

Abstract
Rationale: Idiopathic pulmonary fibrosis (IPF) and other idiopathic interstitial pneumonias (IIPs) have similar clinical and radiographic features, but their histopathology, response to therapy, and natural history differ. A surgical lung biopsy is often required to distinguish between these entities.
Objectives: We sought to determine if clinical variables could predict a histopathologic diagnosis of IPF in patients without honeycomb change on high-resolution computed tomography (HRCT).
Methods: Data from 97 patients with biopsy-proven IPF and 38 patients with other IIPs were examined. Logistic regression models were built to identify the clinical variables that predict histopathologic diagnosis of IPF.
Measurements and Main Results: Increasing age and average total HRCT interstitial score on HRCT scan of the chest may predict a biopsy confirmation of IPF. Sex, pulmonary function, presence of desaturation, or distance walked during a 6-minute walk test did not help discriminate pulmonary fibrosis from other IIPs.
Conclusions: Clinical data may be used to predict a diagnosis of IPF over other IIPs. Validation of these data with a prospective study is needed.
Keywords: idiopathic pulmonary fibrosis, idiopathic interstitial pneumonia, diagnosis, computed tomography of the chest
AT A GLANCE COMMENTARY.
Scientific Knowledge on the Subject
Idiopathic interstitial pneumonias (IIPs), including idiopathic pulmonary fibrosis (IPF), share many clinical and radiographic features. However, their histopathology, response to therapy, and natural history differ. A surgical lung biopsy is often required to differentiate early IPF from other non-IPF IIPs.
What This Study Adds to the Field
Age and total HRCT interstitial score on an HRCT scan of the chest may predict a diagnosis of biopsy-confirmed IPF over non-IPF IIPs. This study provides clinical variables that may be used to predict IPF over non-IPF IIPs, thus saving some patients from a surgical lung biopsy.
Idiopathic interstitial pneumonias (IIPs), including idiopathic pulmonary fibrosis (IPF), are characterized by chronic dyspnea, interstitial infiltrates on radiographs, reduced lung volumes, and impaired diffusion capacity of the lung (DlCO) (1). There is considerable overlap in the clinical features (2) of the IIPs, although the histopathologic features differ (1). An accurate diagnosis is critical as the response to treatment and prognosis differs between IIPs, with IPF having the worst prognosis (3–5). Difficulty in diagnostic separation between these entities has been emphasized by several groups (6–8), highlighting the need to optimize the diagnostic evaluation.
The diagnosis of IPF is confirmed when appropriate clinical and radiographic features are seen in combination with a surgical lung biopsy showing a pattern of usual interstitial pneumonia (UIP). In an appropriate clinical setting, a diagnosis of IPF can be made without a surgical lung biopsy when definitive high-resolution computed tomography (HRCT) scan features are present (8–10). Definitive features of UIP on HRCT include subpleural reticulation with a basal predominance and honeycombing with an absence of extensive ground glass abnormality, micronodules, discrete cysts, mosaic attenuation/air trapping, or consolidation (8–11). Unfortunately, many patients do not have definitive radiographic features of UIP, leaving a wide differential diagnosis and the requirement for a surgical lung biopsy. IPF is ultimately diagnosed in a large proportion of these patients based on the histopathologic finding of UIP (3). Unfortunately, many patients who present with fibrotic lung disease are elderly and have important comorbidities that increase the risk of a surgical lung biopsy (12–15). We hypothesized that a combination of clinical and radiological features could be used to accurately predict a diagnosis of IPF confirmed by surgical lung biopsy. Some of the results of these studies have been previously reported in the form of an abstract (16, 17).
METHODS
This is a retrospective study of 135 patients with biopsy-proven IPF/UIP (n = 97) or other IIPs (n = 38) from our database who did not have honeycomb change on HRCT. A total of 644 patients with evidence of fibrotic lung disease were enrolled sequentially into protocols investigating the etiology, diagnosis, and treatment of fibrotic lung disease from 1995 to 2006 (Figure 1). Patients without a surgical lung biopsy or in whom there was evidence of underlying connective tissue disease (ie, clinical diagnosis and/or specific serology and/or clear symptoms of connective tissue disease) or other cause of interstitial disease were excluded. Data were extracted for those patients with HRCT, pulmonary function testing (PFT), and 6-minute walk test (6MWT) within 6 months of the lung biopsy. Patients with evidence of pulmonary fibrosis but without honeycombing on HRCT (HRCT interstitial score <2) were then selected for data analysis (see below). A final diagnosis was established using a clinical/radiological/pathological diagnostic schema that we have previously described (18).
Figure 1.

CONSORT diagram illustrating the selection of patients included in the data analysis. CTD-ILD = connective tissue disease–related interstitial lung disease; HRCT = high-resolution computed tomography; IIP = idiopathic interstitial pneumonia; IPF = idiopathic pulmonary fibrosis; PFT = pulmonary function tests; 6MWT = 6-minute walk test.
HRCT scans were scored by two expert thoracic radiologists using a semiquantitative scale that assesses the degree of ground glass attenuation (HRCT alveolar score) and fibrotic change (HRCT interstitial score) (19) (Table 1 and Figure 2). Radiologists were blinded to the diagnosis. Agreement between the radiologists was very good (kappa = 0.727; 95% confidence interval [CI], 0.641–0.813). It is important to recognize that an HRCT interstitial score of less than 2 in all lobes identifies patients without any honeycomb change on HRCT. Pulmonary function studies and 6MWT were performed as described (20, 21). 6MWT data include the distance walked and whether the patient's oxygen saturation fell below 88% during the test.
TABLE 1.
HIGH-RESOLUTION COMPUTED TOMOGRAPHY SCORING SYSTEM (19)
| Score | Alveolar | Interstitial |
|---|---|---|
| 0 | No alveolar disease | No interstitial disease |
| 1 | Ground glass opacity <5% of lobe | Septal thickening but no honeycomb |
| 2 | Ground glass opacity <25% of lobe | Honeycomb change <25% of lobe |
| 3 | Ground glass opacity 25–49% of lobe | Honeycomb change 25–49% of lobe |
| 4 | Ground glass opacity 50–75% of lobe | Honeycomb change 50–75% of lobe |
| 5 | Ground glass opacity >75% of lobe | Honeycomb change >75% of lobe |
Radiologists score each lobe and then take an average of all the lobes for the final HRCT scores.
Figure 2.

Examples of varied high-resolution computed tomography (HRCT) alveolar and HRCT interstitial scores. Each panel is from a different patient. The scores for each panel are for the image that is displayed. Scores are represented as alveolar/interstitial. LLL = left lower lobe; LUL = left upper lobe; RLL = right lower lobe; RUL = right upper lobe.
Baseline demographic data, pulmonary function, 6MWT, and HRCT scores were compared between patients with IPF and non-IPF IIP using the Student t test for continuous variables (22) and the chi-square test for categorical variables (23), except where indicated. Univariate and multivariate logistic models were constructed to determine which variables predict a diagnosis of IPF. The quality of the prediction models was evaluated using receiver operating characteristics (ROC) analysis with the area under the curve calculated (24). The positive predictive values (PPVs), specificities, sensitivities, and negative predictive values (NPVs) for new patients with different risk profiles were examined based on the final model. Statistical analysis was performed with R 2.8.0 software (http://www.r-project.org/).
RESULTS
We examined data from 97 patients with biopsy-proven IPF and 38 patients with other IIPs (nonspecific interstitial pneumonia, 19; hypersensitivity pneumonia, 9; respiratory bronchiolitis interstitial lung disease/desquamative interstitial pneumonia, 9; and cryptogenic organizing pneumonia, 1). Patients with IPF were significantly older, marginally more likely to be female, and had similar smoking histories. They had more fibrosis and less ground glass on HRCT. Pulmonary function and 6MWT results were similar between the groups (Table 2).
TABLE 2.
BASELINE DEMOGRAPHIC AND CLINICAL VARIABLES OF 97 PATIENTS WITH BIOPSY-PROVEN IDIOPATHIC PULMONARY FIBROSIS AND 38 PATIENTS WITH NON–IDIOPATHIC PULMONARY FIBROSIS IDIOPATHIC INTERSTITIAL PNEUMONIA
| IPF (n = 97) | Non-IPF IIP (n = 38) | P Value | |
|---|---|---|---|
| Age, yr | 62 ± 9 | 52 ± 9 | <0.0001 |
| Sex, male/female | 40 (41%)/57 (59%) | 22 (58%)/16 (42%) | 0.08 |
| Smoking status, never/ever-smoker | 32 (35%)/60 (65%)* | 15 (41%)/22 (59%)† | |
| HRCT scores | |||
| HRCT alveolar score | 1.72 ± 0.95 | 2.29 ± 1.32 | 0.02 |
| HRCT interstitial score | 0.91 ± 0.29 | 0.54 ± 0.39 | <0.001 |
| Pulmonary function | |||
| FVC, % predicted | 65 ± 16 | 70 ± 19 | 0.15 |
| DlCO, % predicted | 48 ± 17 | 50 ± 17 | 0.57 |
| 6MWT | |||
| Distance walked, ft | 911 ± 473 | 895 ± 578 | 0.92 |
| Desaturation < 88%‡ | 45 (46%) | 6 (16%) | 0.47 |
Definition of abbreviations: DlCO = diffusion capacity for carbon monoxide; HRCT = high-resolution computed tomography; IIP = idiopathic interstitial pneumonia; IPF = idiopathic pulmonary fibrosis; 6MWT = 6-minute walk test.
All patients had an HRCT interstitial score less than 2 in all lobes.
Missing data for five patients.
Missing data for one patient.
Number of patients whose oxygen saturation fell below 88% during the 6MWT.
Univariate logistic regression models of demographic, HRCT, pulmonary function, and 6MWT variables were generated to determine which variables predict a diagnosis of IPF. Older patient age, lower HRCT alveolar scores, and higher HRCT interstitial scores were significant predictors of an IPF diagnosis (Table 3). Multivariate logistic regression models were used to determine variables that individually contribute toward prediction of IPF. From these analyses, increasing age (OR, 1.09 per year; 95% CI, 1.04–1.14; P = 0.0007) and increasing average HRCT interstitial score (OR, 10.44 per unit increase in score; 95% CI, 3.12–34.91; P = 0.0001) best predicted a diagnosis of IPF.
TABLE 3.
UNIVARIATE LOGISTIC REGRESSION MODELS OF VARIABLES PREDICTING A DIAGNOSIS OF IDIOPATHIC PULMONARY FIBROSIS
| OR (95% CI) | P Value | |
|---|---|---|
| Age | 1.11 (1.06, 1.16) | <0.0001 |
| Male sex | 1.96 (0.92, 4.19) | 0.08 |
| Ever smoker | 1.28 (0.58, 2.80) | 0.54 |
| HRCT score | ||
| HRCT alveolar score | 0.61 (0.43, 0.87) | 0.007 |
| HRCT interstitial score | 17.20 (5.41, 54.70) | <0.0001 |
| Pulmonary function | ||
| FVC % predicted | 0.18 (0.02, 1.59) | 0.12 |
| DLCO % predicted | 0.48 (0.04, 6.01) | 0.57 |
| 6MWT Variables | ||
| Desaturation <88% | 0.63 (0.20, 1.93) | 0.41 |
| Distance per 1,000 ft | 1.06 (0.35, 3.26) | 0.91 |
Definition of abbreviations: CI = confidence interval; DlCO = diffusion capacity for carbon monoxide; HRCT = high-resolution computed tomography scan of the chest; OR = odds ratio; 6MWT = 6-minute walk test.
Age and HRCT interstitial scores were examined in detail for their ability to predict a diagnosis of IPF. Increasing age was a powerful predictor of IPF. Age of at least 70 years was associated with a PPV for IPF of at least 95% (Table 4). When HRCT interstitial score was combined with age the ability to predict the presence of IPF improved and could be expanded to even younger patients (Table 5). The likelihood ratio for this model is 40.73 (P < 0.001). For example, a patient aged 50 years or older with an HRCT interstitial score of at least 0.8 had a PPV of IPF of at least 97%. ROC analyses were performed for the model with age alone and the model with age combined with HRCT interstitial score (data not shown). The areas under the ROC curves were 0.77 and 0.84, respectively, for these two models, with P < 0.0001 for improved prediction based on the model including HRCT interstitial score. These data demonstrate the potential clinical usefulness of semiquantitative HRCT scoring in aiding in the diagnosis of IPF.
TABLE 4.
POSITIVE PREDICTIVE VALUE, SPECIFICITY, SENSITIVITY, AND NEGATIVE PREDICTIVE VALUE WHEN CLASSIFYING PATIENTS WITH IDIOPATHIC PULMONARY FIBROSIS BASED ON BEING AT LEAST AS OLD AS THE AGE INDICATED
| Age (yr) | PPV | Specificity | Sensitivity | NPV |
|---|---|---|---|---|
| 30 | 72 | 0 | 100 | NA |
| 35 | 72 | 5 | 99 | 67 |
| 40 | 74 | 11 | 98 | 67 |
| 45 | 74 | 16 | 95 | 55 |
| 50 | 78 | 34 | 92 | 62 |
| 55 | 83 | 58 | 80 | 54 |
| 60 | 87 | 76 | 61 | 43 |
| 65 | 91 | 89 | 43 | 38 |
| 70 | 95 | 97 | 21 | 32 |
| 75 | 100 | 100 | 6 | 29 |
| 80 | 100 | 100 | 1 | 28 |
Definition of abbreviations: NA = not applicable; NPV = negative predictive value; PPV = positive predictive value.
Data expressed as percentages.
TABLE 5.
POSITIVE PREDICTIVE VALUE, SPECIFICITY, SENSITIVITY, AND NEGATIVE PREDICTIVE VALUE WHEN CLASSIFYING PATIENTS WITH IDIOPATHIC PULMONARY FIBROSIS BASED ON BEING AT LEAST AS OLD AS THE AGE INDICATED AND HAVING A HIGH-RESOLUTION COMPUTED TOMOGRAPHY INTERSTITIAL SCORE AT LEAST AS HIGH AS INDICATED
| Age | HRCT Interstitial Score | PPV | Specificity | Sensitivity | NPV |
|---|---|---|---|---|---|
| 30 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 72 | 0 | 100 | NA | |
| 0.6 | 73 | 5 | 99 | 67 | |
| 0.8 | 76 | 24 | 97 | 75 | |
| 1.0 | 85 | 61 | 88 | 66 | |
| 35 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 72 | 0 | 100 | NA | |
| 0.6 | 74 | 11 | 98 | 67 | |
| 0.8 | 82 | 50 | 92 | 70 | |
| 1.0 | 90 | 79 | 73 | 54 | |
| 40 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 72 | 0 | 100 | NA | |
| 0.6 | 76 | 24 | 97 | 75 | |
| 0.8 | 86 | 66 | 84 | 61 | |
| 1.0 | 97 | 97 | 32 | 36 | |
| 45 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 73 | 5 | 99 | 65 | |
| 0.6 | 81 | 42 | 94 | 73 | |
| 0.8 | 92 | 84 | 70 | 52 | |
| 1.0 | 100 | 100 | 7 | 30 | |
| 50 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 73 | 8 | 98 | 60 | |
| 0.6 | 85 | 61 | 88 | 66 | |
| 0.8 | 97 | 97 | 32 | 36 | |
| 1.0 | 100 | 100 | 1 | 28 | |
| 55 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 74 | 13 | 97 | 63 | |
| 0.6 | 87 | 68 | 81 | 59 | |
| 0.8 | 100 | 100 | 8 | 30 | |
| 1.0 | 100 | 100 | 1 | 28 | |
| 60 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 76 | 24 | 97 | 75 | |
| 0.6 | 92 | 84 | 70 | 52 | |
| ≥0.8 | 100 | 100 | 1 | 28 | |
| 65 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 78 | 32 | 96 | 75 | |
| 0.6 | 96 | 95 | 45 | 40 | |
| ≥0.8 | 100 | 100 | 1 | 28 | |
| 70 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 82 | 50 | 92 | 70 | |
| 0.6 | 100 | 100 | 24 | 34 | |
| ≥0.8 | 100 | 100 | 1 | 28 | |
| 75 | 0.2 | 72 | 0 | 100 | NA |
| 0.4 | 85 | 61 | 88 | 66 | |
| 0.6 | 100 | 100 | 7 | 30 | |
| ≥0.8 | 100 | 100 | 1 | 28 | |
| 80 | 0.2 | 0 | 0 | 100 | NA |
| 0.4 | 66 | 66 | 84 | 61 | |
| ≥0.6 | 100 | 100 | 1 | 28 |
Definition of abbreviations: HRCT = high-resolution computed tomography; NA = not applicable; NPV = negative predictive value; PPV = positive predictive value.
Data expressed as percentages.
For user convenience, we provide a probability of IPF score (Table 6), which predicts the PPV, NPV, sensitivity, and specificity of a diagnosis of IPF based on a patient's age and HRCT interstitial score using the formula:
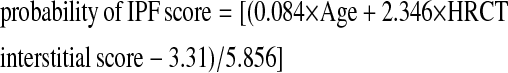 |
(truncate negative values as 0; positive values >1 as 1)
TABLE 6.
PROBABILITY OF IDIOPATHIC PULMONARY FIBROSIS SCORES* AND THEIR CORRESPONDING TEST CHARACTERISTICS FOR THE PRESENCE OF IDIOPATHIC PULMONARY FIBROSIS CALCULATED FROM A PATIENT'S AGE AND AVERAGE HIGH-RESOLUTION COMPUTED TOMOGRAPHY INTERSTITIAL SCORE
| Score | PPV | Specificity | Sensitivity | NPV |
|---|---|---|---|---|
| 0 | 72 | 0 | 100 | NA |
| 0.1 | 73 | 5 | 99 | 67 |
| 0.2 | 77 | 26 | 96 | 71 |
| 0.3 | 86 | 65 | 83 | 61 |
| 0.4 | 96 | 95 | 45 | 40 |
| 0.5 | 100 | 100 | 3 | 29 |
| ≥0.6 | 100 | 100 | 1 | 28 |
Score = [(0.084 × Age + 2.346 × HRCT interstitial score − 3.31)/5.856] (truncate negative values as 0; positive values >1 as 1)
Definition of abbreviations: NA = not applicable; NPV = negative predictive value; PPV = positive predictive value.
The distribution of the probability of IPF scores for patients in this study is shown in Figure 3.
Figure 3.

Histogram illustrating the frequency of the probability of idiopathic pulmonary fibrosis (IPF) scores for patients with a biopsy-proven diagnosis of IPF (n = 97) and non-IPF (n = 38). Solid columns, IPF; striped columns, non-IPF.
The probability of IPF score can be used to estimate the probability of IPF for an individual patient. For example, if a patient has an average HRCT interstitial score of 0.8 and is 50 years old, the probability of IPF score would be 0.5 corresponding to a PPV of 86% for the presence of IPF. If the same patient is 70 years old, the probability of IPF score would be 0.8 and the PPV for the presence of IPF is 100%.
DISCUSSION
Differentiating between IPF and non-IPF IIPs is clinically relevant and in some cases can be accomplished without a surgical lung biopsy (9, 10). In this study we examined the ability of clinical variables to predict a histopathologic pattern of UIP in a group of patients without a diagnostic HRCT study—patients who traditionally would require a surgical lung biopsy to obtain a definitive diagnosis. We identified increased age and HRCT interstitial score as important discriminators between IPF and non-IPF IIPs. These data provide practical, important information in the evaluation of patients with suspected IPF. If validated, these data will allow the clinician to estimate the probability that a patient with suspected IPF will have IPF confirmed by surgical lung biopsy by the simple measure of patient's age and average HRCT interstitial score. As older patients often have comorbidities and increased risk of operative morbidity we believe these data will be useful in helping patients and caregivers weigh the risks, benefits, and usefulness of surgical lung biopsy for the diagnosis of suspected IPF or non-IPF IIP. If validated, these data may also allow for a broader inclusion of patients into clinical trials (i.e., those patients unable to tolerate a surgical lung biopsy and with consistent but not definite HRCT criteria for the diagnosis of IPF).
The probability of IPF was increased in older patients consistent with published incidence and prevalence data (25). We extend these data by defining specific test characteristics (sensitivity, specificity, PPV, and NPV) for finding UIP/IPF at biopsy compared with non-IPF IIP for specific age values. Using age as a sole predictor a cutoff of 75 years was associated with a 100% predictive value of confirming IPF by surgical lung biopsy; 70 years was nearly as good, with a PPV of 95%. These findings are consistent with evolving pathobiologic paradigms of IPF as a degenerative disease of aging (26, 27). Our finding that increased age is a strong predictor of IPF in patients with relatively early radiographic manifestations of disease (no honeycomb change) lends some support for this concept.
It is established that HRCT can accurately predict the presence of UIP at biopsy when features of honeycombing in a basilar, peripheral distribution (without a predominance of ground glass or other features to suggest an alternative diagnosis) are present (1, 10, 28, 29). Our data extend these finding by demonstrating that even modest amounts of fibrosis, without honeycombing, can be highly predictive of the presence of IPF when combined with the patient's age.
We were unable to identify any physiologic variables that helped predict the presence of IPF. In aggregate, patients with IPF tend to be older and have more pronounced physiologic derangements (21, 28–30); however, there is a significant overlap between these features between patients with IPF and non-IPF IIP (30). This overlap likely explains the lack of predictive ability for these variables.
We found the best predictive model for finding IPF was an easy-to-calculate probability of IPF score that combined age with the HRCT interstitial score. Using this score, patients 55 years of age or older with even modest amounts of fibrosis (average HRCT interstitial scores of 0.8–1.0) were associated with a PPV of 100% for confirming IPF at surgical lung biopsy. Review of the data in Table 6 suggests that a cutoff probability of IPF score could be derived, above which there would be a reasonable PPV and NPV for a diagnosis of IPF based on age and HRCT interstitial score. However, these data should be confirmed with a validation cohort before the probability of IPF score is used clinically. Until then, patients who do not meet clinical criteria for the diagnosis of IPF following the American Thoracic Society/European Respiratory Society guidelines (28) should undergo an open lung biopsy to establish the diagnosis.
Strengths of this study include the inclusion of a diverse group of patients typical of those who present to the clinician with suspected IIP as well as a detailed clinical, radiographic, and pathologic approach to diagnosis (18). This study is limited by the lack of a validation cohort, by involving only a single center, and by using radiologists who are experienced in the scoring of HRCT studies. The prevalence of IPF in our cohort was 79%, higher than that found in other studies (10, 31). How these data apply to other academic and community practices needs further study. Until emerging genomic and proteomic tools to accurately diagnose IIPS become available, refining current clinical-radiographic-physiologic assessments is needed to improve diagnostic accuracy. This diagnostic accuracy will also be crucial in future clinical trials because the underlying pathogenetic mechanisms and responses to specific treatment modalities are likely to be distinctly different in IPF versus non-IPF IIPs.
In conclusion, we have shown that a higher HRCT interstitial score and older age are predictive of a diagnosis of IPF. In patients without honeycomb change on HRCT, older age and modest amounts of fibrosis are highly predictive of a diagnosis of IPF. Physiological variables, including FVC, DlCO, and 6MWT variables, are not predictive of an IPF diagnosis. Validation of these findings, preferably with a prospective multicentre study, is needed before they can be applied broadly.
Supported in part by National Institute of Health NHLBI grant P50 HL56402 (G.B.T., F.J.M, K.R.F), NHLBI 2 K24 HL04212 (F.J.M.), 1K23 HL68713 (K.R.F.), R01 HL091743-01 (K.R.F.), and 1K23 HL093351 (M.K.H.). Dr. Fell was supported by the Alberta Heritage Foundation for Medical Research.
Originally Published in Press as DOI: 10.1164/rccm.200906-0959OC on January 7, 2010
Conflict of Interest Statement: C.D.F. has received advisory board fees from Actelion ($1,001–$5,000) and lecture fees paid by GlaxoSmithKline and AstraZeneca (up to $1,000). F.J.M. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. L.X.L. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. S.M. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. M.K.H. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. E.A.K. has received an industry-sponsored grant from GE ($10,001–50,000). B.H.G. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. J.M. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. W.D.T. has received lecture fees from CBCE Biomedical Education ($1,001–$5,000) and Pilot Program, France Foundation ($1,001–$5,000), and has been an expert witness for plaintiff and defense attorneys ($10,001–$50,000). T.V.C. is employed by Gilead Sciences. G.B.T.does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. K.R.F. has received consultancy fees from Gilead, Fibrogen, Boehringer Ingelheim, GlaxoSmithKline, and Nepharm ($1,001–$5,000) and has received lecture fees from Sepracor, Ortho-McNeil, GlaxoSmithKline, Boehringer Ingelheim, and Pfizer ($1,001–$5,000) and an industry-sponsored grant from Intermune, Johnson and Johnson ($50,001–$100,000).
References
- 1.American Thoracic Society/European Respiratory Society. International multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002;165:277–304. [DOI] [PubMed] [Google Scholar]
- 2.Martinez FJ. Idiopathic interstitial pneumonias: usual interstitial pneumonia versus nonspecific interstitial pneumonia. Proc Am Thorac Soc 2006;3:81–95. [DOI] [PubMed] [Google Scholar]
- 3.Flaherty K, Toews G, Travis W, Colby T, Kazerooni E, Gross B, Jain A, Strawderman R III, Paine R III, Flint A, et al. Clinical significance of histological classification of idiopathic interstitial pneumonia. Eur Respir J 2002;19:275–283. [DOI] [PubMed] [Google Scholar]
- 4.Jegal Y, Kim DS, Shim TS, Lim CM, Do Lee S, Koh Y, Kim WS, Kim WD, Lee JS, Travis WD, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005;171:639–644. [DOI] [PubMed] [Google Scholar]
- 5.Bjoraker J, Ryu J, Edwin M, Myers J, Tazelaar H, Schoreder D, Offord K. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998;157:199–203. [DOI] [PubMed] [Google Scholar]
- 6.Veeraraghavan S, Latsi P, Wells A, Pantelidis P, Nicholson A, Colby T, Haslam P, Renzoni E, du Bois R. BAL findings in idiopathic nonspecific interstitial pneumonia and usual interstitial pneumonia. Eur Respir J 2003;22:239–244. [DOI] [PubMed] [Google Scholar]
- 7.Flaherty K, Travis W, Colby T, Toews G, Kazerooni E, Gross B, Jain A, Strawderman R III, Flint A, Lynch J III, et al. Histologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med 2001;164:1722–1727. [DOI] [PubMed] [Google Scholar]
- 8.Flaherty K, Thwaite E, Kazerooni E, Gross B, Toews G, Colby T, Travis W, Mumford J, Murray S, Flint A, et al. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax 2003;58:143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunninghake G, Lynch D, Galvin J, Muller N, Schwartz D, King T Jr, Lynch J III, Hegele R, Waldron J Jr, Colby T, et al. Radiologic findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia. Chest 2003;124:1215–1223. [DOI] [PubMed] [Google Scholar]
- 10.Hunninghake G, Zimmerman M, Schwartz D, King T Jr, Lynch J, Hegele R, Waldron J, Colby T, Muller N, Lynch D, et al. Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2001;164:193–196. [DOI] [PubMed] [Google Scholar]
- 11.Lynch DA, Travis WD, Muller NL, Galvin JR, Hansell DM, Grenier PA, King TE Jr. Idiopathic interstitial pneumonias: CT features. Radiology 2005;236:10–21. [DOI] [PubMed] [Google Scholar]
- 12.Utz JP, Ryu JH, Douglas WW, Hartman TE, Tazelaar HD, Myers JL, Allen MS, Schroeder DR. High short-term mortality following lung biopsy for usual interstitial pneumonia. Eur Respir J 2001;17:175–179. [DOI] [PubMed] [Google Scholar]
- 13.Lettieri CJ, Veerappan GR, Helman DL, Mulligan CR, Shorr AF. Outcomes and safety of surgical lung biopsy for interstitial lung disease. Chest 2005;127:1600–1605. [DOI] [PubMed] [Google Scholar]
- 14.Falcoz PE, Conti M, Brouchet L, Chocron S, Puyraveau M, Mercier M, Etievent JP, Dahan M. The thoracic surgery scoring system (Thoracoscore): risk model for in-hospital death in 15,183 patients requiring thoracic surgery. J Thorac Cardiovasc Surg 2007;133:325–332. [DOI] [PubMed] [Google Scholar]
- 15.Kreider ME, Hansen-Flaschen J, Ahmad NN, Rossman MD, Kaiser LR, Kucharczuk JC, Shrager JB. Complications of video-assisted thoracoscopic lung biopsy in patients with interstitial lung disease. Ann Thorac Surg 2007;83:1140–1144. [DOI] [PubMed] [Google Scholar]
- 16.Fell CD, Han M, Kazerooni EA, Gross BH, Travis WD, Colby TV, Toews GB, Martinez FJ, Flaherty KR. Predicting IPF vs. NSIP without a surgical lung biopsy [abstract]. Chest 2007;132:428c–429c. [Google Scholar]
- 17.Flaherty KR, Gay SE, Travis WD, Colby TV, Kazerooni E, Gross BG, Toews GB, Martinez FJ. Advanced age is associated with a diagnosis of IPF compared to NSIP [abstract]. Am J Respir Crit Care Med 2006;173:A104. [Google Scholar]
- 18.Flaherty KR, King TE Jr, Raghu G, Lynch JP III, Colby TV, Travis WD, Gross BH, Kazerooni EA, Toews GB, Long Q, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004;170:904–910. [DOI] [PubMed] [Google Scholar]
- 19.Kazerooni E, Martinez F, Flint A, Jamadar D, Gross B, Spizarny D, Cascade P, Whyte R, Lynch J III, Toews G. Thin-section CT obtained at 10-mm increments versus limited three-level thin-section CT for idiopathic pulmonary fibrosis: correlation with pathologic scoring. AJR Am J Roentgenol 1997;169:977–983. [DOI] [PubMed] [Google Scholar]
- 20.Gay SE, Kazerooni EA, Toews GB, Lynch JP III, Gross BH, Cascade PN, Spizarny DL, Flint A, Schork MA, Whyte RI, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998;157:1063–1072. [DOI] [PubMed] [Google Scholar]
- 21.Flaherty KR, Andrei A-C, Murray S, Fraley C, Colby TV, Travis WD, Lama V, Kazerooni EA, Gross BH, Toews GB, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006;174:803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gosset WSS. (pseud). The probable error of a mean. Biometrika 1908;6:1–25. [Google Scholar]
- 23.Pearson K. On the criterion that a given system of deviations from the probable in the case of a correlated system of variables is such that it can be reasonably supposed to have arisen from random sampling. Philosophical Magazine, Series 5 1900;50:157–175. [Google Scholar]
- 24.Zweig MH, Campbell G. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin Chem 1993;39:561–577. [PubMed] [Google Scholar]
- 25.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006;174:810–816. [DOI] [PubMed] [Google Scholar]
- 26.Cronkhite JT, Xing C, Raghu G, Chin KM, Torres F, Rosenblatt RL, Garcia CK. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med 2008;178:729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thannickal VJ, Loyd JE. Idiopathic pulmonary fibrosis: a disorder of lung regeneration? Am J Respir Crit Care Med 2008;178:663–665. [DOI] [PubMed] [Google Scholar]
- 28.American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International Consensus Statement. American Thoracic Society (ATS) and the European Respiratory Society (ERS). Am J Respir Crit Care Med 2000;161:646–664. [DOI] [PubMed] [Google Scholar]
- 29.Raghu G, Mageto Y, Lockhart D, Schmidt R, Wood D, Godwin J. The accuracy of the clinical diagnosis of new-onset idiopathic pulmonary fibrosis and other interstitial lung disease: a prospective study. Chest 1999;116:1168–1174. [DOI] [PubMed] [Google Scholar]
- 30.Flaherty K, Martinez F, Travis W, Lynch J III. Nonspecific interstitial pneumonia (NSIP). Semin Respir Crit Care Med 2001;22:423–433. [DOI] [PubMed] [Google Scholar]
- 31.Thomeer MJ, Costabel U, Rizzato G, Poletti V, Demedts M. Comparison of registries of interstitial lung diseases in three European countries. Eur Respir J 2001;18:114S–118S. [PubMed] [Google Scholar]