

Abstract
Opioid-induced glial activation and its proinflammatory consequences have been associated with both reduced acute opioid analgesia and the enhanced development of tolerance, hyperalgesia and allodynia following chronic opioid administration. Intriguingly, recent evidence demonstrates that these effects can result independently from the activation of classical, stereoselective opioid receptors. Here, a structurally disparate range of opioids cause activation of signaling by the innate immune receptor Toll Like Receptor 4 (TLR4), resulting in proinflammatory glial activation. In the present series of studies, we demonstrate that the (+)-isomers of methadone and morphine, which bind with negligible affinity to classical opioid receptors, induced upregulation of proinflammatory cytokine and chemokine production in rat isolated dorsal spinal cord. Chronic intrathecal (+)-methadone produced hyperalgesia and allodynia, which were associated with significantly increased spinal glial activation (TLR4 mRNA and protein) and the expression of multiple chemokines and cytokines. Statistical analysis suggests that a cluster of cytokines and chemokines may contribute to these nociceptive behavioral changes. Acute intrathecal (+)-methadone and (+)-morphine were also found to induce microglial, interleukin-1 and TLR4/MD-2 dependent enhancement of pain responsivity. In silico docking analysis demonstrated (+)-naloxone sensitive docking of (+)-methadone and (+)-morphine to human MD-2. Collectively, these data provide the first evidence of the pro-nociceptive consequences of small molecule xenobiotic activation of spinal TLR4 signaling independent of classical opioid receptor involvement.
Keywords: Cytokine, Chemokine, Analgesia, Tolerance, Microglia, (+)-Morphine, (+)-Methadone
Introduction
Since Wybran et al. (1979) demonstrated a clear link between the peripheral immune and opioid systems, a great deal of work has characterized peripheral opioid immunomodulation. However, it has only been in the past decade that opioid immunomodulation of glia, a major class of immunocompetent cells of the central nervous system, has been examined. It is clear from in vitro studies that opioids directly influence microglial and astrocyte function. Morphine activates cultured microglia as indicated by retraction of processes, membrane ruffling, activation of ERK1/2, and enhanced chemotaxis and phagocytosis (Dobrenis et al., 1995, Lipovsky et al., 1998, Takayama and Ueda, 2005). Morphine increases microglial production of nitric oxide (Magazine et al., 1996, Stefano, 1998) and proinflammatory cytokines (Peterson et al., 1998). In addition, morphine sensitizes (“primes”) microglia in vitro to over-respond to subsequent stimuli, thereby generating exaggerated release of these neuroexcitatory substances (Chao et al., 1994). In astrocyte cultures, morphine upregulates activation markers (Narita et al., 2006) and increases proinflammatory cytokine and chemokine production (El-Hage et al., 2005).
In vivo, studies of opioid-induced glial activation have focused on glial modulation of pain. Glial activation and opioid-induced proinflammatory cytokine actions oppose acute opioid analgesia, and to contribute to the development of morphine tolerance, opioid-induced hyperalgesia, opioid-induced allodynia and withdrawal induced allodynia (for review see Watkins et al. (2009)). The pharmacological characteristics of opioid-induced glial activation are important to understand, as antagonizing these non-neuronal events potentiates analgesia and blocks the development of the unwanted opioid-induced nociceptive side-effects noted above (Shavit et al., 2005, Hutchinson et al., 2008a, Hutchinson et al., 2008b).
It is now understood that proinflammatory cytokines oppose acute and chronic opioid analgesia, in a microglial-, nitric oxide- and p38 MAPK-dependent fashion (Cui et al., 2006, Hutchinson et al., 2007). The neuron-to-microglia signal, fractalkine, also appears to be involved in opposition of opioid analgesia, both acutely (Hutchinson et al., 2008a) and chronically (Johnston et al., 2004). However, the critical question of the mechanism(s) underlying proinflammatory glial activation remained unclear until recently (Hutchinson et al., 2009). We have demonstrated that novel toll-like receptor 4/myeloid differentiation factor-2 (TLR4/MD-2) activity of a structurally disparate range of opioids results in glial activation with a proinflammatory phenotype (Hutchinson et al., 2009). Importantly, the TLR4/MD-2 activity was demonstrated to be non-stereoselective, such that both the classical opioid receptor active (−)-isomers and the classical opioid receptor inactive (+)-isomers of opioids displayed TLR4/MD-2 activity (Hutchinson et al., 2009) with a low eudismic ratio. Therefore, upon administration of clinically utilized (−)-isomer opioids, both the beneficial neuronal opioid receptor actions (i.e., analgesia) and the detrimental TLR4-mediated glial actions (i.e., opposition to analgesia, enhancement of tolerance, etc.) are triggered.
As noted above, the evidence to date indicate that glial TLR4/MD-2 proinflammatory signaling is activated non-stereoselectively by opioids, including morphine and methadone. If this is indeed true, this makes the strong prediction that (+)-morphine and (+)-methadone should, despite having no action at classical opioid receptors, exert predictable behavioral and immunological effects resulting from their previously published (Hutchinson et al., 2009) proinflammatory actions via glial TLR4. Therefore, the aim of the present studies was to characterize the behavioral and molecular consequence of the opioid inactive (+)-isomers of morphine and methadone and the potential proinflammatory changes following acute and chronic administration.
Materials and Methods
Subjects
Pathogen-free adult male Sprague–Dawley rats (300–375 g; Harlan Labs, Madison, WI) were used in all experiments (n=6 per group for each experiment). Rats were housed in temperature (23±3 °C) and light (12 h:12 h light:dark cycle; lights on at 0700) controlled rooms with standard rodent chow and water available ad libitum. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Colorado at Boulder.
Drugs
Opioid inactive (+)-methadone base was obtained from the National Institute on Drug Abuse (Research Triangle Park, NC, USA). Opioid inactive (+)-morphine and (+)-naloxone were kindly gifted by Dr Kenner Rice (NIDA and NIAAA, Bethesda, MD, USA). Endotoxin-free solutions of recombinant met-human interleukin-1 (IL-1) receptor antagonist (IL-1ra) were purchased from Amgen (Thousand Oaks, CA, USA). (−)-Naloxone, naltrindole, naloxonazine, nor-binaltorphimine (norBNI) and minocycline were purchased from Sigma (St. Louis, MO, USA). Where applicable, drugs were prepared, and are reported, as free base concentrations. The endotoxin-free status of solutions was confirmed by LAL assay.
Catheter implantation
For acute intrathecal drug delivery (Experiments 3 and 4), the catheters were preloaded with drugs at the distal end in a total volume of no greater than 25 µl. The catheters were 90 cm in length, allowing remote drug delivery without touching or otherwise disturbing the rats during the testing. The surgery for chronic indwelling intrathecal catheters (Experiment 2) was exactly the same as for the acute intrathecal catheter, except that after the catheter was led subcutaneously to the nape of the neck the catheter was heat sealed with approximately 3 cm of the catheter protruding to allow easy access for subsequent dosing. These catheters were not preloaded with drug and were instead filled with injection saline. Acute and chronic intrathecal drug experiments began 2 h and 7 days after surgery, respectively.
The method of constructing and implanting the indwelling intrathecal catheters was based on that described previously (Milligan et al., 1999, Hutchinson et al., 2008a). Briefly, intrathecal catheters were implanted under anesthesia (isoflurane; Phoenix Pharmaceuticals, St. Joseph, MO, USA) by threading sterile polyethylene-10 tubing (PE-10 Intramedic Tubing; Becton Dickinson Primary Care Diagnostics, Sparks, MD, USA) guided by an 18-gauge needle between the L5 and L6 vertebrae. The catheter was inserted 8.8 cm beyond the exterior end of the needle such that the proximal catheter tip lay over the lumbosacral enlargement. The needle was removed and the catheter was sutured to the superficial musculature of the lower back and the exterior end led subcutaneously to exit through a small incision at the nape of the neck.
Behavioral assessments
Hargreaves tests for analgesia and hyperalgesia
Rats received at least three 60 min habituations over successive days to the test environment prior to behavioral testing. Latencies for behavioral response to radiant heat stimuli applied to the plantar surface of each hind-paw and tail were assessed using a modified Hargreaves test (Hargreaves et al., 1988). All testing was conducted blind with respect to group assignment. Pilot studies determined that intrathecal catheter surgery did not affect baseline responses after 2 h or 7 days recovery from surgery, compared to latencies recorded prior to surgery. Briefly, baseline withdrawal times were calculated from an average of 2 consecutive withdrawal latencies of the tail and the left and the right hind-paws, measured at 15-min intervals. Withdrawal times for the short baseline latency Hargreaves stimuli at baseline ranged from 3 to 4 s, and a cut-off time of 10 s was imposed to avoid tissue damage. This short baseline latency Hargreaves stimulus was used for studies of analgesia as this intensity was appropriate for documenting lengthening of response latencies in response to drugs that reduce pain sensitivity. Latencies for the long baseline latency Hargreaves stimuli at baseline ranged from 8 to 10 s, and a cut-off time of 20 s was imposed to avoid tissue damage. This long baseline latency Hargreaves stimulus was used for studies of hyperalgesia as this intensity was appropriate for documenting shortening of response latencies in response to drugs that enhance pain sensitivity. In each case, the order of paw and tail testing varied randomly. Nociceptive assessments for acute administration experiments were then made at 0 (immediately following remote drug delivery), 5 min, 15 min and every 10 min thereafter until completion of the experiment only using the short baseline latency Hargreaves stimuli. For the chronic drug delivery experiments short and long baseline latency Hargreaves stimuli were employed on alternating time points, and in these experiments the rats were tested pre dose and 2 h post dose on days 1, 4 and 7 of dosing. For twice daily injections the nociceptive response to the morning dose was assessed.
Von Frey test for mechanical allodynia
Rats received at least three 60 min habituations to the test environment prior to behavioral testing. Response thresholds to calibrated light pressure stimuli applied to the plantar surface of the paws was measured using the von Frey test (Chaplan et al., 1994). The test was performed using 0.406–15.136 gm calibrated Semmes-Weinstein monofilaments (von Frey hairs; Stoelting, Wood Dale, IL, USA) as described in detail previously (Milligan et al., 2000). Briefly, rats were first assessed for baseline response thresholds (average of three consecutive withdrawal assessments) from each paw at 15 min intervals, and the average response threshold from both feet was calculated. All testing was conducted blind with respect to group assignment. The behavioral responses were used to calculate the absolute threshold, by fitting a Gaussian integral psychometric function using a maximum-likelihood fitting method (Harvey, 1986, Treutwein and Strasburger, 1999), as described in detail previously (Milligan et al., 2000). Allodynia was assessed pre- and post-intrathecal drug delivery on days 1 and 4 and pre-dose on day 7 of chronic dose regimens. In experiment 1 where the von Frey test was used, the same animals that were tested on von Frey also received Hargreaves testing prior to and following the Hargreaves time course.
Dorsal spinal cord and cerebrospinal fluid (CSF) collection
Under pentobarbital anesthesia (50 mg/kg), CSF was collected by threading sterile PE-10 tubing guided by an 18-gauge needle between the L5 and L6 vertebrae on the side contralateral to the chronically implanted catheter. The catheter used to collect CSF was inserted rostrally 8.8 cm from the exterior end of the needle such that the catheter tip lay over the lumbosacral enlargement. Approximately 20–30 µl of CSF was withdrawn and immediately flash-frozen in liquid nitrogen. Rats were then transcardially perfused with chilled saline. The lumbosacral enlargement of the spinal cord was exposed by laminectomy, and after verifying intrathecal catheter placement by visual inspection, the lumbosacral spinal cord was then dissected free and placed on an ice-chilled glass plate. The dorsal aspects of these tissues were dissected, divided into left and right halves, and separately flash-frozen in liquid nitrogen. Left and right halves of the dorsal spinal cord were randomly assigned to mRNA or protein quantification. The entire procedure required a maximum of 15–20 min per animal. Samples were stored at −80°C until the time of assay.
In vitro lumbar dorsal spinal cord preparation
An in vitro assay was used to test whether opioid inactive (+)-isomers of methadone and morphine induced the release of cytokines and/or chemokines in a fashion similar to that previously described for (−)-morphine (Johnston et al., 2004; Hutchinson et al., 2008a). The lumbar spinal cord was rapidly exposed from unanaesthetized decapitated rats using aseptic technique, and then the cord was placed in a 70% ethanol rinse. The dorsal spinal cord was isolated and rinsed with sterile HBSS, and a 1.75 cm section of the lumbar enlargement was isolated. Tissue was placed in 25 µl of incubation medium (DMEM, supplemented with 2 mM L-glutamine, 100 U penicillin, 100 µg streptomycin and 10 mM HEPES buffer (Sigma)), inside a sterile modified 500 µl Eppendorf centrifuge tube. The tube was modified by removal of the snap cap and creation of a 2 mm wide slit down one side to within 7 mm of the base. Once the tissue was inserted into the tube, dorsal side up, media and drug were added such that the total volume was 200 µl. In parallel to our previously published studies of (−)-morphine in this model (Johnston et al., 2004; Hutchinson et al., 2008a), tissues were incubated with 100 µM (+)-morphine, 100 µM (+)-methadone or saline (vehicle) and incubated for 3 hr at 37°C, 5% CO2-95% air. After incubation, supernatants were collected and assayed for various rat cytokines and rat chemokines using a custom multiplex system described below (IL-1β, IL-6, IL-10, Fractalkine, GRO/KC, MIP-1α, MCP-1, RANTES and TNF-α).
Cytokine, chemokine, and TLR4 quantification
Procedures for tissue processing for protein quantification were similar to those described in detail previously (Hutchinson et al., 2008a). Mulitplex protein quantification (Thermo Scientific SearchLight Multiplex Sample Testing Service, Woburn, MA, USA) was utilized to quantify 16 rat analytes from a single sample. Specifically the cytokines: tumor necrosis factor-α (TNF-α), granulocyte macrophage colony stimulating factor (GMCSF), interferon -γ(IFN-γ), interleukin-10 (IL-10), interleukin-1α (IL-1α), interleukin-1β (IL-1β), interleukin-2 (IL-2), interleukin-4 (IL-4) and interleukin-6 (IL-6); and chemokines: GRO/KC (CXCL1), monocyte chemotatic protein-1 (MCP-1, CCL2), macrophage inflammatory protein-1α (MIP-1α), macrophage inflammatory protein-2 (MIP-2), macrophage inflammatory protein-3α (MIP-3α), Regulated upon Activation Normal T-cell Expressed and Secreted (RANTES), were quantified from one half of the dorsal spinal cord at the level of the lumbar enlargement and CSF collected over this region. TLR4 protein was examined by Western blot described previously (Hutchinson et al., 2008a). Data were expressed relative to the density of the TLR4 positive control run on each gel. Dorsal spinal cord samples from the remaining hemisphere were prepared for real-time reverse transcription-PCR cytokine mRNA quantification. Both protein and mRNA were analyzed blind with respect to group assignment. The following targets were investigated and expressed relative to the levels of the housekeeping gene GAPDH (M17701; forward: GTTTGTGATGGGTGTGAACC; reverse: TCTTCTGAGTGGCAGTGATG); IL-1 (M98820; forward: GAAGTCAAGACCAAAGTGG; reverse: TGAAGTCAACTATGTCCCG), TNF-α (D00475; forward: CTTCAAGGGACAAGGCTG; reverse: GAGGCTGACTTTCTCCTG), IL-6 (NM_012589; forward: ACTTCACAGAGGATACCAC; reverse: GCATCATCGCTGTTCATAC), IL-1 converting enzyme (D85899; forward: TGGTCTTGTGACTTGGAGG; reverse: CCTTTCAGTGGTTGGCATC) and Toll-like receptor 4 (NM_019178; forward: CAGAGGAAGAACAAGAAGC; reverse: CCAGATGAACTGTAGCATTC). The choice of mRNAs analyzed here is based on their analysis in our previous (−)-opioid studies (Hutchinson et al., 2009), so to allow comparisons with these prior data, and the availability of antagonists that would allow for mechanistic studies of changes observed, as appropriate. Primers were purchased from Proligo (Boulder, CO, USA).
In silico docking simulation
In Silico docking simulation methods previously outlined were employed to examine the docking of (+)-methadone, (+)-morphine and (+)-naloxone to TLR4 and MD-2 (Hutchinson et al., 2009). Briefly, the complexed human TLR4 and MD-2 pdb file was obtained from RCSB Protein Data Bank database (PDBID: 3fxi). Modified pdb files were inputted into AutoDock 4.0 (http://autodock.scripps.edu), hydrogens added, and resaved in pdbqt format. (+)-Methadone, (+)-morphine and (+)-naloxone structures were gathered using PubChem isomeric SMILES then converted to .pdb using a structure file generator (http://cactus.nci.nih.gov/services/translate/). Initially, the in silico docking of ligands to the entire TLR4 MD-2 dimer complex was conducted (AutoGrid center set 3.438, −7.805, 2.034; 126 grid points expanding in all directions; GA running number of 100, Max Evals 5 × 106 and 1.0 Å spacing). These data demonstrated that the ligands docked with human MD-2 independent of human TLR4 interactions. Therefore, all the ligands were docked to MD-2 alone with greater resolution (AutoGrid center set 27.991, 0.851, 19.625; 126 grid points expanding in all directions; GA running number of 100, Max Evals 5 × 106 and 0.375 Å spacing). All dockings were executed with Lamarkian genetic algorithms. The lowest energy and highest interaction docking conformation were visualized for (+)-naloxone, (+)-methadone and (+)-morphine. The (+)-naloxone conformation was then saved as a combined MD-2/(+)-naloxone pdf file and the (+)-methadone and (+)-morphine in silico dockings repeated on the new combined MD-2/(+)-naloxone complex to determine the change in docking owing to the presence of already docked (+)-naloxone.
Statistics
The analgesic responses were calculated as the % of maximal possible effect (%MPE) using the following equation (Carmody, 1995). For short baseline latency stimuli Hargreaves tests a cut off of 10 s was used whilst for long baseline latency stimuli a 20 s cut off was employed. Statistical significance was assessed using a two-way repeated ANOVA with Bonferroni post hoc test when comparing the analgesic responses of intrathecal treatments vs their vehicle controls.
Statistical mediation analysis of nociceptive behaviors (change from baseline in tolerance, hyperalgesia and allodynia) by dorsal spinal cord and CSF cytokine and chemokine levels was conducted. The data were first standardized to remove the inherent variability between different cytokine and chemokine levels and the behavioral response values. Hierarchical clustering was then used to analyze the absolute correlations between the cytokine and chemokine levels from dorsal spinal cord and CSF and nociceptive behaviors using the R Project package pvclust and 1000 bootstraps (Suzuki and Shimodaira, 2006). The cytokines and chemokines that clustered with behaviors in (+)-methadone treatment group were identified and the standardized values were summed. Multivariate analysis was conducted using treatment as a fixed factor controlling for the summed cytokine cluster variable.
Analyses and calculations were conducted with Excel 2003 SP2 (Microsoft, Redmond, CA, USA), Prism 4.03 (GraphPad, San Diego, CA, USA), R Project (Vienna, Austria)(R Development Core Team, 2006) and SPSS 14 for Windows (SPSS Inc., Chicago, IL, USA). Data are presented as mean ± SEM. Significance was set at P < 0.05.
Results
Experiment 1: Non-classical opioid-induced proinflammation in lumbar dorsal spinal cord results from acute exposure to (+)-opioids in vitro: glial activation and proinflammatory cytokine/chemokines
To define whether non-classical opioid-induced glial activation and release of proinflammatory mediators occurs in response to (+)-opioids as we have previously shown for (−)-isomer opioids (Johnston et al., 2004; Hutchinson et al., 2008a), we first ascertained whether a proinflammatory response could occur following acute exposure of relevant spinal tissue to (+)-opioids in vitro, as we have previously observed for (−)-morphine (Johnston et al., 2004; Hutchinson et al., 2008a). Lumbar dorsal spinal cord sections from naïve rats were incubated for 3 hr with 100 µM of the opioid-inactive (+)-morphine or (+)-methadone versus vehicle as was conducted previously for (−)-isomer opioids (Hutchinson et al., 2008a). Supernatants from these cultures were then analyzed for cytokines and chemokines, and the tissue assessed for glial activation (TLR4 expression (Raghavendra et al., 2004, Tanga et al., 2004)). Significant elevations in IL-1β, CX3CL1 (fractalkine), GRO/KC, MIP-1α, MCP-1 and TNF-α were quantified in the supernatants (P < 0.05) whilst IL-10, RANTES and IL-6 were unchanged (P > 0.05; Figure 1). Protein levels of the glial activation associated marker TLR4 in the dorsal spinal cord of these tissues was also assessed and found to be significantly elevated (P < 0.05) compared to media controls (media: 54 ± 6 %, (+)-methadone: 83 ± 6 %, (+)-morphine: 86 ± 6 %, relative to the gel TLR4 positive control, see methods above).
Figure 1. Non-classical opioid induction of proinflammatory cytokines and chemokines by (+)-morphine and (+)-methadone from lumbar dorsal spinal cord in vitro.

(+)-Morphine and (+)-methadone induce significant increases in proinflammatory cytokine and chemokine release were quantified in the supernatants from lumbar dorsal spinal cord sections incubated for 180 min in vitro with 100 µM of each drug compared to media alone (* P < 0.05). n=6 per treatment.
Experiment 2: Non-classical opioid-induced proinflammation is induced in lumbar dorsal spinal cord by chronic exposure to (+)-opioids in vivo: enhancement of nociceptive responses, glial activation, and elevations of proinflammatory cytokine mRNA and protein expression by intrathecal (+)-methadone
Our previous data demonstrate that intrathecal administration of the natural (−)-isomers of morphine or methadone (15 µg) for seven days caused significant glial activation and elevated proinflammatory cytokine and chemokine production (Hutchinson et al., 2008a). Combining those findings with the results with (+)-opioid isomers in Experiment 1 predicts, given the similarity in changes in responses to (+)- and (−)-opioid isomers, that similar proinflammatory cytokine-driven behavioral responses would be expected following chronic (+)-methadone administration as previously observed with (−)-opioids. Specifically, we have previously demonstrated that (−)-opioid-induced glial activation was associated with progressive loss of analgesic efficacy (tolerance) and the progressive development of exaggerated nociception (hyperalgesia and allodynia). Given that (+)-methadone lacks significant opioid receptor binding affinity (Codd et al., 1995, Kristensen et al., 1995), we would not expect to detect any analgesia, but rather only enhanced nociceptive responses.
In all experiments, both hindpaw withdrawal and tailflick data were collected and were consistent in the results found. For simplicity, only tailflick data are shown. Animals were chronically implanted with intrathecal catheters and received once daily intrathecal (+)-methadone (15 µg) or saline, with behavioral testing to determine the nociceptive responsiveness to short baseline latency radiant heat stimuli (Figure 2A; Hargreaves test for analgesia), long baseline latency radiant heat stimuli (Figure 2B; Hargreaves test for hyperalgesia) and low threshold mechanical stimuli (Figure 2C; von Frey test for allodynia). (+)-Methadone produced no analgesia on the first administration confirming its lack of opioid receptor activity (P > 0.05; Figure 2A and B). Chronic intrathecal administration of (+)-methadone resulted in the development of significant hyperalgesia by the seventh day that could be detected using both short and long baseline latency stimuli on the Hargreaves test (P < 0.05; Figure 2A and B). A corresponding progressive development of significant allodynia by day 4 and maintained at day 7 was also quantified using the von Frey test (P < 0.001; Figure 2C).
Figure 2. Chronic intrathecal administration of opioid-inactive (+)-methadone causes significant hyperalgesia and allodynia and associated glial activation and elevated proinflammatory mediator transcription and translation.

Animals were administered once daily 15 µg (+)-methadone or saline (in 1 µl with a 25 µl flush) intrathecally for 7 days via an indwelling catheter. On days 1, 4 and 7 animals were tested for short baseline latency Hargreaves stimuli (A) to assess the presence of any analgesia and long baseline latency Hargreaves stimuli (B) to assess the development of hyperalgesia over a 2 h time course following drug administration. Panels A and B display the area under the analgesia time course on day 1, day 4 and day 7 for (+)-methadone (black bars) and saline (open bars). No significant analgesia resulted from the administration of the opioid inactive (+)-methadone. Instead significant reductions in withdrawal latencies developed across the treatment demonstration hyperalgesia on both short (A) and long (B) baseline latency stimuli Hargreaves tests. Mechanical allodynia (C) was assessed using von Frey filaments 60 min prior and 2 h after administration on day 1 and day 4 and only 60 min prior to drug administration on day 7 for (+)-methadone (⬆) or saline (✦). Tissues from these animals were collected for analysis in Panels D, E and F. D: Relative mRNA expression in lumbar dorsal spinal cord for TLR4, IL-1, IL-6 and TNF-α and IL-1 converting enzyme (ICE) following 7 days intrathecal (+)-methadone (black bar) or saline (open bar) administration. E: CSF protein levels following 7 days intrathecal (+)-methadone (black bar) or saline (open bar) administration. F: Lumbar dorsal spinal cord protein levels following 7 days intrathecal (+)-methadone (black bar) or saline (open bar) administration. * P < 0.05; ** P < 0.01; *** P < 0.001. n = 6 per group
CSF (protein) and dorsal spinal cord tissue (protein and mRNA) cytokine levels were quantified from these same animals following nociceptive testing on day 7. The same panel of analytes was examined as we have previously employed following chronic intrathecal (−)-morphine and (−)-methadone administration (Hutchinson et al., 2008a). Significant elevations in dorsal spinal cord IL-1 mRNA and IL-6 mRNA were observed following chronic (+)-methadone treatment compared to saline vehicle (P < 0.05), whilst TNF-α and IL-1 converting enzyme mRNAs were not significantly different from saline at this timepoint (P > 0.05; Figure 2D). TLR4 mRNA (Figure 2D; P < 0.05) and protein levels (saline: 85 ± 3 % vs (+)-methadone: 102 ± 3 %; P < 0.05) were increased by (+)-methadone administration demonstrating a significant elevation of this glial activation associated marker expression, as microglia and some astrocytes, but not neurons, express TLR4 (Tanga et al., 2005). Analysis of CSF protein demonstrated that (+)-methadone treatment caused significant elevations of chemokines (CX3CL1, GRO/KC, MIP-1α) as well as cytokines (IL-1β, IL-6, TNF-α, and IL-10) (P < 0.05; Figure 2E). Corresponding increases were observed in dorsal spinal cord IL-1β, IL-1α, IL-6, and TNF-α levels in response to (+)-methadone (P < 0.05; Figure 2F). (+)-Methadone treatment did not significantly alter dorsal spinal cord GM-CSF, GRO/KC, IFN-γ, IL-10, IL-2, IL-4, MCP-1, MIP-1α, MIP-2, MIP-3α or RANTES levels, and for CSF the levels of GM-CSF, IFN-γ, IL-1α, IL-2, IL-4, MCP-1, MIP-2, MIP-3α and RANTES remained constant (P > 0.05).
As we have previously demonstrated for proinflammatory opposition of acute (−)-morphine and (−)-methadone analgesia, multiple cytokines and chemokines can work cooperatively to produce a behavioral response (Hutchinson et al., 2008a). Therefore, an analysis that allowed examination of the sum of these factors was required. A cluster analysis, using bootstrapping to increase the power of the data set owing to its degrees of freedom limitations, of all behavioral (delta baseline to day 7), protein and mRNA data was performed to ascertain which molecular endpoints co-varied with each behavior as we have done successfully previously (Hutchinson et al., 2008a). This method of analysis identified six molecular endpoints from the CSF and eight molecular endpoints from the dorsal spinal cord tissue that reliably clustered with nociceptive behaviors from (+)-methadone treated animals. Specifically, in CSF: CX3CL1 (fractalkine), GRO/KC, IL-1β, IL-6, MIP-1α and MIP-2 proteins reliably clustered with behavior. In dorsal spinal cord tissue, IL-1β, IL-1α, IL-2, IL-4, IL-6, IFN-γ proteins and TLR4 (protein and mRNA) clustered significantly with the nociceptive behaviors. The sum of the standardized CSF and dorsal spinal cord chemokine, cytokine and glial activation associated marker values identified significant (+)-methadone treatment effects compared to saline treated animals (P < 0.05). A multivariate analysis with treatment as a fixed factor revealed that when no co-variate was specified there was a significant (+)-methadone treatment effect on each of the nociceptive behaviors when compared to saline control (P < 0.05). However, when the sum of cluster values was specified as a co-variate the treatment effect was no longer significant as expected (P > 0.6). These data suggest that these cytokines and chemokines identified by cluster analysis may be involved as at least partial mediators of the change in the development of allodynia and hyperalgesia following chronic intrathecal (+)-methadone treatment.
Experiment 3: Acute intrathecal (+)-methadone administration induces an IL-1 dependent but masked κ opioid analgesia, which is possibly dependent on microglial and TLR4/MD-2 signaling activation
The results of Experiments 1 and 2, combined with the our previous report that (−)-morphine and (−)-methadone analgesia are each opposed by acute opioid-induced IL-1 action (Hutchinson et al., 2008a), led us to investigate the acute spinal actions of (+)-methadone so to explore whether (+)-methadone may enhance pain via IL-1. The same intrathecal doses and experimental design we have previously employed to demonstrate the presence of the IL-1 pro-nociceptive action in response to (−)-opioids was again utilized here (Hutchinson et al., 2008a). Acute intrathecal administration of 15 µg of (+)-methadone produced no significant analgesia (P > 0.05; Figure 3A) owing to its very low opioid receptor binding affinity (Codd et al., 1995, Kristensen et al., 1995). Intrathecal IL-1ra was administered at 105 min following intrathecal (+)-methadone administration. This time point corresponds to the time of intrathecal IL-1ra administration following dissipation of (−)-methadone analgesia in previous studies; notably, this is a time at which we have previously reported that intrathecal IL-1ra "unmasked" robust ongoing analgesia (Hutchinson et al., 2008a). Therefore, spinal exposure to (+)-methadone prior to intrathecal IL-1ra administration followed the same procedure as we previously used for (−)-methadone and (−)-morphine (Hutchinson et al., 2008a). Intrathecal IL-1ra at 105 min following intrathecal (+)-methadone produced a significant “rebound” analgesia lasting longer than 50 min (P < 0.05; Figure 3A). Importantly, intrathecal vehicle administration following intrathecal (+)-methadone, acute intrathecal saline administration followed by intrathecal IL-1ra or intrathecal vehicle produced no significant change in nociception (Figure 3A).
Figure 3. Intrathecal interleukin-1 receptor antagonist "unmasks" κ opioid analgesia following acute intrathecal (+)-methadone administration which is dependent on microglial and TLR4 activation.

A: Intrathecal injection of 15 µg of (+)-methadone (in 1 µl with a 10 µl flush; ⬆) produces no significant analgesia (5 min to 95 min compared to vehicle treated animals or saline plus IL-1ra treated animals; O; P > 0.05). However, following intrathecal IL-1ra (100 µg in 1 µl with a 10 µl flush; ⬆) significant analgesia is unmasked (115 min to 165 min; P < 0.05). B: Intrathecal co-administration of 15 µg of (+)-methadone (15 µg (+)-methadone in 1 µl) and IL-1ra (100 µg in 1 µl with 10 µl flush; ⬆) produced no significant analgesia, even following a further intrathecal IL-1ra (100 µg in 1 µl with a 10 µl flush; ⬆). C: Intrathecal co-administration of 15 µg of (+)-methadone (15 µg (+)-methadone in 1 µl) and minocycline (a microglial activation attenuator; 100 µg in 3 µl at time of catheter implant and then 33.3 µg in 1 and with 10 µl flush at time 0; ⬆) produced no significant analgesia, even following a further intrathecal IL-1ra (100 µg in 1 µl with a 10 µl flush; ⬆). D: Intrathecal co-administration of 15 µg of (+)-methadone (15 µg (+)-methadone in 1 µl) and (+)-naloxone (a novel TLR4 signaling inhibitor; 60 µg in 1 µl at with 10 µl flush; ⬆) produced no significant analgesia, even following a further intrathecal IL-1ra (100 µg in 1 µl with a 10 µl flush; ⬆). E: Intrathecal injection of 15 µg of (+)-methadone (in 1 µl with a 10 µl flush; ⬆) produces no significant analgesia. Following intrathecal co-administration of IL-1ra and (−)-naloxone (a non-specific opioid receptor antagonist; 100 µg IL-1ra in 1 µl and 20 µg (−)-naloxone in 1 µl with a 10 µl flush; ⬆) no further analgesia was unmasked. F: Intrathecal injection of 15 µg of (+)-methadone (in 1 µl with a 10 µl flush; ⬆) produces no significant analgesia. Following intrathecal co-administration of IL-1ra and naloxonazine (a specific µ1 opioid receptor antagonist; 100 µg IL-1ra in 1 µl and 5 µg naloxonazine in 1 µl with a 10 µl flush; ⬆) significant analgesia is unmasked (115 min to 165 min; P < 0.05). G: Intrathecal injection of 15 µg of (+)-methadone (in 1 µl with a 10 µl flush; ⬆) produces no significant analgesia. Following intrathecal co-administration of IL-1ra and naltrindole (a specific δ opioid receptor antagonist; 100 µg IL-1ra in 1 µl and 5 µg naltrindole in 1 µl with a 10 µl flush; ⬆) significant analgesia is unmasked (115 min to 165 min; P < 0.05). F: Intrathecal injection of 15 µg of (+)-methadone (in 1 µl with a 10 µl flush; ⬆) produces no significant analgesia. Following intrathecal co-administration of IL-1ra and norBNI (a specific κ opioid receptor antagonist; 100 µg IL-1ra in 1 µl and 5 µg norBNI in 1 µl with a 10 µl flush; ⬆) no significant analgesia is unmasked. n = 6 per group.
One possibility is that the (+)-methadone-induced IL-1 response was responsible for the parallel induction of the unmasked analgesia. In order to determine the origins and potential mechanism of this unmasked analgesia, IL-1ra was co-administered intrathecally with (+)-methadone at time 0, which produced no significant change in nociception suggesting that some duration of (+)-methadone exposure alone was required to recruit the unmasked analgesia (P > 0.05; Figure 3B). Interestingly, intrathecal IL-1ra administration 105 min following (+)-methadone plus IL-1ra intrathecal administration corresponding to the intrathecal IL-1ra administration in Figure 3A failed to produce any rebound analgesic response (P > 0.05). The failure of intrathecal IL-1ra to produce any rebound analgesia suggests that (+)-methadone-induced IL-1 action during the first 100 minutes of the exposure is required for the IL-1ra rebound analgesia to be produced (Figure 3B). The cellular mechanisms behind the (+)-methadone-induced IL-1 rebound analgesia were also examined using the putative microglial inhibitor minocycline (Figure 3C) and the novel TLR4/MD-2 signaling inhibitor (+)-naloxone (Figure 3D). Minocycline co-administration with (+)-methadone produced no analgesia (P > 0.05). Prior minocycline treatment blocked subsequent IL-1ra unmasked analgesia (P > 0.05; Figure 3C). (+)-Naloxone co-administration with (+)-methadone produced no analgesia (P > 0.05). Prior (+)-naloxone treatment blocked subsequent IL-1ra unmasked analgesia (P > 0.05; Figure 3D).
The opioid nature of the rebound analgesia observed in Figure 3A was examined by intrathecal co-administering of the opioid receptor antagonist (−)-naloxone (20 µg) with IL-1ra 105 min following intrathecal (+)-methadone (Figure 3E). Intrathecal (−)-naloxone significantly antagonized the IL-1ra rebound analgesia (P < 0.05; Figure 3E). In order to further characterize the opioid nature of this response, further selective opioid receptor antagonist studies were conducted. Intrathecal co-administration of the µ1 opioid receptor antagonist naloxonazine (5 µg; Watanabe et al. (2005)) with IL-1ra 105 min following (+)-methadone administration failed to significantly antagonize the IL-1ra rebound analgesia (P > 0.05; Figure 3F). Interestingly, intrathecal co-administration of the δ opioid receptor antagonist naltrindole (5 µg; Watanabe et al. (2005)) in the same fashion as naloxonazine was also unable to antagonize the IL-1ra rebound analgesia (P > 0.05; Figure 3G). However, intrathecal co-administration of the κ opioid receptor antagonist nor-BNI (5 µg; Watanabe et al. (2005)) with IL-1ra 105 min following (+)-methadone administration did significantly antagonize the IL-1ra rebound analgesia (P < 0.05; Figure 3H), suggesting a role of an endogenous κ opioid receptor signal in the TLR4- and microglial-dependent IL-1ra unmasked rebound analgesia.
Taken together, the data from Experiment 3 do not support that (+)-methadone is binding to kappa opioid receptors directly but rather that the proximal effect of (+)-methadone is to activate glia (via TLR4) and that this (+)-methadone-induced glial activation causes the release of IL-1. It is the release of IL-1 that appears to induce the downstream release of an endogenous kappa opioid. That conclusion is based on: (a) prevention of a kappa opioid analgesia by the TLR4 antagonist (+)-naloxone, (b) prevention of a kappa opioid analgesia by the glial inhibitor minocycline, (c) prevention of a kappa opioid analgesia by IL-1 receptor antagonist when co-administered with (+)-methadone (i.e., to prevent IL-1 from stimulating the release of an endogenous kappa opioid). These data do not support methadone binding kappa receptors since none of the manipulations above would have prevented kappa opioid analgesia if (+)-methadone binding to kappa receptors were to occur.
Experiment 4: Acute intrathecal (+)-morphine administration induces IL-1 dependent hyperalgesia, which is possibly dependent on microglial and TLR4/MD-2 signaling activation
In order to assess the behavioral specificity of this non-classical opioid response, we also examined the action of the opioid inactive isomer of morphine, which is structurally disparate to that of (+)-methadone. (+)-Morphine possesses even less opioid activity than the minimal amount that (+)-methadone still retains (Codd et al., 1995). Preliminary behavioral studies demonstrated that (+)-morphine did not display the same characteristics as (+)-methadone in two important areas. Firstly, using the short latency Hargreaves test sensitive to analgesia, (+)-morphine actually induced a decrease in withdrawal latencies with no detectable effect of subsequent IL-1ra administration. Therefore, the behavioral testing following (+)-morphine administration was changed to the long Hargreaves latency test to allow reliable quantification of reductions in nociceptive thresholds.
Intrathecal administration (+)-morphine caused significant hyperalgesia which was reversible by IL-1ra administration (Figure 4B) but not by vehicle (Figure 4A). Co-administration of (+)-morphine with IL-1ra blocked the development of hyperalgesia (Figure 4C) demonstrating the significant contribution of IL-1 to the hyperalgesia. Co-administration of (+)-morphine with minocycline also attenuated the development of hyperalgesia demonstrating the likely need for microglial cell activation (Figure 4D). Finally, co-administration of (+)-naloxone with (+)-morphine significantly blocked the (+)-morphine hyperalgesia (Figure 4E), thereby implicating TLR4/MD-2 in the process.
Figure 4. Intrathecal (+)-morphine induces interleukin-1 induced hyperalgesia which is dependent on microglial and TLR4 activation.

A: Intrathecal injection of 15 µg of (+)-morphine (in 1 µl with a 10 µl flush; ■) produces significant hyperalgesia (15 min to 55 min compared to vehicle treated animals O or saline plus IL-1ra treated animals; P > 0.05). This hyperalgesia is not affected by subsequent intrathecal saline administration (65 min to 125 min; P < 0.05). B: Intrathecal injection of 15 µg of (+)-morphine (in 1 µl with a 10 µl flush; ■) produces significant hyperalgesia (25 min to 55 min compared to vehicle treated animals O or saline plus IL-1ra treated animals; P > 0.05). However, following intrathecal IL-1ra (100 µg in 1 µl with a 10 µl flush; ■) significant reversal of the hyperalgesia occurs (65 min to 125 min; P > 0.05). C: Intrathecal co-administration of 15 µg of (+)-morphine (15 µg (+)-morphine in 1 µl) and IL-1ra (100 µg in 1 µl with 10 µl flush; ■) produced no significant hyperalgesia, with no significant effect of a further intrathecal IL-1ra (100 µg in 1 µl with a 10 µl flush; ■). D: Intrathecal co-administration of 15 µg of (+)-morphine (15 µg (+)-morphine in 1 µl) and minocycline (a microglial activation attenuator; 100 µg in 3 µl at time of catheter implant and then 33.3 µg in 1 and with 10 µl flush at time 0; ■) produced no significant hyperalgesia, with no significant effect following a further intrathecal IL-1ra (100 µg in 1 µl with a 10 µl flush; ■). E: Intrathecal co-administration of 15 µg of (+)-morphine (15 µg (+)-morphine in 1 µl) and (+)-naloxone (a novel TLR4 signaling inhibitor; 60 µg 1 µl and with 10 µl flush; ■) produced no significant hyperalgesia, with no significant effect following a further intrathecal IL-1ra (100 µg in 1 µl with a 10 µl flush; ■). n = 6 per group
These data distinguish the effects of (+)-morphine and (+)-methadone (see Experiment 3, above) in that only (+)-methadone induces the release of a kappa opioid, that initially offsets the pain-enhancing effects of (+)-methadone-induced IL-1 release. As hyperalgesia and allodynia do develop in response to repeated (+)-methadone, the pain enhancing effects of (+)-methadone can overcome the pain suppressive effects of the kappa opioid over time. In contrast, (+)-morphine, for currently unknown reasons, does not induce the release of a kappa opioid so that only pain enhancement is observed.
Experiment 5: (+)-Methadone and (+)-morphine dock in silico to human MD-2 in a (+)-naloxone sensitive fashion
The (+)-naloxone sensitivity of the (+)-methadone and (+)-morphine-induced behavioural responses in Experiments 3 and 4 indicate the likely involvement of TLR4/MD-2 signalling in the response, owing to the established TLR4/MD-2 signalling inhibitory actions of (+)-naloxone (Hutchinson et al., 2008c, Hutchinson et al., 2009). To assess this further, and to extend our prior in silico modelling of opioid interactions with TLR4/MD-2, the in silico docking of (+)-methadone and (+)-morphine to human TLR4 and MD-2 was assessed in the presence and absence of (+)-naloxone. In silico modelling was done to explore whether this approach would provide further evidence for the in vivo action of (+)-naloxone blocking (+)-morphine and (+)-methadone induced pronociceptive behavior. Both (+)-methadone and (+)-morphine docked preferentially to the LPS binding domain of MD-2 (Figure 5A and B). The prior in silico binding of the optimal conformation of (+)-naloxone to MD-2 substantially modified the docking of (+)-methadone and (+)-morphine, but in two different ways. The preferred docking conformation for (+)-methadone overlapped that of (+)-naloxone (Figure 5C and E) and as such competitively blocked the docking of (+)-methadone to the original conformation substantially modifying the residues it interacted with. In contrast, the preferred docking conformations of (+)-naloxone and (+)-morphine did not over lap (Figure 5D and F). However, the presence of the MD-2 docked (+)-naloxone caused a substantial change in (+)-morphine binding and sequestered it away from its original conformation, substantially modifying the residues it interacted with, likely modifying its TLR4/MD-2 activation potential.
Figure 5. (+)-Methadone and (+)-morphine dock in silico to human MD-2 in a (+)-naloxone sensitive fashion.
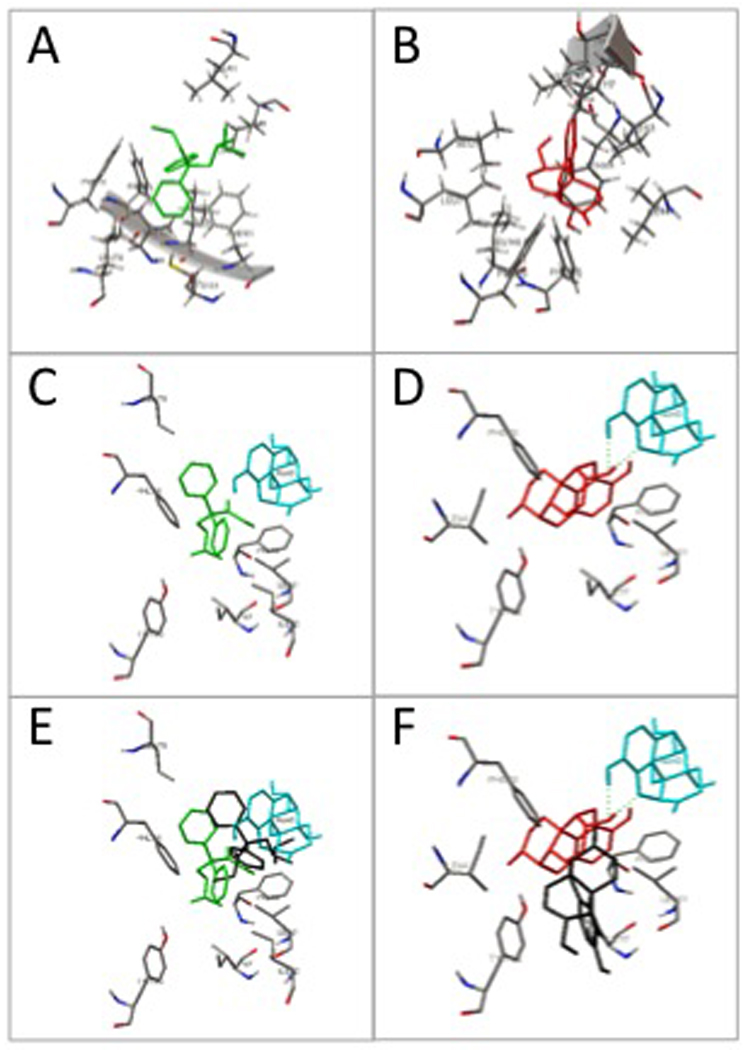
The optimal docking conformations for (+)-methadone (panel A; green) and (+)-morphine (panel B; red) alone are shown following docking in silico to human MD-2 with the MD-2 interaction residues displayed. The prior in silico docking of (+)-naloxone (turquoise) to MD-2 substantially modified (+)-methadone (panel C; green) and (+)-morphine (panel D; red) preferred docking conformation when compared to their docking conformations alone (panels E and F, conformations of docking to MD-2 alone shown in black).
Discussion
The studies presented here support and extend our prior publications (Hutchinson et al., 2008a, Hutchinson et al., 2009) in demonstrating the behavioral and proinflammatory consequences of non-stereoselective opioid activation of TLR4/MD-2 signalling (owing to the [+]-naloxone-sensitivity of the responses). The use of the (+)-isomers of morphine and methadone enabled examination of the proinflammatory actions of opioids on at minimum microglia (owing to minocycline-sensitivity of the responses) in the absence of neuronal µ opioid receptor involvement (owing to the stereoselectivity of neuronal opioid receptors). In addition, the use of these (+)-opioid isomers revealed that this non-classical action of opioids drives clinically relevant negative consequences of opioid administration. Supporting the conclusion that the TLR4/MD-2 effects are involved is the fact that the unnatural (+)-opioids induce the upregulation of a glial activation associated marker, upregulate the production and release of proinflammatory chemokines and cytokines, and enhance nociception in a (+)-naloxone-sensitive fashion. These data demonstrate the action of (+)-methadone and (+)-morphine in producing such effects despite the fact that they are functionally inactive at classical opioid receptors due to their limited opioid receptor binding affinity (Jacquet et al., 1977, Codd et al., 1995, Kristensen et al., 1995). Acute in vitro exposure of lumbar dorsal spinal cord sections to (+)-methadone and (+)-morphine resulted in reliable elevations in proinflammatory cytokine and chemokine release, in addition to acute elevations in the glial activation associated marker TLR4 (Tanga et al., 2004). Chronic intrathecal (+)-methadone administration produced robust hyperalgesia and allodynia that was associated with significant elevations of several chemokines and cytokines as well as TLR4 in dorsal lumbar spinal cord and release of these chemokines and cytokines into surrounding CSF. Mediation statistical analysis (Baron and Kenny, 1986) demonstrated that the sum of these proinflammatory changes was related to the changes in nociception following chronic (+)-methadone administration. The anti-inflammatory cytokine IL-10 was also elevated but apparently not sufficiently to overcome the cumulative proinflammation of the other cytokines and chemokines. Acute intrathecal (+)-methadone administration also produced enhanced IL-1, microglial and TLR4/MD-2 dependent actions. Specifically, intrathecal IL-1ra administration following intrathecal (+)-methadone produced an endogenous κ opioid-mediated rebound analgesia that was dependent on prior (+)-methadone-induced IL-1 action in a minocycline- and (+)-naloxone-sensitive fashion. That is, if IL-1 receptors were blocked, microglial activation was attenuated, or the proinflammatory of TLR4/MD-2 signaling inhibited, throughout (+)-methadone exposure, no rebound analgesia occurred. Interestingly, (+)-methadone and (+)-morphine did differ in their behavioral responses. (+)-Morphine induced hyperalgesia in an IL-1ra, minocycline- and (+)-naloxone-sensitive fashion, without any rebound analgesia, suggesting there are differences between the downstream consequences of these two compounds following intrathecal administration. While not pursued in the present paper, the robust changes in chemokines also noted in response to (+)-opioids are the subject of ongoing investigation.
In silico TLR4/MD-2 docking simulation results are important supporting data for the (+)-naloxone behavioral response sensitivity observed in Experiments 3 and 4. We have previously published that opioids non-stereoselectively activate human TLR4 receptor signaling in vitro and that (+)-naloxone is capable of inhibiting this signaling (Hutchinson et al., 2009). In support of these in vitro data both (+)-methadone and (+)-morphine docked in silico preferentially to the LPS binding domain of MD-2 (Park et al., 2009). Importantly, the presence of (+)-naloxone in its preferred docking conformation in the same domain caused substantial modification of the docking conformations, but in two distinct ways. Firstly, (+)-methadone docking was modified by the presence of (+)-naloxone owing to the spatial overlapping docking conformations. In contrast, (+)-morphine and (+)-naloxone docking conformations did not overlap, yet the preferred (+)-morphine docking conformation in the presence of (+)-naloxone was substantially modified as (+)-naloxone sequestered the (+)-morphine to a different location. Importantly, in both cases the presence of (+)-naloxone caused a significant change in the MD-2 residues that (+)-methadone and (+)-morphine interacted with. Studies are ongoing in an attempt to identify the critical MD-2 residues that are critical to initiating TLR4/MD-2 signaling. Given our previous publications demonstrating morphine and methadone non-stereoselectively activate TLR4/MD-2 signaling in vitro, and (−)-morphine and (−)-methadone induced TLR4/MD-2-dependent CNS proinflammation, one can conclude these small molecules may be non-stereoselectively activating TLR4/MD-2 signaling. However, in silico docking demonstrated that the theoretical docking of the different opioid stereoisomers to the LPS binding pocket of MD-2 is not identical. These multiple binding conformations could be anticipated given the far greater size of the binding domain within MD2 and TLR4 relative to classical neurotransmitter receptors, as well as known “promiscuity” of the ligand recognition and pattern recognition role of TLR4 and MD-2. A similar signaling activation efficacy/potency of different isomers has also been recognized, for example, for a few select ligands for beta-2 adrenergic receptors (e.g., (−) and (+)-terbutaline) and alpha adrenergic receptors (e.g., (−)- and (+)-dobutamine) (Casy, 1993).
The term “opioid-induced” proinflammatory glial activation, when used to imply mediation by classical opioid receptors, cannot be extended to the (+)-isomers of morphine or methadone due to their lack of activity at these receptors. Therefore, this suggests that “xenobiotic-induced” proinflammatory glial activation may be a more appropriate term, as we have highlighted previously (Hutchinson et al., 2009), as this would refer broadly to chemicals found in an organism but which are not endogenous. This recognition that proinflammatory glial activation can occur from a range of structurally disparate exogenous small molecules has precedence in the literature. For example, Narita et al. have demonstrated that methamphetamine induces glial activation (Narita et al., 2006), Wu et al. have also found that (+)-opioids activate glia (Wu et al., 2005), and Fernandez-Lizarbe et al. demonstrated alcohol induced glial activation (Fernandez-Lizarbe et al., 2009). More recently, we have documented that this xenobiotic profile extends to some tricyclic antidepressants (Hutchinson, et al., in review). The activation of TLR4/MD-2 signaling provides a tantalizing potential mechanism linking proinflammatory glial activation by disparate xenobiotics and possible common drug-induced adverse effect profiles. This is especially pertinent in cases where clinically employed pharmacotherapies produce unwanted glial events when they are separate from the primary indication, especially if the glial activation produces unwanted side effects or even diminishes the efficacy of the pharmacotherapy, as is the case for opioids.
The current chronic intrathecal (+)-methadone data compare very favorably with those we have previously published for the (−)-isomers of methadone and morphine (Hutchinson et al., 2008a). In both cases, chronic intrathecal administration of either the classical opioid receptor active (−)-isomer or the classical opioid receptor inactive (+)-isomer of opioid analgesics produce significant hyperalgesia and allodynia by the seventh day. The change in spinal proinflammatory mediators induced by chronic (+)-methadone versus (−)-morphine and (−)-methadone is also of interest. All three treatments cause significant elevations in the same proinflammatory mRNA and protein endpoints in both dorsal spinal cord and surrounding CSF. These data suggest that these proinflammatory responses to chronic intrathecal administration of these three compounds may be mediated via the same mechanism, with similar efficacy, despite the non-stereoselectivity that negates the involvement of classical opioid receptors as a common mediator. Agreeing with this conclusion are the cluster and mediation analyses of the (+)-methadone data. Cluster analysis of the chronic intrathecal (+)-methadone data set identified similar molecular endpoints correlated with behavioral changes as was found for (−)-methadone or (−)-morphine cluster analysis (Hutchinson et al., 2008a). These three different compounds, with disparate pharmacology, have now been shown to produce a very similar pattern of proinflammatory changes and behavioral consequences providing further evidence suggestive that the molecular endpoints identified here may prove to be pivotal to the behavioral changes. Furthermore, these data provide more evidence to the growing literature of the non-classical opioid activation of glia (Wu et al., 2005, Wu et al., 2006a, Wu et al., 2006b).
The rats in the chronic administration studies received once daily intrathecal (+)-methadone, hence "mini-withdrawals" between successive drug doses would have occurred as was originally proposed. It has previously been suggested that the “mini-withdrawals” experienced in the inter-dosing interval may play an important role in exacerbating the negative nociceptive outcomes during chronic opioid administration (Gutstein, 1996). It has been assumed that these "mini-withdrawals" were occurring in neurons, rather than non-neuronal cells. However, in the present study, these "mini-withdrawals" would not have been mediated via neuronal opioid receptors owing to the lack of (+)-methadone opioid receptor binding affinity. Therefore, this implicates the possible action TLR4/MD-2 in producing the behavioral consequences of these “mini-withdrawals”, rather than classical opioid receptors, as has been previously assumed (Gutstein, 1996).
We have previously observed that endogenous IL-1 opposes acute intrathecal (−)-morphine analgesia within 5 minutes of (−)-morphine administration. While the rapidity of the (+)-methadone or (+)-morphine-induced IL-1 actions was not addressed in of the present experiments, we have demonstrated that even the actions of the opioid inactive isomer of methadone and morphine are modulated by endogenous IL-1. As we have previously reported for (−)-opioid isomers (Hutchinson et al., 2008a), the effects of IL-1 were again unmasked here by IL-1ra administration. For (+)-methadone, IL-1ra unmasked a norBNI-sensitive κ opioid receptor-mediated rebound analgesia. For (+)-morphine, IL1-ra administration confirmed the role of IL-1 in mediating the hyperalgesia. Interestingly, for both (+)-methadone and (+)- morphine, the responses were dependent on the action of endogenous IL-1 following intrathecal administration since co-administration of IL-1ra attenuated the subsequent behaviors. Moreover, the effects intrathecal minocycline and (+)-naloxone suggest that microglia and TLR4/MD-2 signaling may be important in the IL-1 response. The behavioral differences following (+)-morphine and (+)-methadone demonstrate that whilst glial responses are involved in the neurochemical responses they may not be the only events occurring. Two behavioral differences were of note. Firstly, (+)-morphine induced acute hyperalgesia, whilst (+)-methadone did not. Secondly, IL-1ra administration following (+)-methadone administration resulted in rebound analgesia whilst it did not for (+)-morphine. Since in vitro studies demonstrated the same profile of proinflammation from dorsal spinal cord sections, these behavioral differences cannot be explained purely by this cytokine and chemokine response. (+)-Methadone has very different pharmacology compared to (+)-morphine such as displaying NMDA receptor antagonistic activity (Codd et al., 1995), thereby implicating other systems working in parallel that are interdependent to produce this endogenous analgesia. The physiological significance of this κ opioid receptor-mediated rebound analgesia is unclear at present.
These data support our previous work (Hutchinson et al., 2009) demonstrating the possible significant behavioral consequences of xenobiotic TLR4/MD-2 signaling activation. These data provide further evidence for the previously masked parallel proinflammatory events that occur following administration of classical (−)-isomer opioid analgesics resulting in the opposition of the beneficial classical neuronal opioid receptor analgesia. Moreover, we provide data supporting that it is likely possible to attenuate the detrimental TLR4/MD-2 signaling activation. Further work is required to fully understand the interplay between IL-1 and endogenous κ opioid systems as this system may play a role in regulating basal nociception under some situations.
Acknowledgements
International Association for the Study of Pain International Collaborative grant, American Australian Association Merck Company Foundation Fellowship, National Health and Medical Research Council CJ Martin Fellowship (ID 465423) and NIH Grants DA015642, DA017670, DA024044 and DE017782. This work was partially supported by the by the NIH Intramural Research Programs of the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism. We thank Avigen (Alameda, CA, USA) for the gift of the HEK293-TLR4 cell line.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baron RM, Kenny DA. The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J Pers Soc Psychol. 1986;51:1173–1182. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- Carmody J. Avoiding fallacies in nociceptive measurements. Pain. 1995;63:136. doi: 10.1016/0304-3959(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Chao CC, Gekker G, Sheng WS, Hu S, Tsang M, Peterson PK. Priming effect of morphine on the production of tumor necrosis factor-alpha by microglia: implications in respiratory burst activity and human immunodeficiency virus-1 expression. J Pharmacol Exp Ther. 1994;269:198–203. [PubMed] [Google Scholar]
- Casy AF. The Steric Factor in Medicinal Chemistry: Dissymmetric Probes of Pharmacological Receptors. Oxford: Plenum Press; 1993. [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Codd EE, Shank RP, Schupsky JJ, Raffa RB. Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther. 1995;274:1263–1270. [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Dobrenis K, Makman MH, Stefano GB. Occurrence of the opiate alkaloid-selective mu3 receptor in mammalian microglia, astrocytes and Kupffer cells. Brain Res. 1995;686:239–248. doi: 10.1016/0006-8993(95)00452-v. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Pascual M, Guerri C. Critical Role of TLR4 Response in the Activation of Microglia Induced by Ethanol. J Immunol. 2009;183:4733–4744. doi: 10.4049/jimmunol.0803590. [DOI] [PubMed] [Google Scholar]
- Gutstein HB. The effects of pain on opioid tolerance: how do we resolve the controversy? Pharmacol Rev. 1996;48:403–407. discussion 409–411. [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Harvey LO. Efficient estimation of sensory thresholds. Behav Res Methods Instr Comp. 1986;18:623–632. [Google Scholar]
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. ScientificWorldJournal. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N, Baker EM, Jekich BM, Wieseler JL, Somogyi AA, Martin D, Poole S, Judd CM, Maier SF, Watkins LR. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun. 2008a;22(8):1178–1789. doi: 10.1016/j.bbi.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Northcutt AL, Chao LW, Kearney JJ, Zhang Y, Berkelhammer DL, Loram LC, Rozeske RR, Bland ST, Maier SF, Gleeson TT, Watkins LR. Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun. 2008b;22(8):1248–1256. doi: 10.1016/j.bbi.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008c;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, Watkins LR. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. 2009;24(1):83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquet YF, Klee WA, Rice KC, Iijima I, Minamikawa J. Stereospecific and nonstereospecific effects of (+)- and (−)-morphine: evidence for a new class of receptors? Science. 1977;198:842–845. doi: 10.1126/science.199942. [DOI] [PubMed] [Google Scholar]
- Johnston IN, Milligan ED, Wieseler-Frank J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P, Fleshner M, Leinwand L, Maier SF, Watkins LR. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J Neurosci. 2004;24:7353–7365. doi: 10.1523/JNEUROSCI.1850-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen K, Christensen CB, Christrup LL. The mu1, mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1995;56:PL45–PL50. doi: 10.1016/0024-3205(94)00426-s. [DOI] [PubMed] [Google Scholar]
- Lipovsky MM, Gekker G, Hu S, Hoepelman AI, Peterson PK. Morphine enhances complement receptor-mediated phagocytosis of Cryptococcus neoformans by human microglia. Clin Immunol Immunopathol. 1998;87:163–167. doi: 10.1006/clin.1998.4518. [DOI] [PubMed] [Google Scholar]
- Magazine HI, Liu Y, Bilfinger TV, Fricchione GL, Stefano GB. Morphine-induced conformational changes in human monocytes, granulocytes, and endothelial cells and in invertebrate immunocytes and microglia are mediated by nitric oxide. J Immunol. 1996;156:4845–4850. [PubMed] [Google Scholar]
- Milligan ED, Hinde JL, Mehmert KK, Maier SF, Watkins LR. A method for increasing the viability of the external portion of lumbar catheters placed in the spinal subarachnoid space of rats. J Neurosci Methods. 1999;90:81–86. doi: 10.1016/s0165-0270(99)00075-8. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Mehmert KK, Hinde JL, Harvey LO, Martin D, Tracey KJ, Maier SF, Watkins LR. Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res. 2000;861:105–116. doi: 10.1016/s0006-8993(00)02050-3. [DOI] [PubMed] [Google Scholar]
- Narita M, Miyatake M, Narita M, Shibasaki M, Shindo K, Nakamura A, Kuzumaki N, Nagumo Y, Suzuki T. Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology. 2006;31:2476–2488. doi: 10.1038/sj.npp.1301007. [DOI] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Molitor TW, Chao CC. The opioid-cytokine connection. J Neuroimmunol. 1998;83:63–69. doi: 10.1016/s0165-5728(97)00222-1. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2006. [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Shavit Y, Wolf G, Goshen I, Livshits D, Yirmiya R. Interleukin-1 antagonizes morphine analgesia and underlies morphine tolerance. Pain. 2005;115:50–59. doi: 10.1016/j.pain.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Stefano GB. Autoimmunovascular regulation: morphine and anandamide and ancodamide stimulated nitric oxide release. J Neuroimmuno. 1998;83:70–76. doi: 10.1016/s0165-5728(97)00223-3. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- Takayama N, Ueda H. Morphine-induced chemotaxis and brain-derived neurotrophic factor expression in microglia. J Neurosci. 2005;25:430–435. doi: 10.1523/JNEUROSCI.3170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int. 2004;45:397–407. doi: 10.1016/j.neuint.2003.06.002. [DOI] [PubMed] [Google Scholar]
- Treutwein B, Strasburger H. Fitting the psychometric function. Percept Psychophys. 1999;61:87–106. doi: 10.3758/bf03211951. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Nakayama D, Ito K, Watanabe C, Mizoguchi H, Fujimura T, Murayama K, Kawamura S, Sato T, Sakurada C, Sakurada T, Sakurada S. A Tyr-W-MIF-1 analog containing D-Pro2 acts as a selective mu2-opioid receptor antagonist in the mouse. J Pharmacol Exp Ther. 2005;312:1075–1081. doi: 10.1124/jpet.104.075697. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF. The "toll" of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci. 2009;30:581–591. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HE, Sun HS, Cheng CW, Tseng LF. p38 Mitogen-activated protein kinase inhibitor SB203580 reverses the antianalgesia induced by dextro-morphine or morphine in the mouse spinal cord. Eur J Pharmacol. 2006a;550:91–94. doi: 10.1016/j.ejphar.2006.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HE, Sun HS, Terashivili M, Schwasinger E, Sora I, Hall FS, Uhl GR, Tseng LF. dextro-and levo-morphine attenuate opioid delta and kappa receptor agonist produced analgesia in mu-opioid receptor knockout mice. Eur J Pharmacol. 2006b;531:103–107. doi: 10.1016/j.ejphar.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Wu HE, Thompson J, Sun HS, Terashvili M, Tseng LF. Antianalgesia: stereoselective action of dextro-morphine over levo-morphine on glia in the mouse spinal cord. J Pharmacol Exp Ther. 2005;314:1101–1108. doi: 10.1124/jpet.105.087130. [DOI] [PubMed] [Google Scholar]
- Wybran J, Appelboom T, Famaey JP, Govaerts A. Suggestive evidence for receptors for morphine and methionine-enkephalin on normal human blood T lymphocytes. Journal of Immunology. 1979;123:1068–1070. [PubMed] [Google Scholar]