

Abstract
Bovine spinal dysmyelination (BSD) is a recessive congenital neurodegenerative disease in cattle (Bos taurus) characterized by pathological changes of the myelin sheaths in the spinal cord. The occurrence of BSD is a longstanding problem in the American Brown Swiss (ABS) breed and in several European cattle breeds upgraded with ABS. Here, we show that the disease locus on bovine chromosome 11 harbors the SPAST gene that, when mutated, is responsible for the human disorder hereditary spastic paraplegia (HSP). Initially, SPAST encoding Spastin was considered a less likely candidate gene for BSD since the modes of inheritance as well as the time of onset and severity of symptoms differ widely between HSP and BSD. However, sequence analysis of the bovine SPAST gene in affected animals identified a R560Q substitution at a position in the ATPase domain of the Spastin protein that is invariant from insects to mammals. Interestingly, three different mutations in human SPAST gene at the equivalent position are known to cause HSP. To explore this observation further, we genotyped more than 3,100 animals of various cattle breeds and found that the glutamine allele exclusively occurred in breeds upgraded with ABS. Furthermore, all confirmed BSD carriers were heterozygous, while all affected calves were homozygous for the glutamine allele consistent with recessive transmission of the underlying mutation and complete penetrance in the homozygous state. Subsequent analysis of recombinant Spastin in vitro showed that the R560Q substitution severely impaired the ATPase activity, demonstrating a causal relationship between the SPAST mutation and BSD.
Keywords: Inbreeding, Hereditary disease, Animal model, Spastin, Dysmyelination
Introduction
Domestic animals provide a valuable resource for studying the genetics of phenotypic variation in quantitative production and health traits as well as for the identification of mutations underpinning hereditary defects [1]. In cattle, the emergence and spreading of genetic diseases caused by inbreeding is a recurrent phenomenon, demonstrating that international trading of semen from elite bulls and artificial insemination accelerates dispersal of deleterious alleles [2, 3]. An efficient mean to control genetic diseases is to implement DNA-based testing for the causative mutation in breeding programs in order to avoid at-risk matings. A prominent example is the globally distributed complex vertebral malformation syndrome in the Holstein Friesian population, where the origin of the undesirable gene mutation has been traced back to a specific founder animal [4, 5]. Bovine spinal dysmyelination (BSD) is one of several neurodegenerative disorders in cattle, and the recessive mutation underlying BSD was widely spread in Europe because American Brown Swiss carriers were used to upgrade European cattle breeds such as the Danish Red Dairy breed [6]. The main clinical signs of BSD are manifested immediately at birth and include, most notably, lateral recumbency with slight to moderate opisthotonos, body tremor, and spastic extension of the limbs (Fig. 1a) [7–9]. General muscle atrophy resulting from denervation occurs to variable degrees and is most obvious in the hind limbs. Attempts to rise and limb movements are absent; however, the animals remain alert to their surroundings, and spinal reflexes are normal or slightly increased. The morphological cause of the phenotype is bilateral symmetrical hypo- and demyelination of axons in the cervical and thoracic segments of the spinal cord. The lesions are particularly prominent in the ascending gracile funiculus, the ascending dorsal spinocerebellar tracts, and in the descending sulcomarginal tracts (Fig. 1b) [8, 9]. Other characteristic features are a reduced number of axons and astrogliosis in the affected tracts. In contrast, the nerve cell bodies in the gray matter of the spinal cord are unaffected, and myelination defects are not observed in any other parts of the nervous system.
Fig. 1.
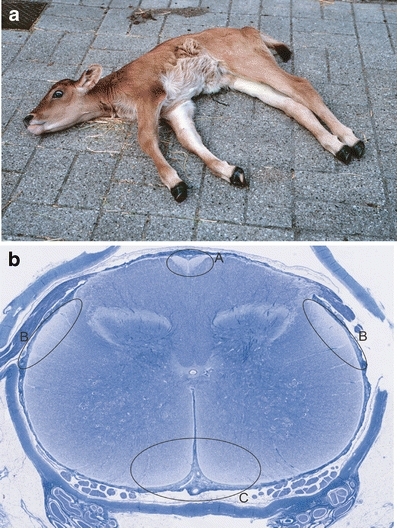
Clinical and pathological characteristics of bovine spinal dysmyelination. a Phenotypic appearance of a calf affected with bovine spinal dysmyelination. b Whole slide digital microscopic image of cervical spinal cord stained with Luxol fast blue. Lack of myelin is seen as reduced blue staining in the ascending gracile funiculus (a), the ascending dorsolateral spinocerebellar tracts (b), and the descending sulcomarginal tracts (c)
We have previously mapped the BSD disease locus to a narrow interval on bovine chromosome 11 [6], but strong candidate genes were not identified in the unfinished, early version of the bovine genome sequence. Recently, a new assembly of the bovine genome sequence (Btau_4.0, released October 2007) showed that the disease locus contains the SPAST gene, which in its mutated form is known to cause hereditary spastic paraplegia (HSP) in humans. HSP is a disorder of the central nervous system characterized by lower extremity weakness, spasticity, and progressive gait disturbance. SPAST encodes a microtubule-severing enzyme, and disease mutations are morphologically associated with progressive axonopathy of spinal neurons. A priori SPAST seemed unlikely as a candidate gene for BSD for two reasons. First, HSP associated with mutations in SPAST is inherited as a dominant trait in humans, which is different from the recessive mode of inheritance of BSD in cattle. Second, affected calves display very severe symptoms of spinal dysmyelination immediately after birth, whereas the clinical features of human HSP are mild initially and usually do not appear before the second to fourth decade of life [10]. It was therefore unexpected when our sequence analysis of SPAST in affected animals identified a disease-causing mutation that results in an arginine to glutamine change in the highly conserved ATPase domain of Spastin.
Materials and methods
SNP genotyping
We applied the ABI Prism TaqMan Allelic Discrimination Assay to type 3137 individuals for the G/A mutation in the SPAST gene. The reactions were prepared by using TaqMan Universal Master Mix, 30 ng of genomic DNA, and custom-made primers and minor groove binder probes from Applied Biosystems in a 10-µl reaction volume. The polymerase chain reaction (PCR) amplification was performed under the following conditions: 10 min 95°C enzyme activation followed by 40 cycles at 95°C for 15 s and 60°C for 1 min (annealing/extension). Allele discrimination was accomplished by running end point detection using the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA) and the Sequence Detection System version 2.2 software.
GST-Spastin constructs, expression, and purification
First strand cDNA was synthesized from approximately 4 µg total RNA and 0.5 µg oligo(dT)12-18 primer using the superscript II RNase H-reverse transcriptase system (Invitrogen). A cDNA fragment encoding wild-type Spastin between amino acid positions 227 and 614 was obtained by BamHI digestion of a PCR product, which was generated using the forward primer 5′CTGGGATCCGAAAGTGGAGCTGTTCCAAAAAGA3′ and the reverse primer 5′GACGGATCCTTACACAGTGGTATCTCCAAAGTCCT3′. PCR contained 1.5 mM MgCl2, 0.2 mM dNTP, 0.5 μM primer, and 0.2 U Phusion DNA polymerase (Finnzymes) in a total volume of 10 µL. The PCR fragment was cloned into the unique BamHI site of the pGEX-6P-3 vector to generate an in-frame fusion protein with glutathione S-transferase (GST). Site-directed mutagenesis (QuickChange, Stratagene) using the complementary primers 5′GATGCAGCACTGGGTCCTATCCAAGAACTGAAACCAGAACAAGTGA3′ and 5′TCACTTGTTCTGGTTTCAGTTCTTGGATAGGACCCAGTGCTGCATC3′ was performed to generate a vector that encodes a fusion protein between GST and mutated Spastin harboring the R560Q substitution. Escherichia coli BL21 cells were transformed with wild-type and mutant GST-Spastin fusion constructs. Protein expression was induced in 100 ml LB cultures at an OD600 of 0.8 by addition of isopropyl-D-thioglucoside to a final concentration of 0.5 mM for 4 h at 25°C. Cells were harvested by centrifugation and incubated in 2 ml extraction buffer (140 mM NaCl; 10 mM Na2HPO4; 1,8 mM KH2PO4; 1 mM ATP; 1 mM DTT; and EDTA-free protease inhibitor; pH 7.5) and 1 mg/ml lysozyme for 15 min at 0°C. Cell extracts were prepared by sonication, and GST-Spastin fusion proteins were affinity-purified using glutathione-agarose beads according to manufacturer’s instructions (Clontech). ATPase activities were measured using an EnzChek Phosphate kit (Molecular Probes). The ATPase assays were repeated three times.
Expression analysis
Total RNA was extracted using the Quiagen RNeasy kit with on column DNase digestion. From each tissue, 1.5 µg total RNA was reverse transcribed using an oligo dT primer and Superscript II (Invitrogen) in 20 µl as recommended by supplier. Expression in different tissues was monitored by PCR using 22 cycles in an attempt to measure amplification in the exponential phase of the reaction. PCR products were analyzed on 2% agarose gels stained with ethidium bromide.
DNA sequencing
DNA sequencing was performed using BigDye Terminator v.3.1 Cycle Sequencing Kit (PE Applied Biosystems) according to the recommendations of the manufacturer. The DNA sequences were run on a 3730xl DNA Analyzer (PE Applied Biosystems) and were analyzed using the Sequencher software (version 4.0.5, Gene Codes Corporation).
Results
Candidate genes for BSD
The BSD locus was known from a previous mapping effort, which showed that a 2.6-cM region between microsatellite markers BP38 and BMS1953 on chromosome 11 cosegregated with the disease [6]. The recent assembly of the draft genome sequence of Bos taurus (http://www.ensembl.org; assembly Btau_4.0; 7× sequence coverage) revealed the presence of approximately 32 genes in the critical interval, including the SPAST gene (Fig. 2a). The disease gene responsible for spinal dysmyelination is presumably expressed in the bovine central nervous system. Thus, as a first evaluation of SPAST as a candidate for BSD, the expression was investigated by reverse transcriptase PCR in a selection of tissues (Fig. 2b). The data showed that the SPAST gene was ubiquitously expressed including in the neural tissues, cervical and lumbar spinal cord, cerebellum, and frontal cortex. To further investigate the possible association of SPAST with BSD, we analyzed the cDNA sequence from a BSD carrier for the presence of mutations. RNA was isolated from spinal cord tissue and reverse transcribed into cDNA in order to be used for amplification of cDNA encoding Spastin. Sequence analysis identified three different splice variants. The longest SPAST cDNA encoded 614 amino acid residues, which is highly similar to the full length of Spastin in other species (Fig. 3). Spastin belongs to the AAA protein family (ATPase associated with various cellular activities), which is a group of Walker-type ATPases. The conserved AAA module is located between amino acid residues 327 and 601, and it contains several functional domains essential for ATP binding and hydrolysis, which are the Walker-homology motifs A and B between positions 380–387 and 435–442, respectively, and an arginine finger (496-RR-497) associated with the second region of homology between residues 482 and 498. Phylogenetic analysis of AAA proteins places Spastin in the “meiotic” subgroup or subfamily 7 together with katanin, which is a microtubule-severing protein like Spastin [11].
Fig. 2.
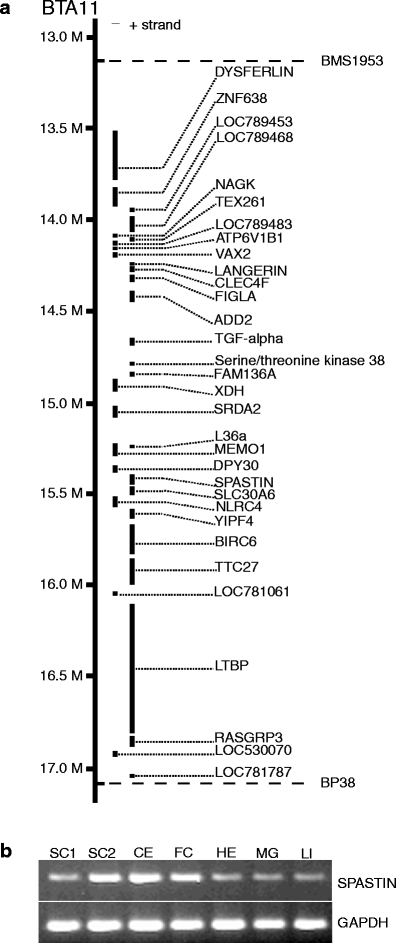
a Physical map of the bovine spinal dysmyelination locus between the linked microsatellite markers BP38 and BMS1953. The location and order of the genes are shown according to the cattle genome assembly Btau_4.0. b Expression analysis by reverse transcription-polymerase chain reaction of SPAST. Tissues analyzed are SC1 spinal cord 1 (cervical), SC2 spinal cord 2 (lumbar), CE cerebellum, FC frontal cortex, HE heart, MG mammary gland, and LI liver
Fig. 3.
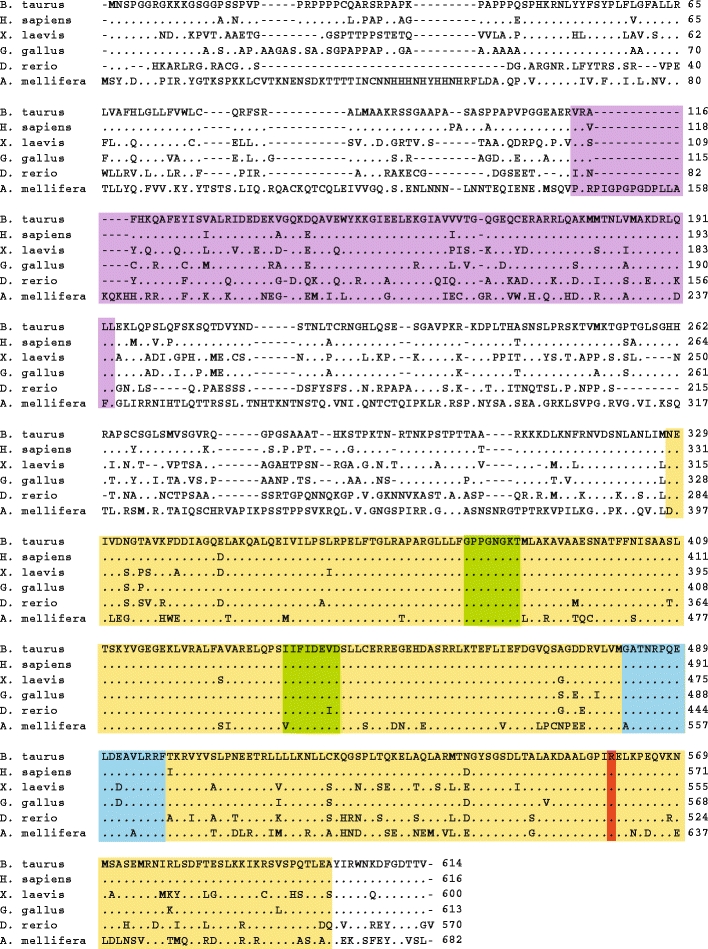
Multiple sequence alignment of Spastin homologues. Key domains in Spastin are color marked: The AAA domain ATPase associated with diverse cellular activities (yellow), MIT microtubule interacting and trafficking domain (purple), Walker A and B motifs (green), and arginine finger and second region of homology (blue). The arginine residue at position 560 is evolutionary conserved from mammals to insects (red)
A potential disease-causing mutation in SPAST
In addition to the full-length cDNA, the sequencing analysis also identified two smaller splice products of which one variant lacked the nucleotides encoded by exon 4, and the other variant lacked two amino acid residues due to alternative splicing of exon 12. Furthermore, two single nucleotide polymorphisms were discovered: a silent polymorphism (A253A) and a missense polymorphism, which replaces the amino acid residue arginine with glutamine at position 560. Sequencing of the corresponding genomic locus established that wild-type and affected calves were homozygous for arginine and glutamine, respectively, whereas carriers were heterozygous (Fig. 4). The R560Q substitution is remarkable for two reasons: First, arginine at this position is completely conserved from insects to mammals, indicating functional importance of this amino acid (Fig. 2); second, several different amino acid substitutions at the equivalent position (the human protein is two amino acid residues longer than the bovine protein) in human Spastin are associated with disease. Thus, mutational analysis of probands of unrelated HSP families has detected two different disease-associated missense mutations, R562Q [12] and R562G [13], and one nonsense mutation, R562X [14].
Fig. 4.
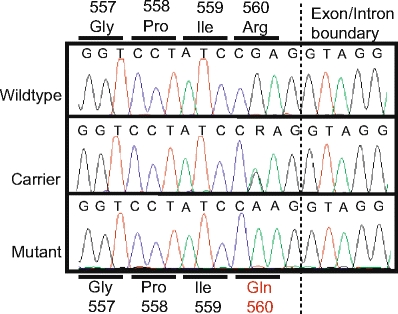
Sequence of the missense mutation causing the R560Q substitution. The chromatograms show the sequence of a genomic fragment spanning the putative disease-causing mutation. Wild-type and affected individuals were homozygous for arginine and glutamine, respectively, whereas individuals with bovine spinal dysmyelination carrier status were heterozygous
Family and population-based genotyping
BSD is a recessive trait, and obligate carriers such as the parents of affected calves must therefore be heterozygous for the glutamine allele, while the calves with BSD must be homozygous. To test this, we genotyped genomic sequences encoding the R560Q variant in 33 calves, which have been diagnosed as BSD affected on the basis of postmortem neuropathological examination, as well as a group of 68 individuals comprising available parents and grandparents. Some of these animals were previously used in the genome-wide screen that led to the identification of the disease locus on BTA11. All affected animals had the purebred American Brown Swiss bull White Cloud Jason’s Elegant as a common ancestor. The data showed that all obligate carriers were heterozygous and that all affected calves were homozygous for the glutamine residue. This result strengthens the conclusion that the R560Q substitution is causative for the disease (Fig. 3).
To test whether the Glu560 allele occurred generally in bovine populations, we analyzed more than 3,100 animals comprising six different cattle breeds for the presence of the R562Q polymorphism (Table 1). The samples were randomly selected from the national cattle DNA bank and without prior knowledge of their carrier or disease status. The data showed that the glutamine allele was unique for breeds cross-bred with American Brown Swiss such as the Danish Red Dairy breed and the Danish Red Holstein (Table 1). Moreover, pedigree analysis showed that heterozygous animals for which the pedigree information was available could be traced back to confirmed BSD carriers. Furthermore, we identified four animals, which were homozygous for the glutamine residue; importantly, registration records showed that these four calves were previously diagnosed as BSD affected based on postmortem examination, and that they have confirmed BSD carriers appearing on both the paternal and maternal sides. We conclude that the glutamine allele occurred exclusively in breeds and families in which the disease segregates.
Table 1.
Distribution of R560Q alleles in different cattle breeds
| Breed | RR | RQ | |
|---|---|---|---|
| Red Danish Dairy | 1,361 | 39 | 4 |
| Danish Red Holstein | 255 | 1 | 0 |
| Holstein Friesian | 913 | 0 | 0 |
| Hereford | 379 | 0 | 0 |
| Limousin | 93 | 0 | 0 |
| Grauvieh | 93 | 0 | 0 |
Population-based samples from six different breeds were genotyped for the presence of the disease-causing amino acid substitution. Samples were selected randomly without prior knowledge of carrier and disease status
The R560Q substitution abolishes the ATPase activity of Spastin
The C-terminal, approximately 285 amino acids of Spastin, contains an AAA ATPase domain (Fig. 3). To determine whether the R560Q substitution affects the ATPase activity, we created recombinant fusion proteins between Spastin and GST, which were expressed in E. coli and purified by affinity chromatography (Fig. 5a). ATP hydrolysis was measured spectrophotometrically using a coupled enzyme assay [15]. We used Spastin lacking amino acids 1–227 because this shortened version has been shown to be more stable and soluble than the full-length protein [16]. ATPase activity of wild-type and mutated Spastin were measured as a function of time using wild-type Spastin denatured by boiling as a control (Fig. 5b). The data demonstrated that the R560Q substitution essentially abolishes the ATPase activity of Spastin, thereby establishing a causal relationship between loss of catalytic activity and BSD.
Fig. 5.
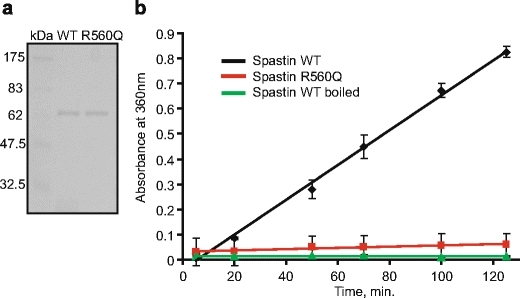
a Coomassie blue stain of a sodium dodecyl sulfate-polyacrylamide gel. Recombinant wild-type and mutant Spastin fused to glutathione S-transferase (GST) were expressed in Escherichia coli and purified by affinity chromatography using glutathione sepharose columns. Lane 1, molecular size marker. Lane 2, wild-type GST-Spastin fusion protein. Lane 3, mutant GST-Spastin protein. b ATPase activity of wild-type and mutated Spastin were measured as a function of time. ATP hydrolysis was measured spectrophotometrically using a coupled enzyme assay. Wild-type Spastin denatured by boiling served as a control
Discussion
HSP is a human disorder of the central nervous system with clinical features usually beginning in the second to fourth decade of life, although first symptoms may appear already in early childhood or as late as in the eighth or ninth decade. In its pure form, the disorder is characterized by lower extremity weakness and spasticity, progressive gait disturbance, hyperreflexia, and loss of vibratory sense, whereas the complicated forms have additional neurological symptoms such as reduced vision, auditory impairment, mental retardation, or dementia. A conspicuous neuropathological feature of pure HSP is decreased staining of myelin in histological sections and a distal-to-proximal degeneration of axons of the neurons in the lateral and anterior corticospinal tracts and the fasciculi gracilis as well as in the spinocerebellar tracts in about 50% of all patients. Peripheral nerves and dorsal root ganglia and posterior roots are usually unaffected [17]. HSP is a genetically heterogeneous disorder with 29 causative genes and loci identified to date, consisting of dominant, recessive, and X-linked forms. Mutations in SPAST are the most common cause of autosomal dominant HSP [18].
The present study provides interesting new insight into the behavior of SPAST mutations. Thus, both HSP and BSD are associated with spasticity and weakness of the lower limbs due to dysfunction of specific nerve fibers of the spinal cord. However, in humans, the vast majority of the SPAST mutations act dominantly, apparently contrasting with the bovine mutation identified in this study, which is clearly recessive [6, 19]. The sole recessive mutation identified to date in humans is a S44L substitution, which has been found in individuals with mild, late onset HSP [20]. The phenotypic effect of dominant SPAST mutations in a homozygous state has not been reported in humans. Interestingly, however, compound heterozygosity for the S44L mutation and an adult onset SPAST mutation, P361L or D470V, is associated with a marked increase in severity of clinical symptoms as well as a decrease in time of onset to infancy or early childhood, which is significantly earlier than for each mutation separately [21, 22].
The difference in modes of inheritance, time of onset, and severity between BSD and HSP is intriguing. In humans, a wide spectrum of DNA alterations that include nonsense, frame-shifts, deletions, insertions, and missense mutations in the SPAST gene result in the dominant form of HSP, which suggest that loss-of-function or haploinsufficiency is a primary pathogenic mechanism. Furthermore, Spastin forms a hexameric complex with itself [16] and interacts with other proteins such as the endosomal protein CHMP1B [23], tubulin [16], and also atlastin [24], which is another HSP disease gene product. This implies that certain missense mutations that destroy the catalytic activity of Spastin but do not affect the ability of Spastin to assemble into protein complexes may act in a dominant-negative manner by reducing the level of fully functional Spastin complexes below a critical threshold. These mechanisms are of likely relevance in the context of the highly variable penetrance of human SPAST mutations, where the age of onset, the severity of symptoms, and rate of disease progression vary considerably both between and within families. This contrasts strongly with the situation in cattle, where all calves are invariably affected already at birth. It is possible that a heterozygous R560Q animal eventually would develop symptoms of the disease if allowed to live out its entire life, but we have no veterinary records to substantiate this. Thus, the natural life span of a cow is about 25 years but usually does not exceed 5–7 years in modern dairy farming.
The affected calves have been subject to detailed clinical and histopathological examination [7–9], and the results from these studies now gain additional value considering that the number of neuropathological studies of human patients with SPAST mutations is limited [17, 25]. Importantly, affected calves exhibit a much more severe disease phenotype than human patients with HSP and also far worse than observed in a loss-of-function mouse model, which develops a late and mild motor defect [26]. The most striking observations were the severe myelin lesions and hypomyelination of the nerve fibers in both ascending and descending tracts in the spinal cord. The oligodendrocytes contained myelin debris in the cytoplasm and displayed necrotic changes characterized by karyorrhexis and vacuolation. Furthermore, ultrastructural examination revealed the presence of slightly swollen axons with accumulations of disoriented microtubules and intermediate filaments, and also highly swollen axons filled with organelles, especially mitochondria and lysosomal bodies. These findings are similar to observations on the loss-of-function mouse model, which also develops focal axonal swellings that are surrounded by thin myelin sheaths, and in addition contain an abnormal accumulation of organelles and disorganized neurofilaments [26]. The phenotype was proposed to arise from impairment of microtubule-dependent transport of organelles and neurofilaments in axons. Thus, Spastin has been shown to target and disassemble microtubules in cells, and recombinant Spastin severs purified microtubules in vitro and in permeabilized cells [27, 28]. This shows that Spastin is implicated in microtubule severing like the homologous katanin protein and suggest that dysregulation of microtubule dynamics may underlie pathogenesis. Interestingly, Spastin has been shown to interact with the CHMP1B protein, which plays a role in intracellular membrane traffic events [23], and mechanisms for axonal transport and trafficking seem to be a unifying theme for several genes associated with HSP [18]. Thus, SPG7-deficient mice are affected by distal axonopathy and have massive accumulation of organelles and neurofilaments in the axons, which are clear evidences of impaired transport [29]. Furthermore, the SPG10 (KIF5A) gene encodes a neuronal kinesin heavy chain protein, which drives anterograde axonal transport of membranous organelles and other cargo [30]. SPG20, encoding spartin, binds the transport vesicle adaptor protein Eps15, which indicates a role in endocytosis and vesicle sorting [31].
The present work establishes a causal relationship between a recessive SPAST mutation and BSD, which is an important step towards development of a gene-based diagnostic test to control spreading of the disease as well as to establish BSD as an animal model of the corresponding human neurological disease. Furthermore, the unanticipated discovery that a condition of congenital axonopathy and severe myelination defects can be caused by SPAST mutations suggests that cases of congenital hypomyelination neuropathy in humans should be investigated for mutations in SPAST.
Acknowledgments
The whole slide digital microscopic image was kindly provided by Visiopharm A/S (Horsholm, Denmark) and Hamamatsu (Herrsching am Ammersee, Germany).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Andersson L, Georges M. Domestic-animal genomics: deciphering the genetics of complex traits. Nat Rev Genet. 2004;5:202–212. doi: 10.1038/nrg1294. [DOI] [PubMed] [Google Scholar]
- 2.Agerholm JS. Inherited disorders in Danish cattle. APMIS. 2007;122(Suppl):1–76. doi: 10.1111/j.1600-0463.2007.apm_v115_s122.x. [DOI] [PubMed] [Google Scholar]
- 3.Charlier C, Coppieters W, Rollin F, Desmecht D, Agerholm JS, Cambisano N, Carta E, Dardano S, Dive M, Fasquelle C, Frennet JC, Hanset R, Hubin X, Jorgensen C, Karim L, Kent M, Harvey K, Pearce BR, Simon P, Tama N, Nie H, Vandeputte S, Lien S, Longeri M, Fredholm M, Harvey RJ, Georges M. Highly effective SNP-based association mapping and management of recessive defects in livestock. Nat Genet. 2008;40:449–454. doi: 10.1038/ng.96. [DOI] [PubMed] [Google Scholar]
- 4.Agerholm JS, Bendixen C, Andersen O, Arnbjerg J. Complex vertebral malformation in holstein calves. J Vet Diagn Invest. 2001;13:283–289. doi: 10.1177/104063870101300401. [DOI] [PubMed] [Google Scholar]
- 5.Thomsen B, Horn P, Panitz F, Bendixen E, Petersen AH, Holm LE, Nielsen VH, Agerholm JS, Arnbjerg J, Bendixen C. A missense mutation in the bovine SLC35A3 gene, encoding a UDP-N-acetylglucosamine transporter, causes complex vertebral malformation. Genome Res. 2006;16:97–105. doi: 10.1101/gr.3690506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nissen PH, Shukri NM, Agerholm JS, Fredholm M, Bendixen C. Genetic mapping of spinal dysmyelination in cross-bred American Brown Swiss cattle to bovine Chromosome 11. Mamm Genome. 2001;12:180–182. doi: 10.1007/s003350010245. [DOI] [PubMed] [Google Scholar]
- 7.Stocker H, Berger PJ, Lutz H, Ossent P. A new hereditary disease in Braunvieh cattle in Switzerland: spinal demyelinization (SDM) in calves that remain in lateral recumbency. Schweiz Arch Tierheilkd. 1996;138:295–300. [PubMed] [Google Scholar]
- 8.Agerholm JS, Hafner A, Olsen S, Dahme E. Spinal dysmyelination in cross-bred brown Swiss calves. Zentralbl Veterinarmed [A] 1994;41:180–188. doi: 10.1111/j.1439-0442.1994.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 9.Hafner A, Dahme E, Obermaier G, Schmidt P, Dirksen G. Spinal dysmyelination in new-born brown Swiss x Braunvieh calves. Zentralbl Veterinarmed [B] 1993;40:413–422. doi: 10.1111/j.1439-0450.1993.tb00158.x. [DOI] [PubMed] [Google Scholar]
- 10.Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F, Davoine CS, Cruaud C, Durr A, Wincker P, Brottier P, Cattolico L, Barbe V, Burgunder JM, Prud’homme JF, Brice A, Fontaine B, Heilig B, Weissenbach J. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet. 1999;23:296–303. doi: 10.1038/15472. [DOI] [PubMed] [Google Scholar]
- 11.Frickey T, Lupas AN. Phylogenetic analysis of AAA proteins. J Struct Biol. 2004;146:2–10. doi: 10.1016/j.jsb.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 12.Svenson IK, Shley-Koch AE, Gaskell PC, Riney TJ, Cumming WJ, Kingston HM, Hogan EL, Boustany RM, Vance JM, Nance MA, Pericak-Vance MA, Marchuk DA. Identification and expression analysis of spastin gene mutations in hereditary spastic paraplegia. Am J Hum Genet. 2001;68:1077–1085. doi: 10.1086/320111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meijer IA, Hand CK, Cossette P, Figlewicz DA, Rouleau GA. Spectrum of SPG4 mutations in a large collection of North American families with hereditary spastic paraplegia. Arch Neurol. 2002;59:281–286. doi: 10.1001/archneur.59.2.281. [DOI] [PubMed] [Google Scholar]
- 14.Fonknechten N, Mavel D, Byrne P, Davoine CS, Cruaud C, Bonsch D, Samson D, Coutinho P, Hutchinson M, McMonagle P, Burgunder JM, Tartaglione A, Heinzlef O, Feki I, Deufel T, Parfrey N, Brice A, Fontaine B, Prud’homme JF, Weissenbach J, Durr A, Hazan J. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet. 2000;9:637–644. doi: 10.1093/hmg/9.4.637. [DOI] [PubMed] [Google Scholar]
- 15.Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci USA. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White SR, Evans KJ, Lary J, Cole JL, Lauring B. Recognition of C-terminal amino acids in tubulin by pore loops in Spastin is important for microtubule severing. J Cell Biol. 2007;176:995–1005. doi: 10.1083/jcb.200610072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDermott C, White K, Bushby K, Shaw P. Hereditary spastic paraparesis: a review of new developments. J Neurol Neurosurg Psychiatry. 2000;69:150–160. doi: 10.1136/jnnp.69.2.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rugarli EI, Langer T. Translating m-AAA protease function in mitochondria to hereditary spastic paraplegia. Trends Mol Med. 2006;12:262–269. doi: 10.1016/j.molmed.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Agerholm JS, Andersen O. Inheritance of spinal dysmyelination in calves. Zentralbl Veterinarmed [A] 1995;42:9–12. doi: 10.1111/j.1439-0442.1995.tb00350.x. [DOI] [PubMed] [Google Scholar]
- 20.Lindsey JC, Lusher ME, McDermott CJ, White KD, Reid E, Rubinsztein DC, Bashir R, Hazan J, Shaw PJ, Bushby KM. Mutation analysis of the spastin gene (SPG4) in patients with hereditary spastic paraparesis. J Med Genet. 2000;37:759–765. doi: 10.1136/jmg.37.10.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chinnery PF, Keers SM, Holden MJ, Ramesh V, Dalton A. Infantile hereditary spastic paraparesis due to codominant mutations in the spastin gene. Neurology. 2004;63:710–712. doi: 10.1212/01.wnl.0000135346.63675.3e. [DOI] [PubMed] [Google Scholar]
- 22.Svenson IK, Kloos MT, Gaskell PC, Nance MA, Garbern JY, Hisanaga S, Pericak-Vance MA, Shley-Koch AE, Marchuk DA. Intragenic modifiers of hereditary spastic paraplegia due to spastin gene mutations. Neurogenetics. 2004;5:157–164. doi: 10.1007/s10048-004-0186-z. [DOI] [PubMed] [Google Scholar]
- 23.Reid E, Connell J, Edwards TL, Duley S, Brown SE, Sanderson CM. The hereditary spastic paraplegia protein spastin interacts with the ESCRT-III complex-associated endosomal protein CHMP1B. Hum Mol Genet. 2005;14:19–38. doi: 10.1093/hmg/ddi003. [DOI] [PubMed] [Google Scholar]
- 24.Evans K, Keller C, Pavur K, Glasgow K, Conn B, Lauring B. Interaction of two hereditary spastic paraplegia gene products, spastin and atlastin, suggests a common pathway for axonal maintenance. Proc Natl Acad Sci USA. 2006;103:10666–10671. doi: 10.1073/pnas.0510863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol. 2004;30:576–584. doi: 10.1111/j.1365-2990.2004.00587.x. [DOI] [PubMed] [Google Scholar]
- 26.Tarrade A, Fassier C, Courageot S, Charvin D, Vitte J, Peris L, Thorel A, Mouisel E, Fonknechten N, Roblot N, Seilhean D, Dierich A, Hauw JJ, Melki J. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum Mol Genet. 2006;15:3544–3558. doi: 10.1093/hmg/ddl431. [DOI] [PubMed] [Google Scholar]
- 27.Errico A, Ballabio A, Rugarli EI. Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum Mol Genet. 2002;11:153–163. doi: 10.1093/hmg/11.2.153. [DOI] [PubMed] [Google Scholar]
- 28.Evans KJ, Gomes ER, Reisenweber SM, Gundersen GG, Lauring BP. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J Cell Biol. 2005;168:599–606. doi: 10.1083/jcb.200409058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferreirinha F, Quattrini A, Pirozzi M, Valsecchi V, Dina G, Broccoli V, Auricchio A, Piemonte F, Tozzi G, Gaeta L, Casari G, Ballabio A, Rugarli EI. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest. 2004;113:231–242. doi: 10.1172/JCI20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reid E, Kloos M, Shley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am J Hum Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bakowska JC, Jenkins R, Pendleton J, Blackstone C. The Troyer syndrome (SPG20) protein spartin interacts with Eps15. Biochem Biophys Res Commun. 2005;334:1042–1048. doi: 10.1016/j.bbrc.2005.06.201. [DOI] [PubMed] [Google Scholar]