absence of a [K+]o threshold for seizure initiation in vivo. Also, brief [K+]o transients of 0.2 to 2.0 mM by focal K+ injection were sufficient to trigger fast network oscillations that lasted several seconds (LeBeau and others 2002).
Together, these ionic models illustrate that elevated [K+]o is clearly sufficient to trigger synchronized oscillatory activity at various frequencies in the hippocampal networks in vitro. Nevertheless, it has remained mostly unclear how the observed epileptiform dynamics correspond to the in vivo situation. This limitation is of heightened concern when the activity-dependent changes of ion concentrations are studied, because it is unknown how the presence of a practically infinite K+ source/sink in the form of the perfused ACSF affects the ion concentration dynamics. Clearly, ion concentrations are not tightly controlled by the perfusion, as activity-dependent [K+]o fluctuations are routinely measured in vitro. Therefore, the interpretation of the above-described findings as absolute levels of ionic concentrations required for initiation of epileptiform activity is difficult to justify in their direct application to the in vivo situation. Although elevated [K+]o and decreased [Ca2+]o are most certainly useful models of the ionic microenvironment during epileptic seizures, manipulations such as the omission of magnesium in the ACSF or the pharmacological blockade of inhibition are more difficult to interpret in terms of their applicability in vivo.
Effects of [K+]o on Neural Activity
The slice preparation provides a relatively controlled environment to study the dependence of intrinsic and synaptic properties on [K+]o. Although we are far from having a complete picture of how the parameters of neurons and synapses depend on [K+]o, it is well established that a [K+]o increase in the range observed in vivo during electrographic seizures depolarizes neurons, decreases input resistance, and—importantly—can activate latent intrinsic bursting mechanisms in cortical pyramidal neurons. The changes in intrinsic firing properties have been best studied in the hippocampus where pyramidal cells in both CA1 and CA3 (Jensen and others 1994; Frohlich and Bazhenov 2006) but not stratum oriens inhibitory interneurons (McBain 1994) can burst in the presence of elevated [K+]o. Further regional and cell-type specific variations can be expected but have not been explored. Elevated [K+]o also affects action potential propagation (Hablitz and Lundervold 1981; Poolos and others 1987; Meeks and Mennerick 2004). For example, activity-dependent increases in [K+]o from postsynaptic cell firing modulate fiber recruitment and action potential propagation in the presynaptic Schaffer collaterals in the hippocampus CA1 (Poolos and others 1987). This effect was first described in the cerebellum (Malenka and others 1981; Kocsis and others 1983). Interestingly, elevated [K+]o affects action potential propagation and thus transmitter release differently in glutamatergic and GABAeric axons in the hippocampus (Meeks and Mennerick 2004).
Increases in [K+]o also directly affect synaptic inhibition. The equilibrium potential of GABA(A)-type receptors, EGABA(A), shifts toward less negative values in elevated [K+]o (Thompson and Gahwiler 1989; Jensen and others 1993) due to the reduced electrochemical driving force for the potassium-chloride co-transporter KCC2 that extrudes chloride from the cytoplasm at low levels of [K+]o (DeFazio and others 2000; Payne and others 2003). Consequently, EGABA(A) may move beyond the membrane potential such that cannel activation produces depolarization instead of the characteristic hyperpolarization (e.g., Fujiwara-Tsukamoto and others 2007). In the case of low intracellular chloride concentration [Cl−]i and elevated [K+]o, KCC2 switches transport direction and aids [K+]o homeostasis by transporting K+ ions back into cells at the price of intracellular Cl− accumulation (Payne 1997; Jarolimek and others 1999; Staley and Proctor 1999; DeFazio and others 2000). In addition to the KCC2-mediated change in equilibrium potential, depolarization of postsynaptic cells by elevated [K+]o increased the GABAergic conductance due to inward rectification of the GABA(A) receptor channels (Jensen and others 1993).
Because KCC2 provides a key link between [K+]o and fast synaptic inhibition, it is particularly interesting that deficient KCC2 expression has been related to different types of cortical hyperactivity. For example, depolarizing GABA(A) currents were found in human epileptic tissue from the subiculum (Cohen and others 2002) and associated with reduced or absent KCC2 expression in subicular pyramidal cells (Palma and others 2006; Huberfeld and others 2007; Munoz and others 2007). Also, impaired KCC2-dependent Cl− extrusion ability was found in epileptic tissue in a model of injury-induced epileptogenesis (Jin and others 2005). Sustained activity in hippocampal slices down-regulated KCC2 expression level by endogenous brain-derived neurotrophic factor action on tyrosine receptor kinase B (TrkB) (Rivera and others 2004). Increased seizure propensity in the neonatal brain may be caused by age-related differences in chloride homeostasis, specifically increased NKCC1 and decreased KCC2 expression levels in comparison to the adult (Dzhala and others 2005). As a result, the activation of GABA(A) receptor channels excites neurons in the developing brain due to elevated [Cl−]i and thus less negative EGABA(A) (Ben-Ari 2002). Nevertheless, the exact role of GABAergic inhibition in seizure-like activity in experimental model systems of neonatal seizures is complex and not fully understood (e.g., Dzhala and Staley 2003; Isaev and others 2005; Isaev and others 2007). In fact, a better understanding of the interaction between [K+]o- and [Cl−]i-mediated dynamics may be key to a better understanding of (neonatal) epileptiform activity.
Regulation of [K+]o
Mechanisms that contribute to [K+]o homeostasis under physiological conditions have been recently reviewed in detail (Kofuji and Newman 2004). Although the relative individual contributions are not fully known for the cortex, it is clear that transporters (Na+/K+ ATPase, KCC2, and NKCC) on both neurons and astrocytes (Kofuji and Newman 2004), passive uptake through inward-rectifying potassium (Kir) channels on astrocytes (Butt and Kalsi 2006), and diffusion in the extracellular space (Lux and Neher 1973; Fisher and others 1976; Nicholson and others 2000) contribute to extracellular potassium homeostasis. The question whether [K+]o is regulated locally (K+ uptake) or whether K+ is moved to sites of low [K+]o by K+ currents through the glial syncytium (spatial buffering) is one of considerable debate and may depend on brain region (Kofuji and Newman 2004). Simultaneous dual glial recordings combined with KSM measurements suggest the presence of spatial buffering in the cortex in vivo during slow sleep oscillations and electrographic paroxysmal activity (Amzica and others 2002). Such nonlocal K+ transport by glial cells may contribute to the spatial propagation of synchronized neural activity (Steriade 2003). The dissection of [K+]o homeostasis into its individual components has been hampered by the fact that activity levels are usually not controlled for, by the lack of specificity of the applied pharmacology, and by the technical pitfalls concerning selectivity of KSMs.
Despite the experimental difficulties listed above, alterations of the potassium homeostasis apparatus represent an appealing hypothesis for explaining the pathophysiology of epilepsy given the role of [K+]o in regulating excitability (Pollen and Trachtenberg 1970). Although early measurements in artificially induced glial scarring remained inconclusive (Heinemann and Dietzel 1984), there is now accumulating evidence for glial dysfunction in epileptic tissue from patients with temporal lobe epilepsies (Binder and Steinhauser 2006). Density and inward rectification of potassium current through glial Kir channel are reduced in patients with temporal lobe epilepsy accompanied by Ammon’s horn sclerosis (Hinterkeuser and others 2000; Schroder and others 2000), as shown in Figure 2A. This finding is in agreement with previous, less direct studies that showed that barium, a Kir channel antagonist, had a reduced effect on [K+]o dynamics in slices from pilocarpine-treated rats and epileptic patients with sclerosis (Gabriel and others 1998; Kivi and others 2000; Jauch and others 2002).
Fig. 2.

Aberration in [K+]o regulation. A, Current densities as a function of resting potential for astrocytes from tissue with Ammon’s horn sclerosis (AHS) and lesion-associated temporal lobe epilepsy (lesion). Reduced densities for AHS group (left). The ratio of current at hyperpolarized and depolarized holding potentials was reduced for the AHS group (reduced inward rectification). Figure adapted from Hinterkeuser and others (2000) © Wiley Blackwell Publishing. B, Reduced barium-sensitive potassium conductance in astrocytes from a mouse model of tuberous sclerosis. Recordings at age before behavioral seizures occur. Figure adapted from Jansen and others (2005) © Wiley Blackwell Publishing. C, Reduced barium-sensitivity of K+-clearance in albumin-exposed cortex that serves as a model for blood-brain barrier disruption. K+ applied by ionophoresis in the presence of pharmacological blocker for Na+-dependent action potentials and fast synaptic transmission. Figure adapted from Ivens and others (2007) distributed under the Creative Commons License. D, [K+]o in response to orthodromic stimulation in hippocampal slice from α-syntrophin-null mice that exhibited aquaporin-4 mislocalization. Slower [K+]o clearance in comparison to wild type. ACSF = artificial cerebrospinal fluid. Figure adapted with permission from Amiry-Moghaddam and others (2003) © National Academy of Sciences, USA.
The Tsc1GFAPCKO mouse model of tuberous sclerosis complex (TSC), a genetic disorder associated with multiple seizure types, was found to exhibit reduced Kir channel expression and accordingly decreased Kir current amplitudes in astrocytes (Jansen and others 2005), as shown in Figure 2B. Decreased glial Kir channel expression and reduced potassium buffering capacity were also found in an animal model of blood-brain barrier disruption (Ivens and others 2007), as illustrated in Figure 2C. A link between altered [K+]o homeostasis mediated by changes in Kir channels on astrocytes and posttraumatic epilepsy has also been suggested (D’Ambrosio and others 1999), although this study has been questioned by some on methodological grounds (Santhakumar and others 2003). Na+/K+ ATPase also contributes to [K+]o clearance. Ouabain application hindered clearance of stimulation-induced [K+]o increases in the olfactory cortex (Ballanyi and others 1987) and hippocampus (D’Ambrosio and others 2002). Although the exact roles of Na+/K+ ATPase with neuronal and glial location have not yet been fully determined, differential affinity for K+ suggests that mostly glial Na+/K+-ATPase is responsible for clearance of activity-dependent changes in [K+]o. In samples from human epileptic patients, overall activity of Na+/K+-ATPase was reduced and K+ sensitivity of glial Na+/K+-ATPase was lost (Grisar and others 1992). Loss of function mutations of the ATP1A2 gene that codes for the α 2 subunit of the Na+/K+ ATPase were found in families with familial hemiplegic migraine and benign familial infantile convulsions (Vanmolkot and others 2003). Benign familial neonatal convulsions are a potassium channelopathy characterized by (partial) loss-of-function mutations in the KCNQ2 and KCNQ3 genes (Bate and Gardiner 1999; Singh and others 2003) that make up the M-type potassium current (Wang and others 1998). Retigabine, a drug that activates KCNQ/Kv7 potassium channels, shows great promise as a novel antiseizure drug (Porter and others 2007).
Recent evidence suggests that the glial water channel aquaporin-4, AQP4 (Amiry-Moghaddam and Ottersen 2003), may also play a role in [K+]o homeostasis. Clearance of elevated [K+]o after orthodromic stimulation was slower and hyperthermia-induced epileptic seizures were of higher intensity in α-syntrophin knockout mice that exhibited disrupted AQP4 localization (Amiry-Moghaddam and others 2003; see also Fig. 2D). Similarly, prolonged seizure duration and extended [K+]o transients during seizures (Padmawar and others 2005) and spreading depression (Padmawar and others 2005; Binder and others 2006) were observed in AQP4−/− knockout mice. Measurements of changes in extracellular volume fraction in neocortical slices by intrinsic imaging combined with KSM measurements (Niermann and others 2001) further support the link between water transport and [K+]o clearance.
In summary, there is an increasing number of studies suggesting an important link between aberrant [K+]o regulation and epileptogenesis in a broad variety of human epilepsies and animal models. Although it is unclear whether the observed alterations are a cause or an effect of epileptiform activity, there is little doubt that the further study of [K+]o may be key to a better understanding of epileptogenesis.
Understanding Potassium Dynamics
The original consideration of [K+]o as a key element in epileptogenesis was mostly based on the question of whether increases in [K+]o are a cause or an effect of epileptiform activity. This conceptual framework seems ill posed, as neural activity and [K+]o are intimately linked through complex feedback loops. Unfortunately, however, feedback dynamics are notoriously hard to study experimentally. Computational models, however, can provide new insights into the dynamics of feedback interactions. We review some of the main modeling results to illustrate the power of computational models in studying the role of [K+]o dynamics in epileptic seizures.
Historically, computational models mostly served the study of [K+]o dynamics by focusing on mechanisms of extracellular K+ clearance. These initial models did not consider the effect of activity-dependent [K+]o increases on neural activity (but see Whisler and Johnston 1978 for a pioneering exception) and therefore avoided the complexity of feedback dynamics (e.g., Vern and others 1977; Gardner-Medwin 1983; Odette and Newman 1988; Dietzel and others 1989). Although the conclusions in these studies were not uniform, these models contributed to the important insight that mechanisms different from diffusion must also be involved in [K+]o regulation (see discussion above). Nevertheless, the exact role of K+diffusion is not yet fully clear. For example, in vitro experiments suggest that K+ diffusion can synchronize otherwise unconnected neural populations (e.g., Lian and others 2001). Models of neuronal coupling via K+ transients in the absence of synaptic transmission (Lebovitz 1996; Park and Durand 2006) support the relevance of extracellular spatial structure. Compartmentalization of the extracellular space and inhomogeneity of potassium channel localization may require more detailed modeling of microenvironments with explicit consideration of electrodiffusion (Qian and Sejnowski 1990).
More recent models include the feedback between neural activity and [K+]o (Kager and others 2000; Kager and others 2002; Bazhenov and others 2004; Frohlich and Bazhenov 2006; Frohlich and others 2006). In these models, individual neurons are endowed with ion channels described by the commonly used conductance-based formalism (Hodgkin and Huxley 1952). This modeling approach allows the detailed quantification of K+ ions entering the extracellular space via ion channels. Additionally, each cell is surrounded by an extracellular compartment that includes a [K+]o regulation apparatus (Na+/K+-ATPase and glial buffer). Ion concentrations are dynamically updated and the corresponding equilibrium potentials computed. The first single-cell models with ion concentration dynamics (Kager and others 2000; Kager and others 2002) included detailed neuronal morphology but a limited number of ion-channel types (reviewed in Somjen 2002). In response to stimulation, these model neurons exhibited sustained after discharges (bursting) in the case of weakened Na+/K+-ATPase capacity. The authors focused on different recovery time-constants of [K+]o and [Na+]i at the time scale of individual burst as the underlying mechanism of these self-sustained “clonic” discharges because the model did not include ion channels mediating intrinsic bursting. In the case of enhanced inward currents in the dendrites, self-sustained prolonged depolarization (“spreading-depression-like”) occurred after a critical [K+]o level (“ignition point”) was reached. The key insight from this model was that the interaction dynamics between ion concentrations and neural activity can lead to self-sustained pathological neural activation even in the case of an isolated cell (in this case, clonic bursting and spreading-depression). From a dynamic system viewpoint, the bursting represents a stable oscillatory state because it lasted infinitely. The spreading-depression-like episode, however, is the result of an unstable positive feedback loop, similar in nature to the original [K+]o accumulation hypothesis. The main limitations of these models are 1) the absence of a more realistic ion channel composition and 2) the lack of network interaction.
These points were addressed by the incorporation of [K+]o regulation mechanisms in standard models of cortical pyramidal cells, PYs, and fast-spiking inhibitory interneurons, INs (Bazhenov and others 2004). In comparison to the models discussed above, these PY and IN models were of simplified morphology (Fig. 3) but had a more comprehensive set of ion channels (Bazhenov and others 2004; Frohlich and Bazhenov 2006) such that intrinsic bursting occurred for elevated [K+]o in agreement with the experimental literature. Specifically, persistent sodium and high-threshold calcium ion channels were critical for the depolarization at the burst onset. This depolarization then caused a burst of action potentials before Ca2+-activated potassium current was sufficiently activated to mediate burst termination (Bazhenov and others 2004; Frohlich and Bazhenov 2006). In this model, prolonged intense stimulation caused [K+]o to increase. After termination of stimulation, a single PY exhibited a transient after-discharge that was structured into two distinct consecutive phases, that is, bursting and tonic firing (Bazhenov and others 2004; Frohlich and others 2006). Yet, in contrast to the single-cell model, a network of PYs and INs exhibited slow state transitions between bursting and tonic firing (Fig. 4A–C) that qualitatively resembled sequences of tonic-clonic discharges during seizures (Frohlich and others 2006). [K+]o increased during tonic firing and decreased during bursting in agreement with the classic in vivo recordings (Moody and others 1974; Sypert and Ward 1974) and more recent in vitro ionic models of tonic-clonic seizures (Jensen and Yaari 1997). Different firing rates and thus different loads on the [K+]o regulation apparatus for these two firing modes explains the different signs of the[K+]o gradient.
Fig. 3.

Schematic representation of computational models of neurons with surrounding extracellular space. The complex morphology of neuronal structure and extracellular space (A) was reduced to a simplified geometrical representation (B). In this model, neurons consisted of two connected compartments (axo-somatic and dendritic compartments). Each neuron was enclosed with a surrounding volume that represented the extracellular space and for which the K+ concentration was computed for determining the K+ equilibrium potential.
Fig. 4.

Slow state transitions in cortical network model. A, The activity of five pyramidal cells (PYs) is structured into alternating epochs of tonic firing and bursting. B, [K+]o increases during tonic firing (positive feedback) but decreases during bursting (negative feedback). C, Sample PY membrane voltage trace. Panels A-C adapted with permission from Frohlich and others (2006) © Society for Neuroscience. D, Open-loop analysis shows bistability between tonic firing and bursting for [K+]o between 5.0 and 5.4 mM (left). This bistability with hysteresis explains the slow state transitions in the closed-loop system (right). PY = pyramidal cells.
The identification and eventual abstraction of dynamic principles of epileptic seizures carries the promise that the broad range of clinical manifestations associated with seizures can eventually be reduced to a few key pathophysiological mechanisms. The differing time scales of action-potential firing and changes in [K+]o(neglecting small amplitude transients following individual action potentials) provide the means to study [K+]o dynamics in computational models by opening the feedback loop (so-called open-loop dynamics, see Box 4). In practical terms, the behavior of the neuron is determined as a function of [K+]o that is treated as a constant parameter (Hahn and Durand 2001; Frohlich and Bazhenov 2006; Frohlich and others 2006). Application of this open-loop analysis (also called bifurcation theory) on the above-discussed single-cell PY model revealed 1) the existence of four distinct activity patterns as a function of [K+]o, that is, silence, tonic firing, bursting, and depolarization block, and 2) a bistability with hysteresis between tonic firing and bursting for elevated [K+]o levels (Frohlich and Bazhenov 2006; Frohlich and others 2006).
Box 4. Understanding [K+]o Feedback Dynamics.
In computational models, feedback interaction between [K+]o and neural activity can be studied by treating [K+]o as a parameter. With this “open-loop” method, the modulation of [K+]o by neural activity is artificially removed and therefore the feedback removed (top). By choosing different values for [K+]o, the corresponding activity patterns for each value of [K+]o can be determined (“open-loop” dynamics). The behavior of the entire system with feedback (“closed-loop” dynamics) is then predicted by stringing together the activity patterns as determined by open-loop analysis (bottom). This approximation is valid because 1) the macroscopic time-course of [K+]o is slow in comparison to the time scale of action potential firing and 2) most effects of changes in [K+]o on neural dynamics are instantaneous.
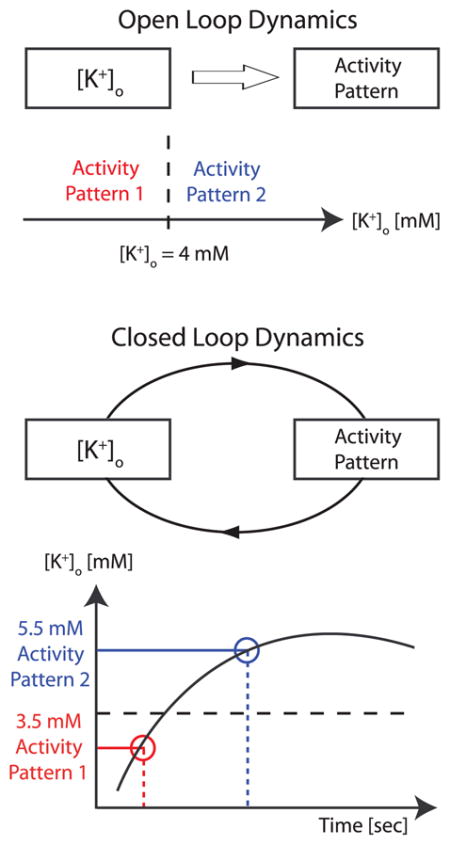
This bistability found by open-loop analysis of the models explains the occurrence of transitions between tonic firing and bursting (Fig. 4D). Specifically, neurons remained in tonic-firing mode while [K+]o increased up to the level where they were forced to switch to bursting mode (upper endpoint of hysteresis). Conversely, neurons remained in bursting mode while [K+]o decreased until the lower end-point of the hysteresis was reached where they were forced to switch back to tonic-firing mode and the next cycle began. In other words, although it was originally assumed that only positive feedback between [K+]o and neural activity occurred during seizures, these modeling results show the alternating occurrence of positive (tonic firing) and negative (bursting) feedback. Thus, these slow transitions were essentially the result of slow alternations between two meta-stable states, tonic firing and bursting. The existence of this bistability and therefore the occurrence of slow state transitions was robust to changes in model parameters but depended on the high-threshold Ca2+conductance. Thus, the relatively detailed nature of the model permitted novel insights into the possible involvement of specific ion channel types in mediating tonic-clonic seizure dynamics. Also of note is the fact that even if a different mechanism were to provide such a bistability with hysteresis between tonic firing and burst mode, we would expect the same slow patterning of the epileptiform activity.
The different responses of the single cell and the network to a potassium transient (transient after-discharge versus sustained tonic-clonic sequences) showed that the nature of the paroxysmal-like activity crucially depended on the network interaction. This finding further emphasizes the importance of network simulations to investigate the role of [K+]o in cortical dynamics. The model predicts in vivo neural dynamics during tonic-clonic seizures that have previously lacked an explanation. Also, the [K+]o time-course is in qualitative agreement with the original [K+]o recordings in vivo (Moody and others 1974; Sypert and Ward 1974). Slow state transitions as discovered in the network model resemble electrographic neocortical seizures that are patterned into epochs of “fast runs” (tonic discharge) and “slow bursting” (Frohlich and others 2006).
Ways Forward
Aberrant [K+]o regulation has recently been reconsidered as an integral part of epileptogenesis. The accumulating evidence for disturbances in [K+]o regulation discussed here warrants renewed research efforts to unravel the role of [K+]o dynamics in physiological brain activity and epileptogenesis. We therefore conclude by outlining an integrated research approach based on recent methodological developments that should render it possible to overcome the limitations of previous generations of [K+]o studies. Specifically, we propose the use of relatively non-invasive optical techniques to control and measure activity of neural populations combined with fluorometric and KSM-based [K+]o measurements to dissect the interaction between neural activity and [K+]o. In conjunction, advanced patch-clamp recording techniques combined with genetic labeling can be used to obtain more specific information about how [K+]o modulates synaptic and intrinsic properties of individual cell types. Below, we outline how in vivo, in vitro, and computational modeling methods can be combined to tackle the challenge of understanding the role of [K+]o in cortical dynamics.
Although [K+]o time-courses during cortical seizures in the anesthetized preparation have been thoroughly documented in the early in vivo studies, we know very little about [K+]o dynamics in the awake and naturally sleeping animal. For example, active cortical states and synchronized sleep oscillations may be associated with significant fluctuations in [K+]o. Crucially, [K+]o recordings in vivo need to be accompanied by simultaneous quantification of neural activity ideally with single unit resolution (Sykova and others 1974; Heinemann and Lux 1975). Such quantification of physiological [K+]o fluctuations will provide an important baseline for the eventual design of therapeutic interventions that enhance the power of the [K+]o regulation apparatus. In as much as the uncoupling of the feedback loop between [K+]o and neural activity will remain a challenge in vivo, the in vitro preparation remains the model system of choice to study the effects of [K+ ]o levels on intrinsic and synaptic properties. So far, most studies of that kind in cortical preparations have focused on the effect of strong [K+]o elevations on hippocampal fields CA3 and CA1. Because substantial [K+]o fluctuations were also found in the neocortex in vivo, it is essential to understand the resulting modulation of neural behavior independent of whether epileptiform activity can be elicited by elevated [K+]o in the neocortical slice. Moreover, because most studies have been restricted to studying intrinsic excitability and synaptic transmission by extracting average pre- and post-synaptic behavior from field recordings, we know very little about how different cell types are affected by changes in [K+]o. Therefore, patch-clamp recordings from dendrites, somata, and axon terminals of identified cells and synaptically coupled neuron pairs will provide crucial new insight into specific effects of different levels of [K+]o.
Recent computational models of neuronal networks with incorporated ion concentration dynamics were successful at providing new insights and predictions concerning cortical seizure dynamics. Therefore, we propose that such models can serve as the essential and so far neglected link between in vitro results on specific properties and in vivo results on global network dynamics. In fact, the complexity of [K+]o dynamics is best tackled with tools derived from systems theory and computational neuroscience that have begun seeing widespread application in many other subfields of neuroscience. We therefore hope that this combined approach of experiments and computational models will eventually lead to the development of a new generation of antiepileptic drugs that specifically target the [K+]o regulation system and therefore might be free from the current limitations of pharmacotherapy for epilepsy (Duncan and others 2006).
Acknowledgments
The authors thank Jay Coggan, Justin Elstrott, and Luke Wylie for helpful comments and discussions. F. F. acknowledges Anita Fröhlich for excellent assistance and support. This work was supported by a grant from NIDCD (5R01DC006306) to M. B.
References
- Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4(12):991–1001. doi: 10.1038/nrn1252. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, et al. Delayed K+clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003;100(23):13615–20. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amzica F, Massimini M, Manfridi A. Spatial buffering during slow and paroxysmal sleep oscillations in cortical networks of glial cells in vivo. J Neurosci. 2002;22(3):1042–53. doi: 10.1523/JNEUROSCI.22-03-01042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amzica F, Steriade M. Neuronal and glial membrane potentials during sleep and paroxysmal oscillations in the neocortex. J Neurosci. 2000;20(17):6648–65. doi: 10.1523/JNEUROSCI.20-17-06648.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballanyi K, Grafe P, ten Bruggencate G. Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol. 1987;382:159–74. doi: 10.1113/jphysiol.1987.sp016361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate L, Gardiner M. Molecular genetics of human epilepsies. Expert Rev Mol Med. 1999;1999:1–22. doi: 10.1017/S1462399499001349. [DOI] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Potassium model for slow (2–3 Hz) in vivo neocortical paroxysmal oscillations. J Neurophysiol. 2004;92(2):1116–32. doi: 10.1152/jn.00529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3(9):728–39. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Binder DK, Steinhauser C. Functional changes in astroglial cells in epilepsy. Glia. 2006;54(5):358–68. doi: 10.1002/glia.20394. [DOI] [PubMed] [Google Scholar]
- Binder DK, Yao X, Verkman AS, Manley GT. Increased seizure duration in mice lacking aquaporin-4 water channels. Acta Neurochir Suppl. 2006;96:389–92. doi: 10.1007/3-211-30714-1_80. [DOI] [PubMed] [Google Scholar]
- Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10(1):33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298(5597):1418–21. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Connors B, Dray A, Fox P, Hilmy M, Somjen G. LSD’s effect on neuron populations in visual cortex gauged by transient responses of extracellular potassium evoked by optical stimuli. Neurosci Lett. 1979;13(2):147–50. doi: 10.1016/0304-3940(79)90032-6. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K(+) in rat hippocampus. J Neurophysiol. 2002;87(1):87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. Impaired K(+) homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J Neurosci. 1999;19(18):8152–62. doi: 10.1523/JNEUROSCI.19-18-08152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFazio RA, Keros S, Quick MW, Hablitz JJ. Potassium-coupled chloride cotransport controls intracellular chloride in rat neo-cortical pyramidal neurons. J Neurosci. 2000;20(21):8069–76. doi: 10.1523/JNEUROSCI.20-21-08069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Lux HD. Relations between slow extracellular potential changes, glial potassium buffering, and electrolyte and cellular volume changes during neuronal hyperactivity in cat brain. Glia. 1989;2(1):25–44. doi: 10.1002/glia.440020104. [DOI] [PubMed] [Google Scholar]
- Duncan JS, Sander JW, Sisodiya SM, Walker MC. Adult epilepsy. Lancet. 2006;367(9516):1087–100. doi: 10.1016/S0140-6736(06)68477-8. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Staley KJ. Excitatory actions of endogenously released GABA contribute to initiation of ictal epileptiform activity in the developing hippocampus. J Neurosci. 2003;23(5):1840–6. doi: 10.1523/JNEUROSCI.23-05-01840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11(11):1205–13. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Fertziger AP, Ranck JB., Jr Potassium accumulation in interstitial space during epileptiform seizures. Exp Neurol. 1970;26(3):571–85. doi: 10.1016/0014-4886(70)90150-0. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Pedley TA, Prince DA. Kinetics of potassium movement in norman cortex. Brain Res. 1976;101(2):223–37. doi: 10.1016/0006-8993(76)90265-1. [DOI] [PubMed] [Google Scholar]
- Frohlich F, Bazhenov M. Coexistence of tonic firing and bursting in cortical neurons. Phys Rev E Stat Nonlin Soft Matter Phys. 2006;74(3 Pt 1):031922. doi: 10.1103/PhysRevE.74.031922. [DOI] [PubMed] [Google Scholar]
- Frohlich F, Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Slow state transitions of sustained neural oscillations by activity-dependent modulation of intrinsic excitability. J Neurosci. 2006;26(23):6153–62. doi: 10.1523/JNEUROSCI.5509-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich F, Timofeev I, Sejnowski T, Bazhenov M. Extracellular potassium dynamics and epileptogenesis. In: Soltesz I, Staley KJ, editors. Computational neuroscience in epilepsy. San Diego: Elsevier; 2007. pp. 419–39. [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Imanishi M, Fukai T, Takada M. Distinct types of ionic modulation of GABA actions in pyramidal cells and interneurons during electrical induction of hippocampal seizure-like network activity. Eur J Neurosci. 2007;25(9):2713–25. doi: 10.1111/j.1460-9568.2007.05543.x. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Eilers A, Kivi A, Kovacs R, Schulze K, Lehmann TN, et al. Effects of barium on stimulus induced changes in extracellular potassium concentration in area CA1 of hippocampal slices from normal and pilocarpine-treated epileptic rats. Neurosci Lett. 1998;242(1):9–12. doi: 10.1016/s0304-3940(98)00012-3. [DOI] [PubMed] [Google Scholar]
- Gardner-Medwin AR. Analysis of potassium dynamics in mammalian brain tissue. J Physiol. 1983;335:393–426. doi: 10.1113/jphysiol.1983.sp014541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JD. The hippocampus. Physiol Rev. 1964;44:561–608. doi: 10.1152/physrev.1964.44.4.561. [DOI] [PubMed] [Google Scholar]
- Grisar T, Guillaume D, Delgado-Escueta AV. Contribution of Na+, K(+)-ATPase to focal epilepsy: a brief review. Epilepsy Res. 1992;12(2):141–9. doi: 10.1016/0920-1211(92)90034-q. [DOI] [PubMed] [Google Scholar]
- Hablitz JJ, Lundervold A. Hippocampal excitability and changes in extracellular potassium. Exp Neurol. 1981;71(2):410–20. doi: 10.1016/0014-4886(81)90099-6. [DOI] [PubMed] [Google Scholar]
- Hahn PJ, Durand DM. Bistability dynamics in simulations of neural activity in high-extracellular-potassium conditions. J Comput Neurosci. 2001;11(1):5–18. doi: 10.1023/a:1011250329341. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Dietzel I. Extracellular potassium concentration in chronic alumina cream foci of cats. J Neurophysiol. 1984;52(3):421–34. doi: 10.1152/jn.1984.52.3.421. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD. Undershoots following stimulus-induced rises of extracellular potassium concentration in cerebral cortex of cat. Brain Res. 1975;93(1):63–76. doi: 10.1016/0006-8993(75)90286-3. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD. Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res. 1977;120(2):231–49. doi: 10.1016/0006-8993(77)90903-9. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion channels of excitable membranes. Sunderland (MA): Sinauer; 2001. [Google Scholar]
- Hinterkeuser S, Schroder W, Hager G, Seifert G, Blumcke I, Elger CE, et al. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. 2000;12(6):2087–96. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117(4):500–44. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, et al. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27(37):9866–73. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaev D, Isaeva E, Khazipov R, Holmes GL. Anticonvulsant action of GABA in the high potassium-low magnesium model of ictogenesis in the neonatal rat hippocampus in vivo and in vitro. J Neurophysiol. 2005;94(4):2987–92. doi: 10.1152/jn.00138.2005. [DOI] [PubMed] [Google Scholar]
- Isaev D, Isaeva E, Khazipov R, Holmes GL. Shunting and hyper-polarizing GABAergic inhibition in the high-potassium model of ictogenesis in the developing rat hippocampus. Hippocampus. 2007;17(3):210–9. doi: 10.1002/hipo.20259. [DOI] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, et al. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130(Pt 2):535–47. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- Jansen LA, Uhlmann EJ, Crino PB, Gutmann DH, Wong M. Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia. 2005;46(12):1871–80. doi: 10.1111/j.1528-1167.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- Jarolimek W, Lewen A, Misgeld U. A furosemide-sensitive K+-Cl- cotransporter counteracts intracellular Cl- accumulation and depletion in cultured rat midbrain neurons. J Neurosci. 1999;19(12):4695–704. doi: 10.1523/JNEUROSCI.19-12-04695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauch R, Windmuller O, Lehmann TN, Heinemann U, Gabriel S. Effects of barium, furosemide, ouabaine and 4,4′-diisothio-cyanatostilbene-2,2′-disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potassium concentration in hippocampal slices from rats and from patients with epilepsy. Brain Res. 2002;925(1):18–27. doi: 10.1016/s0006-8993(01)03254-1. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Azouz R, Yaari Y. Variant firing patterns in rat hippocampal pyramidal cells modulated by extracellular potassium. J Neurophysiol. 1994;71(3):831–9. doi: 10.1152/jn.1994.71.3.831. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Cherubini E, Yaari Y. Opponent effects of potassium on GABAA-mediated postsynaptic inhibition in the rat hippocampus. J Neurophysiol. 1993;69(3):764–71. doi: 10.1152/jn.1993.69.3.764. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Yaari Y. Role of intrinsic burst firing, potassium accumulation, and electrical coupling in the elevated potassium model of hippocampal epilepsy. J Neurophysiol. 1997;77(3):1224–33. doi: 10.1152/jn.1997.77.3.1224. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl- extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93(4):2117–26. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Kager H, Wadman WJ, Somjen GG. Simulated seizures and spreading depression in a neuron model incorporating interstitial space and ion concentrations. J Neurophysiol. 2000;84(1):495–512. doi: 10.1152/jn.2000.84.1.495. [DOI] [PubMed] [Google Scholar]
- Kager H, Wadman WJ, Somjen GG. Conditions for the triggering of spreading depression studied with computer simulations. J Neurophysiol. 2002;88(5):2700–12. doi: 10.1152/jn.00237.2002. [DOI] [PubMed] [Google Scholar]
- Kivi A, Lehmann TN, Kovacs R, Eilers A, Jauch R, Meencke HJ, et al. Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci. 2000;12(6):2039–48. doi: 10.1046/j.1460-9568.2000.00103.x. [DOI] [PubMed] [Google Scholar]
- Kocsis JD, Malenka RC, Waxman SG. Effects of extracellular potassium concentration on the excitability of the parallel fibres of the rat cerebellum. J Physiol. 1983;334:225–44. doi: 10.1113/jphysiol.1983.sp014491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129(4):1045–56. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Heinemann U, Yaari Y. Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. I. Development of seizurelike activity in low extracellular calcium. J Neurophysiol. 1986;56(2):409–23. doi: 10.1152/jn.1986.56.2.409. [DOI] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R. Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol. 1987;57(1):325–40. doi: 10.1152/jn.1987.57.1.325. [DOI] [PubMed] [Google Scholar]
- Kuramoto T, Haber B. The K+liquid ion exchange electrode system: responses to drugs and neurotransmitters. J Neurosci Res. 1981;6(1):37–48. doi: 10.1002/jnr.490060105. [DOI] [PubMed] [Google Scholar]
- LeBeau FE, Towers SK, Traub RD, Whittington MA, Buhl EH. Fast network oscillations induced by potassium transients in the rat hippocampus in vitro. J Physiol. 2002;542(Pt 1):167–79. doi: 10.1113/jphysiol.2002.015933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM. Quantitative examination of dynamic interneuronal coupling via single-spike extracellular potassium ion transients. J Theor Biol. 1996;180(1):11–25. doi: 10.1006/jtbi.1996.0074. [DOI] [PubMed] [Google Scholar]
- Lian J, Bikson M, Shuai J, Durand DM. Propagation of non-synaptic epileptiform activity across a lesion in rat hippocampal slices. J Physiol. 2001;537(Pt 1):191–9. doi: 10.1111/j.1469-7793.2001.0191k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux HD, Neher E. The equilibration time course of (K +) 0 in cat cortex. Exp Brain Res. 1973;17(2):190–205. doi: 10.1007/BF00235028. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kocsis JD, Ransom BR, Waxman SG. Modulation of parallel fiber excitability by postsynaptically mediated changes in extracellular potassium. Science. 1981;214(4518):339–41. doi: 10.1126/science.7280695. [DOI] [PubMed] [Google Scholar]
- McBain CJ. Hippocampal inhibitory neuron activity in the elevated potassium model of epilepsy. J Neurophysiol. 1994;72(6):2853–63. doi: 10.1152/jn.1994.72.6.2853. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Selective effects of potassium elevations on glutamate signaling and action potential conduction in hippocampus. J Neurosci. 2004;24(1):197–206. doi: 10.1523/JNEUROSCI.4845-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody WJ, Futamachi KJ, Prince DA. Extracellular potassium activity during epileptogenesis. Exp Neurol. 1974;42(2):248–63. doi: 10.1016/0014-4886(74)90023-5. [DOI] [PubMed] [Google Scholar]
- Munoz A, Mendez P, DeFelipe J, Alvarez-Leefmans FJ. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia. 2007;48(4):663–73. doi: 10.1111/j.1528-1167.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- Neher E, Lux HD. Rapid changes of potassium concentration at the outer surface of exposed single neurons during membrane current flow. J Gen Physiol. 1973;61(3):385–99. doi: 10.1085/jgp.61.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Chen KC, Hrabetova S, Tao L. Diffusion of molecules in brain extracellular space: theory and experiment. Prog Brain Res. 2000;125:129–54. doi: 10.1016/S0079-6123(00)25007-3. [DOI] [PubMed] [Google Scholar]
- Niermann H, Amiry-Moghaddam M, Holthoff K, Witte OW, Ottersen OP. A novel role of vasopressin in the brain: modulation of activity-dependent water flux in the neocortex. J Neurosci. 2001;21(9):3045–51. doi: 10.1523/JNEUROSCI.21-09-03045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odette LL, Newman EA. Model of potassium dynamics in the central nervous system. Glia. 1988;1(3):198–210. doi: 10.1002/glia.440010305. [DOI] [PubMed] [Google Scholar]
- Padmawar P, Yao X, Bloch O, Manley GT, Verkman AS. K+waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nat Methods. 2005;2(11):825–7. doi: 10.1038/nmeth801. [DOI] [PubMed] [Google Scholar]
- Palma E, Amici M, Sobrero F, Spinelli G, Di Angelantonio S, Ragozzino D, et al. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci U S A. 2006;103(22):8465–8. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EH, Durand DM. Role of potassium lateral diffusion in non-synaptic epilepsy: a computational study. J Theor Biol. 2006;238(3):666–82. doi: 10.1016/j.jtbi.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol. 1997;273(5 Pt 1):C1516–25. doi: 10.1152/ajpcell.1997.273.5.C1516. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26(4):199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Pollen DA, Trachtenberg MC. Neuroglia: gliosis and focal epilepsy. Science. 1970;167(922):1252–3. doi: 10.1126/science.167.3922.1252. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Mauk MD, Kocsis JD. Activity-evoked increases in extracellular potassium modulate presynaptic excitability in the CA1 region of the hippocampus. J Neurophysiol. 1987;58(2):404–16. doi: 10.1152/jn.1987.58.2.404. [DOI] [PubMed] [Google Scholar]
- Porter RJ, Nohria V, Rundfeldt C. Retigabine. Neurotherapeutics. 2007;4(1):149–54. doi: 10.1016/j.nurt.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince DA, Lux HD, Neher E. Measurement of extracellular potassium activity in cat cortex. Brain Res. 1973;50(2):489–95. doi: 10.1016/0006-8993(73)90758-0. [DOI] [PubMed] [Google Scholar]
- Qian N, Sejnowski TJ. When is an inhibitory synapse effective? Proc Natl Acad Sci U S A. 1990;87(20):8145–9. doi: 10.1073/pnas.87.20.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipila S, et al. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24(19):4683–91. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Voipio J, Kaila K, Soltesz I. Post-traumatic hyperexcitability is not caused by impaired buffering of extracellular potassium. J Neurosci. 2003;23(13):5865–76. doi: 10.1523/JNEUROSCI.23-13-05865.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder W, Hinterkeuser S, Seifert G, Schramm J, Jabs R, Wilkin GP, et al. Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia. 2000;41(Suppl 6):S181–4. doi: 10.1111/j.1528-1157.2000.tb01578.x. [DOI] [PubMed] [Google Scholar]
- Singer W, Lux HD. Extracellular potassium gradients and visual receptive fields in the cat striate cortex. Brain Res. 1975;96(2):378–83. doi: 10.1016/0006-8993(75)90751-9. [DOI] [PubMed] [Google Scholar]
- Singh NA, Westenskow P, Charlier C, Pappas C, Leslie J, Dillon J, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126(Pt 12):2726–37. doi: 10.1093/brain/awg286. [DOI] [PubMed] [Google Scholar]
- Skinner JE, Molnar M. Event-related extracellular potassium ion activity changes in frontal cortex of the conscious cat. J Neurophysiol. 1983;49(1):204–15. doi: 10.1152/jn.1983.49.1.204. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Extracellular potassium in the mammalian central nervous system. Annu Rev Physiol. 1979;41:159–77. doi: 10.1146/annurev.ph.41.030179.001111. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Ion regulation in the brain: implications for pathophysiology. Neuroscientist. 2002;8(3):254–67. doi: 10.1177/1073858402008003011. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Ions in the brain: normal function, seizures, and stroke. New York: Oxford University Press; 2004. [Google Scholar]
- Staley KJ, Proctor WR. Modulation of mammalian dendritic GABA(A) receptor function by the kinetics of Cl- and HCO3-transport. J Physiol. 1999;519(Pt 3):693–712. doi: 10.1111/j.1469-7793.1999.0693n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Neuronal substrates of sleep and epilepsy. Cambridge (UK): Cambridge University Press; 2003. [Google Scholar]
- Sykova E, Rothenberg S, Krekule I. Changes of extracellular potassium concentration during spontaneous activity in the mesencephalic reticular formation of the rat. Brain Res. 1974;79(2):333–7. doi: 10.1016/0006-8993(74)90428-4. [DOI] [PubMed] [Google Scholar]
- Sypert GW, Ward AA., Jr Changes in extracellular potassium activity during neocortical propagated seizures. Exp Neurol. 1974;45(1):19–41. doi: 10.1016/0014-4886(74)90097-1. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gahwiler BH. Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl- in hippocampal CA3 neurons. J Neurophysiol. 1989;61(3):512–23. doi: 10.1152/jn.1989.61.3.512. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59(1):259–76. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Vanmolkot KR, Kors EE, Hottenga JJ, Terwindt GM, Haan J, Hoefnagels WA, et al. Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol. 2003;54(3):360–6. doi: 10.1002/ana.10674. [DOI] [PubMed] [Google Scholar]
- Vern BA, Schuette WH, Thibault LE. [K+]o clearance in cortex: a new analytical model. J Neurophysiol. 1977;40(5):1015–23. doi: 10.1152/jn.1977.40.5.1015. [DOI] [PubMed] [Google Scholar]
- Vyskocil F, Kriz N. Modifications of single and double-barrel potassium specific microelectrodes for physiological experiments. Pflugers Arch. 1972;337(3):365–76. [PubMed] [Google Scholar]
- Walker JL. Ion specific liquid ion exchanger microelectrodes. Anal Chem. 1971;43(3):A89–93. [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282(5395):1890–3. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Whisler JW, Johnston D. Epileptogenesis: a model for the involvement of slow membrane events and extracellular potassium. J Theor Biol. 1978;75(3):271–8. doi: 10.1016/0022-5193(78)90334-x. [DOI] [PubMed] [Google Scholar]
- Yaari Y, Konnerth A, Heinemann U. Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. II. Role of extracellular potassium. J Neurophysiol. 1986;56(2):424–38. doi: 10.1152/jn.1986.56.2.424. [DOI] [PubMed] [Google Scholar]