

Abstract
LIM Mineralization Protein-1 (LMP-1) is an intracellular regulator of bone formation and has been shown to be osteoinductive in vitro and in vivo. The effect of LMP-1 on other aspects of bone homeostasis has not been previously studied. In a pilot study we observed that LMP-1 decreased nitric oxide (NO) production in pre-osteoclasts. Here we report a new anti-inflammatory effect of LMP-1 and define its mechanism of action in lipopolysaccharide (LPS)-stimulated RAW 264.7 pre-osteoclasts. We found that LMP-1 significantly inhibited LPS-induced NO production. LMP-1 also effectively inhibited the expression of inducible Nitric Oxide Synthase (iNOS), potently suppressed the transcriptional activity and nuclear translocation of nuclear factor kappa B (NF-κB), and prevented the phosphorylation of inhibitor of kappa B (IκB). Interestingly, LMP-1 had no effect on Receptor-Activator of Nuclear Factor B Ligand (RANKL)-induced activation of NF-κB. Furthermore, LMP-1 had no effect on the LPS-induced phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2), whereas it did attenuate the phosphorylation of c-Jun NH2-terminal kinase (JNK) while enhancing phosphorylation of p38 mitogen-activated protein kinases (p38 MAPK). These results suggest that LMP-1 has an anti-inflammatory effect, and this effect is, at least in part, due to the inhibition of NO production by the suppression of NF-κB activation and selective regulation of mitogen-activated protein kinase (MAPK) pathways.
Keywords: LIM Mineralization Protein-1 (LMP-1), nuclear factor kappa B (NF-κB), Mitogen-activated protein kinases (MAPK), Nitric Oxide (NO), Inflammatory Bone Loss
Introduction
LIM Mineralization Protein-1 (LMP-1) is an intracellular regulator of bone formation and exerts part of its activity by enhancing cellular responsiveness to bone morphogenetic proteins, the key inducers of osteoblast differentiation [1-6]. Although LMP-1 is expressed in many cell types its role in other aspects of bone homeostasis has not been investigated. In a preliminary experiment, we observed that LMP-1 dramatically inhibited Nitric Oxide (NO) production induced by Tumor Necrosis Factor-alpha (TNF-α) or Lipopolysaccharide (LPS) in macrophage/pre-osteoclast cells. This observation let us to hypothesize that LMP-1 might play a role in modulating the response of macrophages to inflammatory stimuli that are known to play a role in inflammatory bone loss. The production of NO by activated macrophages is dependent on the quantity of inducible Nitric Oxide Synthase (iNOS) regulated at the transcriptional level and is also regulated by the nuclear factor kappa B (NF-κB) pathway and mitogen-activated protein kinases (MAPKs)[7, 8]. Therefore, the purpose of this study was to examine the effects of LMP-1 on these key pathways involved with production of NO.
NF-κB is an important transcription factor complex that regulates the expression of many genes responsible for inflammatory bone loss [9-11]. NF-κB is functional as a homo- or hetero-dimer of Rel family proteins (RelA/p65, RelB, cRel, p50, p52). In unstimulated cells, NF-κB is constitutively localized in the cytosol by physical association with its inhibitory protein, inhibitor of kappa B (IκB) [12, 13]. Many stimuli, utilize different receptors and accessory proteins to activate NF-κB via various signaling cascades that all require phosphorylation of IκB [14]. Once IκB is phosphorylated, the NF-κB hetero-dimer is rapidly released from IκB and translocates to the nucleus, where it activates the transcription of target genes [12-14]. In turn, the pro-inflammatory products regulated by NF-κB, such as TNF-α, Interleukin-1beta (IL-1β) and NO can further activate the NF-κB pathway.
Mitogen activated protein kinases (MAPKs) are a group of serine/threonine protein kinases that respond to extracellular stimuli and play an important role in the control of cellular responses to cytokines and stresses [15, 16]. MAPKs have multiple cellular effects; of these phosphorylated c-Jun NH2-terminal kinase (JNK) has been reported to phosphorylate Activator Protein-1 (AP-1) and increase iNOS expression, which is relevant to our study. [7, 17]
To investigate the effects of LMP-1 on macrophage/pre-osteoclast cells we selected the commonly used model of RAW 264.7 macrophages stimulated with LPS, a potent activator of the Toll-Like Receptor 4 (TLR-4)/NF-κB signaling pathway [18-20]. LPS is a pathogen-derived molecule that causes inflammatory bone loss by activation of macrophages to produce pro-inflammatory cytokines such as Interleukin 1-beta (IL-1β), and TNF-α, chemokines, such as Interleukin-8 (IL-8) and Cytokine (C-X-C motif) Ligand 10 CXCL10, as well as NO [18, 21]. In this study we chose to examine the effect of LMP-1 on LPS-induced NO production, as a measure of its effect on NF-κB and MAPKs. The effects of osteoinductive factors such as LMP-1 have not been carefully studied in macrophage/pre-osteoclast cells. Based on our preliminary experiments, we hypothesize that LMP-1 may play a role in blocking the inflammatory response in bone by inhibiting activation of NF-κB and MAPKs.
Materials and methods
Preparation of recombinant TAT-LMP-1 and TAT-LMP-2 proteins
To determine whether the anti-inflammatory effect of LMP-1 requires the unique osteoinductive region, LMP-2 was used as a negative control for LMP-1 in this study. Recently, short peptide sequences, known as protein transduction domains (PTDs), have become increasingly prevalent as tools to internalize molecules that would otherwise remain extracellular [22]. The PTD from the HIV TAT protein has been widely used and characterized [23]. To facilitate cellular entry of LMP-1, we prepared a cDNA construct containing an N-terminal HIV-TAT derived cationic peptide tag. The TAT domain, when covalently linked, is reported to deliver protein cargo across cell membranes in all the studied cell types[23]. TAT-LMP-1 and TAT-LMP-2 were prepared according to our previously published protocol [24]. Briefly, the full-length cDNA of LMP-1 or LMP-2 was cloned into the TAT-HA vector. BL21 (DE3) competent cells (Novagen, Madison, WI) were transformed with plasmid by standard methods, and the correct clones were confirmed by restriction analysis followed by DNA sequencing. The recombinant LMP-1 and LMP-2 proteins were purified by size exclusion chromatography followed by metal affinity chromatography. Purity of the LMP-1 and LMP-2 proteins used in the current study was higher than 90% as determined by densitometric analysis of a scanned Coomassie stained gel; protein quantification was performed with assay reagent (Bio-Rad, Hercules, CA) using BSA as the standard.
Cell culture
RAW 264.7, a mouse macrophage-like cell line which can be induced to differentiate into osteoclasts, was obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were grown in Dulbecco's modified Eagle medium (DMEM, Gibco Life Technologies, Rockville, MD) with 10% non heat inactivated fetal bovine serum (Atlanta Biologicals, Norcross, GA), 100 U/ml penicillin, and 100 ug/ml streptomycin (Gibco Life Technologies, Rockville, MD) and incubated at 37 °C in 5% CO2 humidified air. For the MTT assay and reporter assay, cells were seeded at 1×104 cells/cm2; for other experiments, cells were seeded at 4×104 cells/cm2.
Preliminary experiments using Fluorescence Activated Cell Sorting (FACS) analysis of FITC-TAT-LMP-1 treated cells determined that greater than 90% of RAW 264.7 cells were transduced with the protein after 30 min treatment with doses used in these experiments. To determine whether reduced NO production could be explained by reduced iNOS gene transcription, the cells were incubated with TAT-LMP-1 (0.05, 0.1, 0.25 nM) or TAT-LMP-2 (0.25 nM) for 30 min prior to treatment with LPS (10 pmol/ml, gift of Dr.David Stephens) for 24 h. To assess the effect of LMP-1 on nuclear translocation of NF-κB, the cells were incubated with TAT-LMP-1 (0.05, 0.1, 0.25 nM) or TAT-LMP-2 (0.25 nM) for 30 min prior to treatment with LPS (10 pmol/ml) for 3 h. To identify the profile of LPS-induced phosphorylation of IκB protein, the cells were incubated with LPS (10 pmol/ml) for 3, 5, 7, 9, 11, 13, 15 and 30 min respectively. To determine the effect of TAT-LMP-1 on LPS-induced phosphorylation of IκB protein, the cells were incubated with TAT-LMP-1 (0.05, 0.1, 0.25 nM) or TAT-LMP-2 (0.25 nM) for 30 min prior to treatment with LPS (10 pmol/ml) for 9 min. To determine the effect of TAT-LMP-1 on LPS-induced phosphorylation of MAPKs, the cells were incubated with TAT-LMP-1 (0.05, 0.1, 0.25 nM) or TAT-LMP-2 (0.25 nM) or different specific inhibitors at the concentration of 10 μM (SB203580 inhibits p38; U0126 inhibits ERK1/2; and SP600125 inhibits JNK, Calbiochem, San Diego, CA) for 30 min prior to treatment with LPS (10 pmol/ml) for 30 min.
MTT assay
The effect of LMP-1 and LPS on cell viability was determined by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromid) assay. RAW 264.7 pre-osteoclasts (1×104 cells/cm2) were seeded into 96-well flat bottom plates in 100 μl culture medium and incubated overnight. The cells were then incubated with various concentrations of TAT-LMP-1 (0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, 10 nM) for 30 min. After the transduction, the cells were incubated with or without 10 pmol/ml of LPS for 4 or 24 h. After the treatment, 10 μl of the MTT reagent (final concentration 0.5 mg/ml) was added into each well and the plates were incubated for 4 h in a humidified atmosphere (37°C and 5% CO2). An aliquot (100 μl) of the solubilization solution (DMSO) was added to each well and plates were shaken for 30 min. The absorbance of the samples was measured at 570 nm with a reference wavelength at 690 nm using a microplate reader (TECAN, Salzburg, Austria).
Nitric oxide release quantified as nitrite accumulation
LPS is commonly used to study inflammatory responses in macrophages. In our preliminary study, LPS caused more robust secretion of NO from RAW 264.7 cells compared with the response to TNF-α and was, consequently, chosen as the inflammatory stimulus for these studies. To determine the inhibitory effect of TAT-LMP-1 on LPS-induced NO production, RAW 264.7 pre-osteoclasts seeded at 4×104 cells/cm2 were incubated with TAT-LMP-1 (0.05, 0.1, 0.25 nM) or TAT-LMP-2 (0.25 nM) for 30 min. After the transduction, the cells were treated with LPS (10 pmol/ml) for 24 h. The nitrite accumulation in the supernatant was assessed using the Griess reaction and reflected NO production [25]. Briefly, each 100 μl of culture supernatant was mixed with an equal volume of Griess reagent (0.1% N-(1- naphthyl)-ethylenediamine, 1% sulfanilamide in 5% phosphoric acid) and the absorbance was measured in an automated microplate reader (TECAN, Salzburg, Austria). A standard curve was produced using known concentrations of sodium nitrite. The amount of nitrite in the supernatants was determined according to the standard curve.
RNA isolation and real time reverse transcription-PCR
Total RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA) as described previously according to the manufacturer's protocol [26]. After isolation, 2 μg of total RNA was reverse transcribed in a 100 μl total volume containing 50 mM KCl, 10 mM Tris-HCl, pH 8.3, 5.5 mM MgCl2, 0.5 mM dNTPs, 0.125 μM random hexamer, 40 units RNase inhibitor, and 125 units MultiScribe (Applied Biosystems, Foster City, CA). In control samples the RNase inhibitor and MultiScribe were omitted. The samples were incubated for 10 min at 25 °C, 30 min at 48 °C, and then 5 min at 95 °C to inactivate the enzyme. Real time PCR was then performed on 5 μl of the resulting cDNA in a total volume of 25 μl containing 12.5 μl of 2X SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), and 0.8 μM each primer. Real Time Polymerase Chain Reaction (RT-PCR) was performed using the 7500 Real time PCR System from Applied Biosystems (Foster City, CA). RT-PCR conditions were as follows: 50 °C for 2 min, 95 °C for 10 min and 95 °C for 15 s, 62 °C for 1 min for 32 cycles. Data were normalized to endogenous 18S rRNA levels using the Applied Biosystems ΔΔCt Method. The iNOS and 18S rRNA primer set was designed and constructed by SuperArray Biosciences (Frederick, MD).
Western blot analysis
Cells were lysed to obtain total protein using Mammalian Protein Extraction Reagent (Pierce Biotechnology, Rockford, IL) or lysed to obtain nuclear protein using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford, IL) according to the manufacturer's protocol. Each sample (10 μg of protein) was mixed with NuPage loading buffer (Invitrogen, Carlsbad, CA) for a total volume of 20 μl and boiled for 5 min. The proteins were separated by electrophoresis under denaturing conditions on NuPage Bis-Tris Pre-Cast gels (Invitrogen, Carlsbad, CA) for 60 min at 200 Volts and transferred onto nitrocellulose membranes (Invitrogen, Carlsbad, CA) for 60 min at 30 Volts. After the transfer, the membranes were incubated in 25 ml of blocking buffer (5% nonfat dry milk in Tris buffered saline (TBS)) for 1h at room temperature. After blocking, membranes were washed three times for 5 min each in 15 ml of TBS with 0.1% Tween-20 (TBST). Washed membranes were incubated with different primary antibodies in TBST overnight at 4 °C as follows: anti-iNOS (1:1000), anti-p65 (1:1000), anti-phospho-IκB-α (1:1000), anti-phospho-p38 (1:1000), anti-phospho-ERK1/2 (1:2000), or anti-phospho-JNK (1:1000). The membranes were also incubated with anti-actin (1:5000), anti-LaminB (1:2000), or anti-IκB-α, anti-p38, anti-ERK1/2, anti-JNK (1:1000) to verify the equal loading of proteins in each lane. Anti-actin antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA); other antibodies were purchased from Cell Signaling Technology (Beverly, MA). After incubation with primary antibody, membranes were washed three times for 5 min each with 15 ml of TBST. Washed membranes were incubated with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies as indicated (1:2000, Cell Signaling Technology, Beverly, MA) in 10 ml of blocking buffer with gentle agitation for 1 h at room temperature. After incubation with secondary antibodies, membranes were washed three times for 5 min each with 15 ml of TBST. Washed membranes were incubated with 5 ml of SuperSignal West Pico Western blot substrate (Pierce Biotechnology, Rockford, IL) with gentle agitation for 4 min at room temperature. Membranes were drained of excess developing solution, wrapped in plastic wrap and exposed to x-ray films. The films were scanned and quantified using ImageJ software (National Institutes of Health Image, US).
Transient transfection and luciferase reporter assay
An iNOS promoter luciferase construct that contains the NF-κB binding element was used to determine the effect of LMP-1 on the iNOS promoter. Cells were plated at a density of 1×104 cells/cm2 and transfected for 4 h with a luciferase reporter construct plasmid mixture of 2.9 μ) iNOS promoter reporter and 0.1 μg pGL4.74 Renilla luciferase control vector. An NF-κB reporter construct containing the consensus NF-κB binding element was used to determine the effect of LMP-1 on activation of NF-κB. The cells were tranfected with a mixture of 1.5 μg NF-κB responsive luciferase reporter plasmid and Renilla luciferase control vector (SuperArray Bioscience Corporation, Frederick, MD) according to the manufacturer's instruction. Superfect tranfection reagent (QIAGEN, Valencia, CA) was used for the transfection. Empty pGL3-BASIC vector (Promega, Madison, WI) was used as the control. After 4 h the transfection reagent was removed and the cells were washed with cold PBS. Cells were then treated with TAT-LMP-1 (0.05, 0.1, 0.25 nM) or TAT-LMP-2 (0.25 nM) for 30 min, followed by LPS or RANKL stimulation for 24 h. Luciferase activity was measured after LPS treatment using the luciferase assay kit (Promega, Madison, WI) according to the manufacturer's instructions on a luminescence reader (Victor™X Light, PerkinElmer Life and Analytical Sciences, Waltham, MA). Data were normalized to Renilla luciferase activity and relative luciferase units were calculated as firefly luciferase activity/Renilla luciferase activity.
Statistical Analysis
Results are reported as the mean of determinations with error bars representing the standard error of the mean (SEM). In all experiments two kinds of comparisons were made. First, a comparison of the response from LPS-treated or untreated cells was made. Secondly, comparisons were made between LPS-induced cells not treated with TAT-LMP-1/TAT-LMP-2 and LPS-induced cells that were treated with various doses of TAT-LMP-1/TAT-LMP-2. Experimental results from Western blots are from one representative experiment. Densitometric and statistical analysis of Western blots was performed on all three replicates of each experiment. Statistical significance was calculated with a One-way analysis of variance (ANOVA) with Levene's homogeneity of variance test, and subsequently Bonferroni Post Hoc test or Dunnett's T3 Post Hoc test based on the comparison to be made and the statistical indication of each test. Analysis was performed using Statistical Products for Social Sciences version 13.0 (SPSS 13.0) for Windows (SPSS Inc, Chicago, IL). Statistical probability of P < 0.05 was considered significant.
Results
LPS and TAT-LMP-1 do not affect cell viability over the concentration range tested in RAW 264.7 macrophages/pre-osteoclasts
The cytotoxic effect of our working concentration of LPS (10 pmol/ml) and TAT-LMP-1 were assessed on RAW 264.7 macrophages/pre-osteoclasts. High doses of TAT-LMP-1 (2.5 and 5 nM) reduced cell viability (P = 0.000 ∼ 0.008). Neither low doses (0.05, 0.1, 0.25, 0.5, 1 nM) of TAT-LMP-1 alone nor low doses of TAT-LMP-1 plus LPS affected cell viability (P = 0.221 ∼ 1.000) (Fig.1). LPS at 10 pmol/ml and low doses of TAT-LMP-1 (0.05, 0.1, 0.25 nM) were used in the subsequent experiments.
Fig.1.
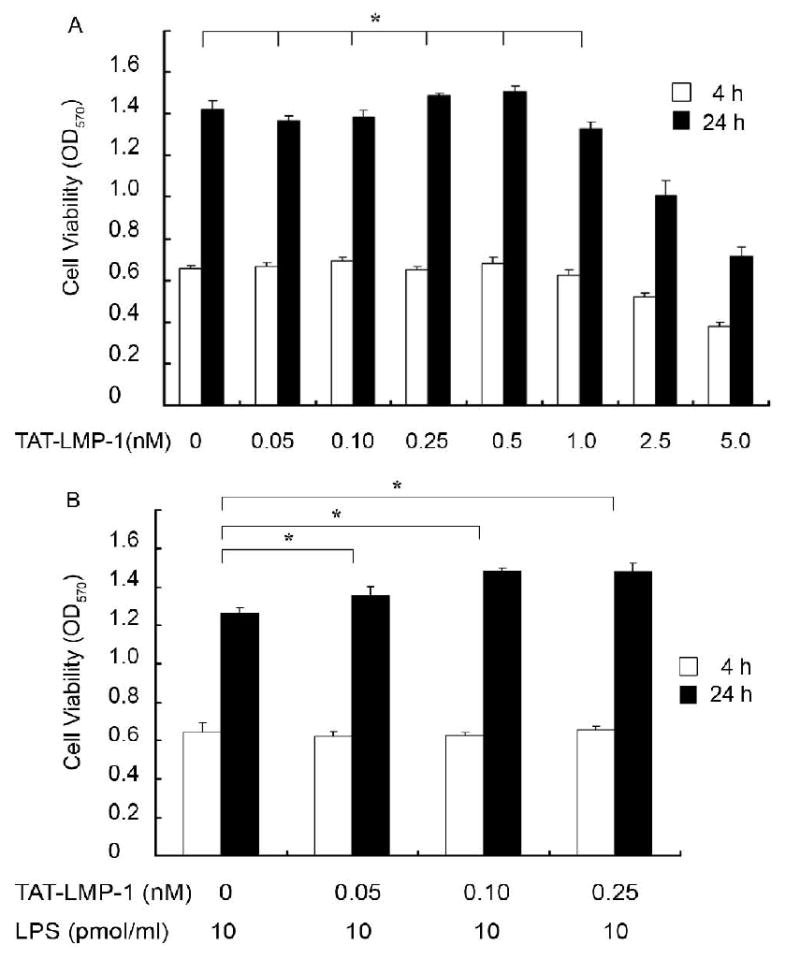
Evaluation of the cytotoxic effect of TAT-LMP-1 alone and LPS plus TAT-LMP-1 on RAW 264.7 macrophages/pre-osteoclasts. The MTT assay was used to assess cytotoxic effects 4 h and 24 h after TAT-LMP-1 or LPS treatment. (A) No significant difference in cell viability was observed in cultures treated with low doses of TAT-LMP-1 compared to the no treatment group. (B) No significant difference in cell viability was observed in cultures treated with LPS plus low doses of TAT-LMP-1 compared to the no treatment group. Values are mean ± SEM of one representative experiment out of three independent experiments performed in triplicate. (*P > 0.05)
LMP-1 inhibits NO production and iNOS gene expression in LPS-induced RAW 264.7 macrophages/pre-osteoclasts
A nitrite assay was used to assess the inhibitory effect of LMP-1 on NO production. The results showed that untreated cells produced 0.97 ± 0.50 μM nitrite. When LPS was added, NO production was dramatically increased to 30.25 ± 1.44 μM (P = 0.0001). Pretreatment with TAT-LMP-1 inhibited LPS-induced NO production in a concentration-dependent manner. NO levels were reduced to 18.14 ± 0.87 μM by 0.05 nM TAT-LMP-1 (P = 0.0001) and to 8.92±0.4 μM by 0.1 nM TAT-LMP-1 (P = 0.0001) after LPS stimulation. TAT-LMP-1 at 0.25 nM completely inhibited NO production to the basal level (P = 0.0001). TAT-LMP-2, a LMP family protein with no osteoinductive effect, did not inhibit LPS induced NO production (Fig.2A).
Fig.2.
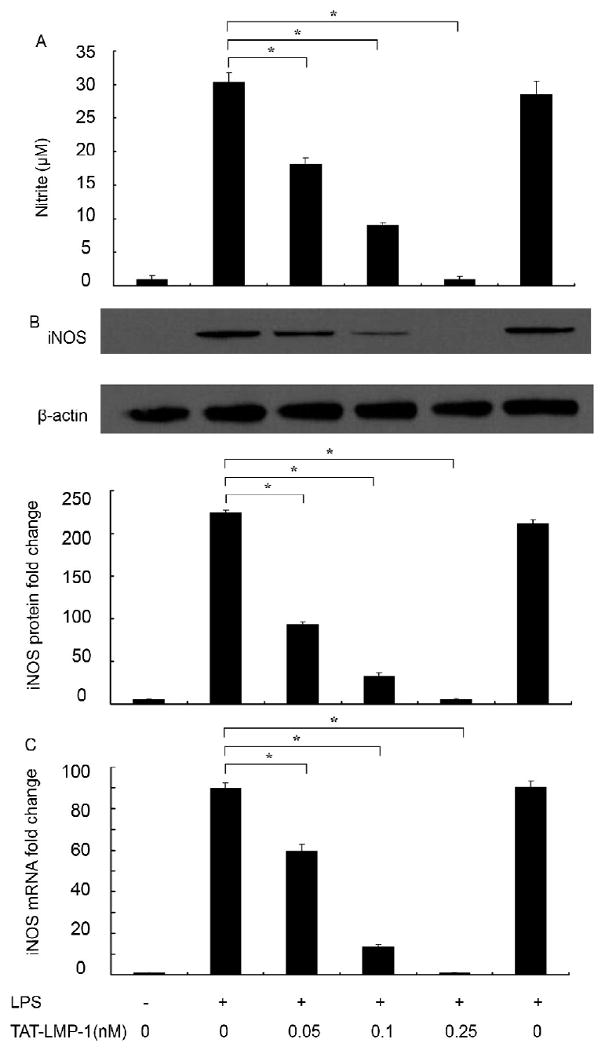
LMP-1 decreases NO production and iNOS expression in LPS-treated RAW 264.7 macrophages/pre-osteoclasts. (A) A Nitrite assay was used to assess NO production. TAT-LMP-1 inhibited LPS-induced NO production in a concentration-dependent manner 24 h after LPS treatment. Data are presented as mean ± SEM from one representative experiment out of three independent experiments performed in triplicate. (B) Expression of iNOS protein was determined by Western blot using specific antibody. β-actin antibody was used to show equal protein loading. TAT-LMP-1 inhibited LPS-induced iNOS protein expression in a concentration-dependent manner. The image presented is from one representative experiment out of three independent experiments. Dentitometric quantification and statistical analysis include the results from three independent experiments. (C) The level of iNOS mRNA was detected by real-time RT-PCR. TAT-LMP-1 inhibited LPS-induced iNOS mRNA expression in a concentration-dependent manner. Data are presented as mean ± SEM of the fold change in mRNA levels from one representative experiment out of three independent experiments performed in triplicate. (*P < 0.05)
To investigate whether the inhibitory effect of TAT-LMP-1 on NO production was due to reduced levels of its main regulating enzyme, iNOS protein levels were measured by Western blot. As shown in Fig.2B, in unstimulated RAW 264.7 macrophages/pre-osteoclasts, the iNOS protein level was undetectable. With LPS treatment, the iNOS protein expression was markedly augmented. Pretreatment of the cells with various doses of TAT-LMP-1, but not TAT-LMP-2, inhibited LPS-induced iNOS protein levels in a concentration-dependent manner. Densitometric analysis showed that TAT-LMP-1 at 0.05 nM reduced the iNOS protein level induced by LPS from 224-fold to 93-fold (P = 0.0001); TAT-LMP-1 at 0.1 nM reduced the iNOS protein level induced by LPS to 33-fold increase (P = 0.0001); TAT-LMP-1 at 0.25 nM reduced the iNOS protein level induced by LPS to the basal level (P = 0.0001). To determine whether reduced protein levels could be explained by reduced transcription of the iNOS gene, we measured iNOS mRNA levels by real time RT-PCR. Consistently, as shown in Fig.2C, in unstimulated RAW 264.7 macrophages/pre-osteoclasts the iNOS mRNA level was undetectable. However, with LPS treatment, the iNOS mRNA expression was markedly increased 90-fold (P = 0.0001), and pretreatment with various doses of TAT-LMP-1 inhibited LPS-induced iNOS gene expression in a concentration-dependent manner. TAT-LMP-1 at 0.05 nM reduced the LPS-induced iNOS mRNA level to 60-fold (P = 0.0001); TAT-LMP-1 at 0.1 nM reduced the LPS-induced iNOS mRNA level to 14-fold (P = 0.0001); TAT-LMP-1 at 0.25 nM reduced the LPS-induced iNOS mRNA level to the basal level (P = 0.0001). LMP-2 had no inhibitory effect on LPS-induced iNOS mRNA levels. These data suggest that TAT-LMP-1 down-regulates LPS-induced iNOS gene expression at the transcriptional level.
LMP-1 inhibits iNOS promoter activation in LPS-induced RAW 264.7 macrophagespre-osteoclasts
To investigate the molecular mechanism of TAT-LMP-1-mediated inhibition of iNOS transcription, activation of the iNOS promoter was determined with an iNOS promoter luciferase reporter construct. LPS treatment for 24 h increased the luciferase expression 27-fold (P = 0.0001) over the basal level in transfected cells. Pretreatment of cells with TAT-LMP-1 significantly inhibited LPS-induced luciferase activity in a concentration-dependent manner. TAT-LMP-1 at 0.1 nM reduced the increase in iNOS reporter luciferase activity to 8-fold (P = 0.0001); TAT-LMP-1 at 0.25 nM reduced the iNOS reporter luciferase activity to the basal level (P = 0.0001). The negative control, TAT-LMP-2, showed no inhibitory effect on iNOS promoter activation (Fig.3).
Fig.3.
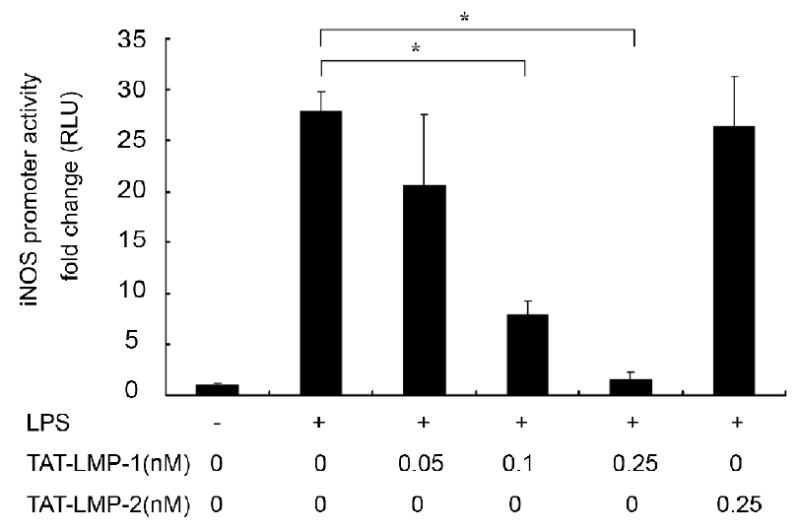
LMP-1 decreases LPS-induced iNOS promoter activity in RAW 264.7 macrophages/pre-osteoclasts. A Luciferase reporter assay was used to assess iNOS promoter activity. Luciferase activity was normalized to Renilla luciferase activity as relative luciferase units (RLU). TAT-LMP-1 inhibited LPS-induced iNOS promoter activity in a concentration-dependent manner. Data are presented as mean ± SEM of the fold change in RLU from one representative experiment out of three independent experiments performed in triplicate. (*P < 0.05)
LMP-1 inhibits NF-κB transcriptional activity in LPS-induced RAW 264.7 macrophages/pre-osteoclasts
We assessed NF-κB transcriptional activity to evaluate the inhibitory effect of LMP-1 on activation of NF-κB with a dual luciferase reporter gene system. LPS treatment for 24 h increased the luciferase expression 235-fold over the basal level in transfected cells (P = 0.0001). Pretreatment of cells with TAT-LMP-1 significantly inhibited LPS-induced luciferase expression in a concentration-dependent manner. TAT-LMP-1 at 0.05 nM reduced the increase in NF-κB reporter luciferase activity to 128-fold (P = 0.001); TAT-LMP-1 at 0.1 nM reduced the increase in NF-κB reporter luciferase activity to 51-fold (P = 0.0001); TAT-LMP-1 at 0.25 nM reduced the NF-κB reporter luciferase activity to the basal level (P = 0.005). The negative control, TAT-LMP-2, showed no inhibitory effect on NF-κB transcriptional activity (Fig.4). The data suggest that TAT-LMP-1 exerts inhibitory effects on NF-κB transcriptional activity activated by LPS.
Fig.4.
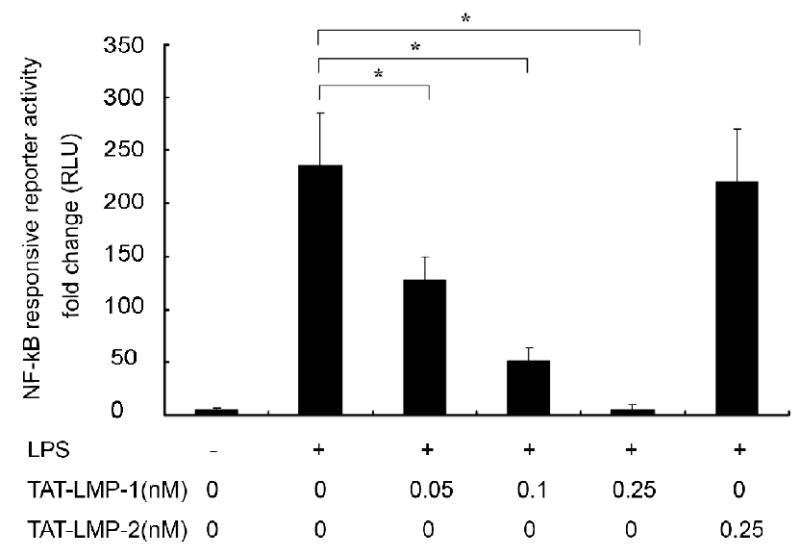
LMP-1 decreases LPS-induced NF-κB transcriptional activity in RAW 264.7 macrophages/pre-osteoclasts. A Luciferase reporter assay was used to assess NF-κB transcriptional activity. Luciferase activity was normalized to Renilla luciferase activity as relative luciferase units (RLU). TAT-LMP-1 inhibited LPS-induced NF-κB transcriptional activity in a concentration-dependent manner. Data are presented as mean ± SEM of the fold change in RLU from one representative experiment out of three independent experiments performed in triplicate. (*P < 0.05)
LMP-1 inhibits NF-κB nuclear translocation in LPS-induced RAW 264.7 macrophages/ pre-osteoclasts
To assess the effect of LMP-1 on LPS-induced nuclear translocation of NF-κB we detected the levels of the p65 subunit in nuclear extracts using Western blots. In untreated cells, p65 was almost undetectable in nuclear extracts. LPS treatment of cells for 3 h increased the levels of p65 in the nucleus dramatically. Pretreatment with TAT-LMP-1 resulted in a significant and concentration-dependent decrease of the p65 protein level in nuclear extracts from LPS-induced cells. The negative control, TAT-LMP-2, showed no inhibitory effect on p65 translocation (Fig.5). Densitometric analysis showed that TAT-LMP-1 at 0.05 nM reduced the p65 protein level in nuclear extracts induced by LPS from a 30-fold increase to an 8-fold increase (P = 0.047); TAT-LMP-1 at 0.1 nM reduced the p65 protein level in nuclear extracts to a 4-fold increase (P = 0.028); TAT-LMP-1 at 0.25 nM reduced the p65 protein level in nuclear extracts to the basal level (P = 0.0001). The data confirmed that the TAT-LMP-1 anti-inflammatory effect is due to inhibition of p65 translocation to the nucleus.
Fig.5.
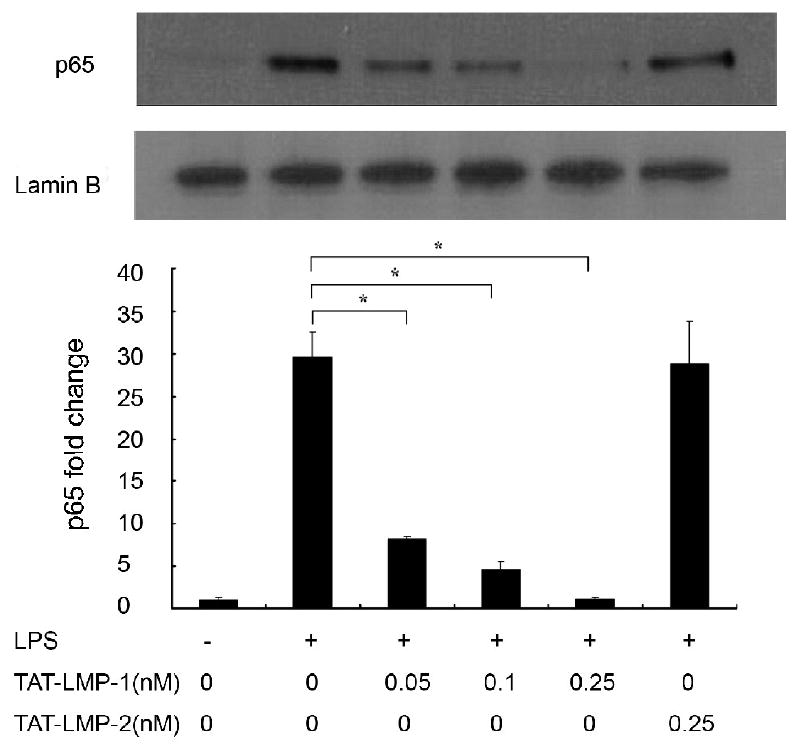
LMP-1 prevents LPS-induced nuclear translocation of NF-κB in RAW 264.7 macrophages/pre-osteoclasts. The protein level of the NF-κB subunit, p65, in nuclear extracts was determined by Western blot 3 h after LPS treatment. Lamin B antibody was used to show equal protein loading. TAT-LMP-1 inhibited LPS-induced p65 protein levels in the nucleus in a concentration-dependent manner. The image presented is from one representative experiment out of three independent experiments. Densitometric quantification and statistical analysis include the results from three independent experiments. (*P < 0.05)
LMP-1 inhibits phosphorylation of IkB in LPS-induced RAW 264.7 macrophages/pre-osteoclasts
An essential step for NF-κB pathway activation and translocation of p65 to the nucleus is phosphorylation of IκB, resulting in its degradation and the subsequent release of p65 from the IκB/NF-κB complex. To determine the mechanism of the inhibitory effect of LMP-1 on the LPS-induced activation of the NF-κB pathway, the effect of TAT-LMP-1 on phosphorylation of IκB was examined by Western blot. A time-course experiment showed that IκBα was maximally phosphorylated 9 min after LPS stimulation (Fig.6A). Pretreatment with TAT-LMP-1 prevented the induced phosphorylation level of IκBα protein at this time point in a dose dependent manner. Densitometric analysis determined that LPS increased IκB phosphorylation 292-fold (P = 0.0001). TAT-LMP-1 at 0.05 nM reduced the LPS-induced increase to 121-fold (P = 0.002); TAT-LMP-1 at 0.1 nM reduced the LPS-induced fold increase 38-fold (P = 0.0001); TAT-LMP-1 at 0.25 nM abolished the LPS-induced phosphorylation of IκB (P = 0.004). The negative control, TAT-LMP-2, showed no inhibitory effect on phosphorylation of IκB (Fig.6B).
Fig.6.
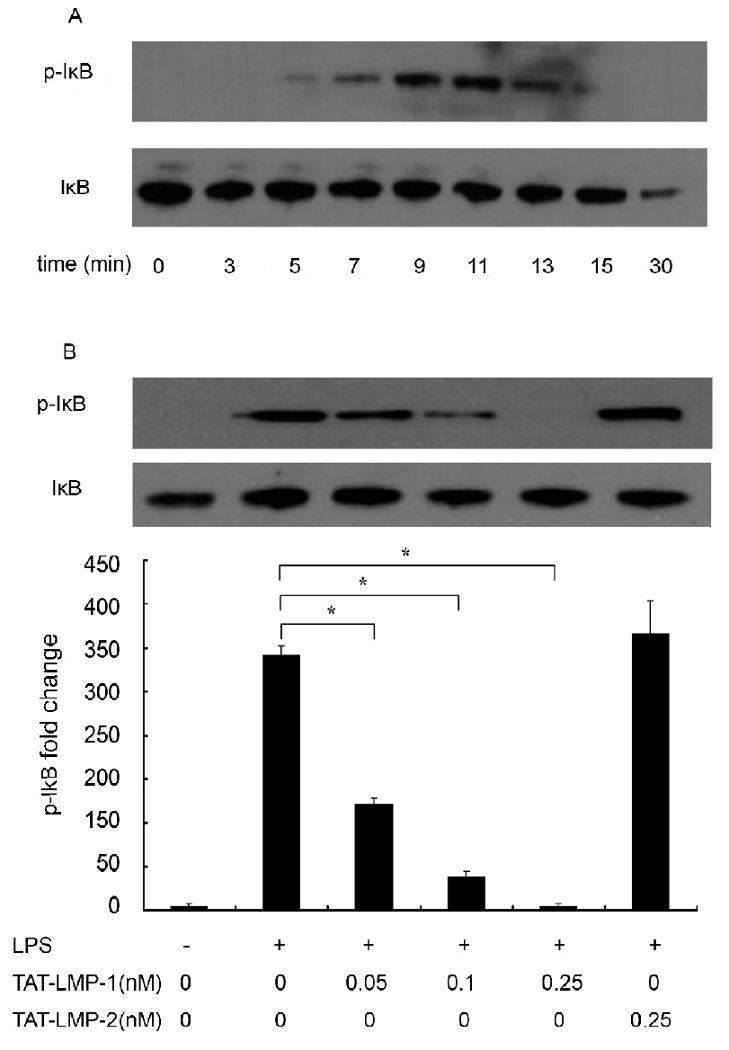
LMP-1 prevents LPS-induced phosphorylation of IκB in RAW 264.7 macrophages/pre-osteoclasts. Protein levels of phospho-IκB and IκB were determined by Western blot. (A) Time course of LPS-induced phosphorylation of IκB in RAW 264.7 cells. (B) TAT-LMP-1 inhibited LPS-induced IκB phosphorylation (9 min) in a concentration-dependent manner. Detection of the IκB level served as a loading control. The image presented is from one representative experiment out of three independent experiments. Densitometric quantification and statistical analysis include the results from three independent experiments. (*P < 0.05)
LMP-1 does not inhibit activation of NF-κB in Receptor Activator of Nuclear Factor Kappa B Ligand (RANKL)-induced RAW 264.7 macrophages/pre-osteoclasts
The above results and our preliminary observation that TAT-LMP-1 inhibited NO production induced by both TNFα and LPS suggested that LMP-1 mediates anti-inflammatory effects by inactivating the NF-κB pathway. To determine whether the inhibition by LMP-1 occurs when the NF-κB pathway is activated by a non-inflammatory stimulus, we compared the effect of TAT-LMP-1 on NF-κB activity induced by LPS or RANKL using the NF-κB reporter assay. We found that LPS enhanced NF-κB reporter activity 116-fold (P = 0.012) while RANKL enhanced NF-κB reporter activity 37-fold (P = 0.018). TAT-LMP-1 did not inhibit activation of the NF-κB reporter by RANKL (P = 0.871), but did inhibit LPS activation of the NF-κB pathway to basal levels (P = 0.012), suggesting that LMP-1 does not universally inhibit NF-κB activation (Fig. 7).
Fig.7.
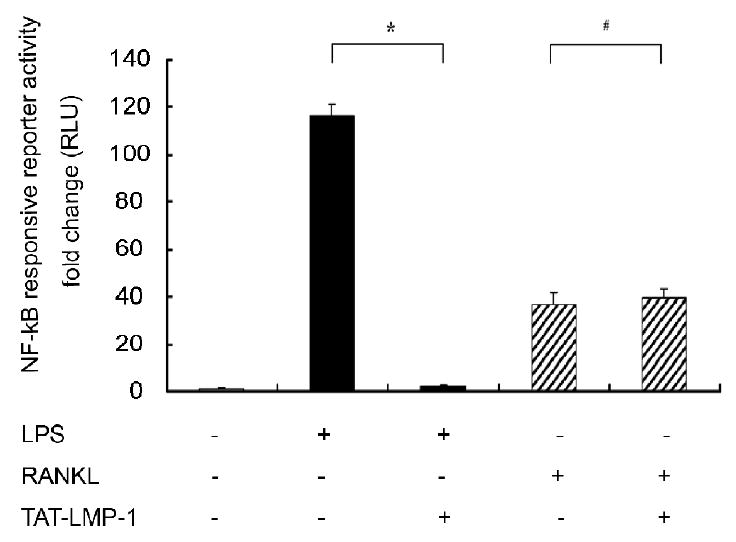
LMP-1 does not inhibit activation of NF-κB in RANKL-induced RAW 264.7 macrophages/pre-osteoclasts. A Luciferase reporter assay was used to assess NF-κB transcriptional activity. Luciferase activity was normalized to Renilla luciferase activity as relative luciferase units (RLU). TAT-LMP-1 inhibited LPS-induced (24 h) NF-κB transcriptional activity in a concentration-dependent manner. Data are presented as mean ± SEM of the fold change in RLU from one representative experiment out of three independent experiments performed in triplicate. (*P < 0.05, #P < 0.05)
LMP-1 selectively regulates activation of MAPKs in LPS-induced RAW 264.7 macrophages/pre-osteoclasts
Since MAPKs are important regulators of inflammatory mediators and LPS is known to activate MAPK pathways in RAW 264.7 macrophages/pre-osteoclasts, we used densitometric scanning of Western blots to examine the effect of LMP-1 on activation of MAPKs induced by 30 min treatment with LPS. The results showed that LPS enhanced phosphorylation of p38, extracellular signal-regulated kinase 1/2 (ERK1/2), and JNK (P = 0.0001). Pretreatment with TAT-LMP-1 did not affect LPS-induced phosphorylation of ERK1/2 (P = 1.000) (Fig.8B), but enhanced phosphorylation of p38 (P = 0.002) (Fig.8A) and inhibited phosphorylation of JNK (P = 0.0001) (Fig.8C). As positive controls MAPK inhibitors SB203580, UO126 and SP600125 were applied prior to LPS treatment to inhibit phosphorylation of p38, ERK1/2 and JNK, respectively. TAT-LMP-2 served as a negative control and had no effect on LPS-induced phosphorylation of all three MAPKs (Fig.8).
Fig.8.
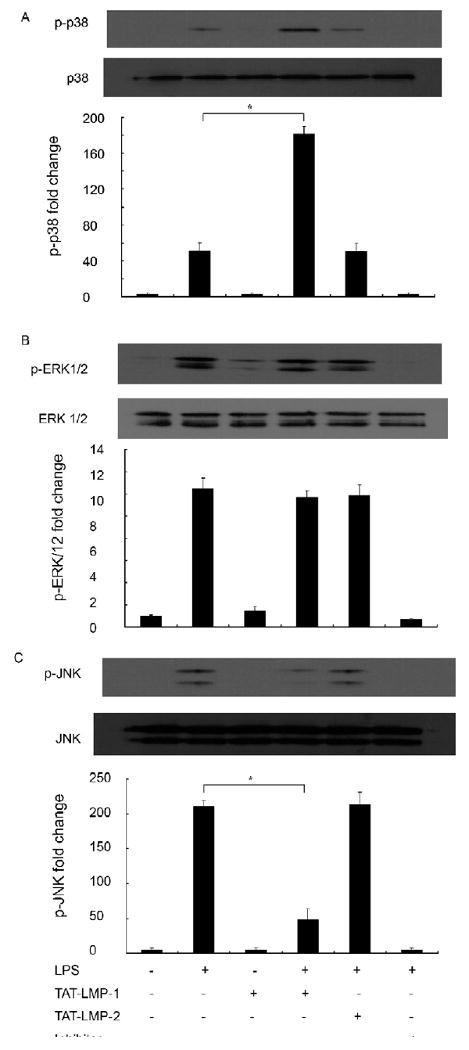
LMP-1 selectively regulates LPS-induced activation of MAPKs in RAW 264.7 macrophages/pre-osteoclasts. Protein levels of phospho-MAPKs were by Western blot after 30 min LPS treatment. (A) LMP-1 enhanced activation of p38 induced by LPS. (B) LMP-1 had no effect on activation of ERK1/2 induced by LPS. (C) LMP-1 suppressed activation of JNK induced by LPS. Detection of levels of the p38, ERK1/2 and JNK served as the loading control. The image presented is from one representative experiment out of three independent experiments. Densitometric quantification and statistical analysis include the results from three independent experiments. (*P < 0.05)
Discussion
LMP-1 is an intracellular regulator of bone formation. It was discovered as a mediator of Bone Morphogenetic Protein-6 (BMP-6)-induced bone formation in fetal calvarial cells [1]. Over-expression of LMP-1 induces osteoblast differentiation in calvarial cultures and culture medium from cells over-expressing LMP-1 mimics the direct effects of LMP-1 when applied to undifferentiated cells [1, 4]. In vivo, leukocytes over-expressing LMP-1 induce ectopic bone formation in rats and spine fusion in rats and rabbits [2, 3, 5]. There are at least three isoforms of LMP (LMP-1, LMP-2, and LMP-3). LMP-1 and LMP-3 are osteoinductive forms and contain a unique 19 amino acid region that is not present in LMP-2, a non-osteoinductive isoform [4]. Our group has shown that at least some of the bone-inducing effects of LMP-1 are mediated by its interaction with the E3 ligase, Smurf1, to enhance cellular responsiveness to BMP-2. This interaction occurs in the unique region of LMP-1/3 [6]. Despite extensive study in osteoblasts, the effects of LMP-1 on macrophages and pre-osteoclasts have not been studied.
In this study we expanded a preliminary observation that LMP-1 dramatically inhibited LPS-induced NO production in macrophages. LPS is a component of the outer-membranes of gram-negative bacteria and can directly mediate inflammatory bone loss [21]. LPS binds to TLR4 on the cell surface of macrophages and causes intracellular responses that activate several signal transduction pathways, including NF-κB and MAPKs. Activation of these pathways triggers the production of inflammatory mediators, such as IL-1β, TNF-α, IL-8, CXCL10, and NO that are intended to defend the host from bacterial infection, but can also mediate inflammatory bone loss via multiple pathways [27, 28]. These inflammation mediators can recruit more osteoclast precursors, induce subsequent osteoclastogenesis, activate osteoclasts, and affect bone formation by osteoblasts [29-32].
Our observation that production of macrophage-derived NO is inhibited by LMP-1 may have implications for reducing inflammatory bone loss. Low concentrations of NO have been shown to potentiate IL-1 induced bone resorption [33]. In addition, NO has been shown to play a key role in sustaining NF-κB activiation in osteoclast precursors [34]. Accordingly we sought to determine whether the LMP-1 inhibition of NO production involved the NF-κB pathway.
NF-κB is a well known transcriptional factor regulating the iNOS gene. In this study, we demonstrated that LMP-1, in a concentration-dependent manner, inhibited LPS-mediated iNOS promoter activation. This suggests that LMP-1 might inhibit iNOS promoter activation by inhibiting its regulatory transcriptional factors. Subsequently, the finding that pre-treatment with LMP-1 prevented the LPS induced IκB phosphorylation and consequent NF-κB release and translocation to the nucleus implies that LMP-1 inhibits iNOS transcription by inhibiting activation of the NF-κB pathway. Furthermore, LPS-stimulated NF-κB transcriptional activity was also shown to be inhibited by LMP-1 in an NF-κB reporter assay. Interestingly, LMP-1 did not inhibit RANKL-induced activation of the NF-κB reporter, implying that LMP-1 specifically inhibits NF-κB activity that has been activated by certain receptor/accessory protein cascades. Thus, LMP-1 may interrupt the positive-feedback loop of the inflammatory response that is responsible for inflammatory bone loss, while having no direct effect on RANKL-induction of NF-κB-dependent osteoclastogenesis. We believe the above effects of LMP-1 require the same 19 amino acid sequence that is responsible for bone induction, since LMP-2, a family member lacking that sequence, did not exert the same effects.
Since MAPKs are also well known to be modulators of the NF-κB pathway, inflammatory responses, and bone formation [35, 36], we assessed the effect of LMP-1 on LPS-induced phosphorylation and activation of MAPKs. Although many stimuli, including LPS, activate all three MAPKs, studies have shown that JNK is the main MAPK involved in inflammatory responses and regulation of NO production [7, 17]. In contrast, inhibiting activation of p38 MAPK does not suppress NO and cytokine production [37]. Interestingly, our results in the current study demonstrate in RAW 264.7 macrophages/pre-osteoclasts that LMP-1 enhances or has no effect on LPS-induced phosphorylation/activation of p38 MAPK and ERK1/2 and suppresses LPS-induced phosphorylation of JNK. These results imply that selective regulation of the signaling cascades of MAPKs might be one of the potential mechanisms by which LMP-1 inhibits NO production. The exact role of each of these MAPK cascades in mediating this effect remains under investigation.
Overall, our findings describe an exciting new anti-inflammatory function for the osteoinductive protein, LMP-1, via its selective inhibition of the NFκB pathway when NFκB is activated by agents, such as LPS, that induce inflammatory responses resulting in bone loss. The fact that LMP-1 does not also inhibit RANKL-induced NF-κB activation suggests that LMP-1 exerts its effects proximal to the IKK complex and is specific for certain receptor/accessory protein cascades. These findings will direct our future studies as to the protein-protein interactions by which LMP-1 inhibits activation of the NF-κB pathway. Additionally, LMP-1 selectively down regulates the JNK signaling pathway. To our knowledge, this is the first demonstration of an intracellular osteoindutive factor that also inhibits inflammatory signals that mediate bone loss. These observations warrant confirmation in other cell types including osteoblasts and chondrocytes. In vivo studies are under investigation to assess the therapeutic potential of LMP-1 in disorders mediated by the NF-κB and MAPK pathways.
Acknowledgments
This work is supported in part by a VA Merit award to Dr. Titus and NIH grant R01 AR53093 awarded to Dr. Boden. This work was performed in facilities at the Atlanta VA Medical Center, Decatur, GA. Dr. Hui Liu is partially supported by the International Program of Project 985, Sun Yat-sen University. We would like to thank Dr. David Stephens, Division of Infectious Diseases, Department of Medicine, Emory University School of Medicine for kindly providing the LPS and Dr. Giovanni Melillo, Science Applications International Corporation, Frederick, Inc for kindly providing the iNOS promoter reporter construct. We would like to thank Dr. M. Neale Weitzmann for helpful discussions, Mesfin Teklemariam for real time RT-PCR and Western blot assistance, and Liping Zhu for cell culture assistance. Dr. Boden has received compensation as a consultant for Medtronic Sofamor Danek and for intellectual property. Emory University and some of the authors have/may receive royalties in the future related to LMP-1. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict of interest policies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boden SD, Liu Y, Hair GA, Helms JA, Hu D, Racine M, Nanes MS, Titus L. LMP-1, a LIM-domain protein, mediates BMP-6 effects on bone formation. Endocrinology. 1998;139:5125–34. doi: 10.1210/endo.139.12.6392. [DOI] [PubMed] [Google Scholar]
- 2.Boden SD, Titus L, Hair G, Liu Y, Viggeswarapu M, Nanes MS, Baranowski C. Lumbar spine fusion by local gene therapy with a cDNA encoding a novel osteoinductive protein (LMP-1) Spine. 1998;23:2486–92. doi: 10.1097/00007632-199812010-00003. [DOI] [PubMed] [Google Scholar]
- 3.Viggeswarapu M, Boden SD, Liu Y, Hair GA, Louis-Ugbo J, Murakami H, Kim HS, Mayr MT, Hutton WC, Titus L. Adenoviral delivery of LIM mineralization protein-1 induces new-bone formation in vitro and in vivo. J Bone Joint Surg Am. 2001;83-A:364–76. doi: 10.2106/00004623-200103000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Hair GA, Boden SD, Viggeswarapu M, Titus L. Overexpressed LIM mineralization proteins do not require LIM domains to induce bone. J Bone Miner Res. 2002;17:406–14. doi: 10.1359/jbmr.2002.17.3.406. [DOI] [PubMed] [Google Scholar]
- 5.Minamide A, Boden SD, Viggeswarapu M, Hair GA, Oliver C, Titus L. Mechanism of bone formation with gene transfer of the cDNA encoding for the intracellular protein LMP-1. J Bone Joint Surg Am. 2003;85-A:1030–9. doi: 10.2106/00004623-200306000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Sangadala S, Boden SD, Viggeswarapu M, Liu Y, Titus L. LIM mineralization protein-1 potentiates bone morphogenetic protein responsiveness via a novel interaction with Smurf1 resulting in decreased ubiquitination of Smads. J Biol Chem. 2006;281:17212–9. doi: 10.1074/jbc.M511013200. [DOI] [PubMed] [Google Scholar]
- 7.Chan ED, Riches DW. Potential role of the JNK/SAPK signal transduction pathway in the induction of iNOS by TNF-alpha. Biochem Biophys Res Commun. 1998;253:790–6. doi: 10.1006/bbrc.1998.9857. [DOI] [PubMed] [Google Scholar]
- 8.Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500:255–66. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 10.Mercurio F, Manning AM. NF-kappaB as a primary regulator of the stress response. Oncogene. 1999;18:6163–71. doi: 10.1038/sj.onc.1203174. [DOI] [PubMed] [Google Scholar]
- 11.Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, Guan K, Krebsbach PH, Wang CY. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 2009;15:682–9. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–6. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 13.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 14.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 15.Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 16.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 17.Tsutsumi R, Ito H, Hiramitsu T, Nishitani K, Akiyoshi M, Kitaori T, Yasuda T, Nakamura T. Celecoxib inhibits production of MMP and NO via down-regulation of NF-kappaB and JNK in a PGE2 independent manner in human articular chondrocytes. Rheumatol Int. 2008;28:727–36. doi: 10.1007/s00296-007-0511-6. [DOI] [PubMed] [Google Scholar]
- 18.Lu SC, Wu HW, Lin YJ, Chang SF. The essential role of Oct-2 in LPS-induced expression of iNOS in RAW 264.7 macrophages and its regulation by trichostatin A. Am J Physiol Cell Physiol. 2009;296:C1133–9. doi: 10.1152/ajpcell.00031.2009. [DOI] [PubMed] [Google Scholar]
- 19.Rajapakse N, Kim MM, Mendis E, Kim SK. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 in lipopolysaccharide-stimulated RAW264.7 cells by carboxybutyrylated glucosamine takes place via down-regulation of mitogen-activated protein kinase-mediated nuclear factor-kappaB signaling. Immunology. 2008;123:348–57. doi: 10.1111/j.1365-2567.2007.02683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ci X, Song Y, Zeng F, Zhang X, Li H, Wang X, Cui J, Deng X. Ceftiofur impairs pro-inflammatory cytokine secretion through the inhibition of the activation of NF-kappaB and MAPK. Biochem Biophys Res Commun. 2008;372:73–7. doi: 10.1016/j.bbrc.2008.04.170. [DOI] [PubMed] [Google Scholar]
- 21.Itoh K, Udagawa N, Kobayashi K, Suda K, Li X, Takami M, Okahashi N, Nishihara T, Takahashi N. Lipopolysaccharide promotes the survival of osteoclasts via Toll-like receptor 4, but cytokine production of osteoclasts in response to lipopolysaccharide is different from that of macrophages. J Immunol. 2003;170:3688–95. doi: 10.4049/jimmunol.170.7.3688. [DOI] [PubMed] [Google Scholar]
- 22.Wadia JS, Dowdy SF. Protein transduction technology. Curr Opin Biotechnol. 2002;13:52–6. doi: 10.1016/s0958-1669(02)00284-7. [DOI] [PubMed] [Google Scholar]
- 23.Becker-Hapak M, McAllister SS, Dowdy SF. TAT-mediated protein transduction into mammalian cells. Methods. 2001;24:247–56. doi: 10.1006/meth.2001.1186. [DOI] [PubMed] [Google Scholar]
- 24.Sangadala S, Okada M, Liu Y, Viggeswarapu M, Titus L, Boden SD. Engineering, cloning, and functional characterization of recombinant LIM mineralization protein-1 containing an N-terminal HIV-derived membrane transduction domain. Protein Expr Purif. 2009;65:165–73. doi: 10.1016/j.pep.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Park E, Quinn MR, Wright CE, Schuller-Levis G. Taurine chloramine inhibits the synthesis of nitric oxide and the release of tumor necrosis factor in activated RAW 264.7 cells. J Leukoc Biol. 1993;54:119–24. doi: 10.1002/jlb.54.2.119. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Titus L, Barghouthi M, Viggeswarapu M, Hair G, Boden SD. Glucocorticoid regulation of human BMP-6 transcription. Bone. 2004;35:673–81. doi: 10.1016/j.bone.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 27.Arenzana-Seisdedos F, Virelizier JL. Interferons as macrophage-activating factors. II. Enhanced secretion of interleukin 1 by lipopolysaccharide-stimulated human monocytes. Eur J Immunol. 1983;13:437–40. doi: 10.1002/eji.1830130602. [DOI] [PubMed] [Google Scholar]
- 28.Chokri M, Freudenberg M, Galanos C, Poindron P, Bartholeyns J. Antitumoral effects of lipopolysaccharides, tumor necrosis factor, interferon and activated macrophages: synergism and tissue distribution. Anticancer Res. 1989;9:1185–90. [PubMed] [Google Scholar]
- 29.Haynes DR. Inflammatory cells and bone loss in rheumatoid arthritis. Arthritis Res Ther. 2007;9:104. doi: 10.1186/ar2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy R, Cooper MS. Bone loss in inflammatory disorders. J Endocrinol. 2009;201:309–20. doi: 10.1677/JOE-08-0568. [DOI] [PubMed] [Google Scholar]
- 31.Udagawa N, Kotake S, Kamatani N, Takahashi N, Suda T. The molecular mechanism of osteoclastogenesis in rheumatoid arthritis. Arthritis Res. 2002;4:281–9. doi: 10.1186/ar431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali T, Lam D, Bronze MS, Humphrey MB. Osteoporosis in inflammatory bowel disease. Am J Med. 2009;122:599–604. doi: 10.1016/j.amjmed.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ralston SH, Ho LP, Helfrich MH, Grabowski PS, Johnston PW, Benjamin N. Nitric oxide: a cytokine-induced regulator of bone resorption. J Bone Miner Res. 1995;10:1040–9. doi: 10.1002/jbmr.5650100708. [DOI] [PubMed] [Google Scholar]
- 34.van't Hof RJ, Armour KJ, Smith LM, Armour KE, Wei XQ, Liew FY, Ralston SH. Requirement of the inducible nitric oxide synthase pathway for IL-1-induced osteoclastic bone resorption. Proc Natl Acad Sci U S A. 2000;97:7993–8. doi: 10.1073/pnas.130511497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kundu JK, Surh YJ. Breaking the relay in deregulated cellular signal transduction as a rationale for chemoprevention with anti-inflammatory phytochemicals. Mutat Res. 2005;591:123–46. doi: 10.1016/j.mrfmmm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 36.Schindeler A, Little DG. Ras-MAPK signaling in osteogenic differentiation: friend or foe? J Bone Miner Res. 2006;21:1331–8. doi: 10.1359/jbmr.060603. [DOI] [PubMed] [Google Scholar]
- 37.Rao KM, Meighan T, Bowman L. Role of mitogen-activated protein kinase activation in the production of inflammatory mediators: differences between primary rat alveolar macrophages and macrophage cell lines. J Toxicol Environ Health A. 2002;65:757–68. doi: 10.1080/00984100290071027. [DOI] [PubMed] [Google Scholar]