

Abstract
Ubiquitin-interacting motifs (UIMs) are an important class of protein domains that interact with ubiquitin or ubiquitin-like proteins. These approximately 20 residue-long domains are found in a variety of ubiquitin receptor proteins and serve as recognition modules towards intracellular targets, which may be individual ubiquitin subunits or polyubiquitin chains attached to a variety of proteins. Previous structural studies of the interactions between UIMs with ubiquitin have shown that UIMs adopt an extended structure of a single α-helix, containing a hydrophobic surface with a conserved sequence pattern that interacts with key hydrophobic residues on ubiquitin. In light of this large body of structural studies, details regarding the presence and roles of structural dynamics and plasticity are surprisingly lacking. In order to better understand the structural basis of ubiquitin-UIM recognition, changes in the structure and dynamics of ubiquitin have been characterized upon binding of a UIM domain from the yeast Vps27 protein. The solution structure of a ubiquitin-UIM fusion protein designed to study these interactions is reported here and found to consist of a well-defined ubiquitin core and a bipartite UIM helix. Moreover, we have studied the plasticity of the docking interface as well as global changes in ubiquitin due to UIM binding at the picosecond to nanosecond and microsecond to millisecond protein motions by NMR relaxation. Changes in generalized order parameters of amide groups show a distinct trend toward increased structural rigidity at the UIM-ubiquitin interface relative to values determined in unbound ubiquitin. Analysis of 15N CPMG relaxation dispersion measurements suggest the presence of two types of motions, one directly related to the UIM-binding interface, the other being induced to distal parts of the protein. This study demonstrates a case where localized interactions among protein domains have global effects in protein motions at timescales ranging from picoseconds to milliseconds.
Keywords: Ubiquitin Interacting Motif, Ubiquitin Fusion Protein, solution NMR structure, conformational exchange, protein recognition dynamics
Introduction
The diverse functions of ubiquitin in the cell are mediated through its association with specific protein domain modules that have been evolutionarily conserved from yeast to humans 1. Among these ubiquitin-binding domains (UBD), the ubiquitin-interacting motif (UIM) 2; 3 is a recurrent theme in ubiquitin-interacting proteins, which are also known as ubiquitin receptors. The biological functions that are regulated by UIM interactions with ubiquitin include a variety of protein trafficking pathways, the regulation of gene expression and ribosomal function, the budding of virus particles and the regulation of DNA replication and repair (reviewed in references 4; 5).
The role of UIMs is to recognize ubiquitin domains, either as individual ubiquitin subunits or polymeric chains, and to provide an interface for processing the linkage information encoded into poly-ubiquitin chain signals which are attached to a variety of cellular proteins. Furthermore, UIMs may promote ubiquitylation of their host proteins, as exemplified for members of the yeast endocytosis machinery 6; 7. Ubiquitylation is the post-translational modification process of covalently linking ubiquitin through its C-terminal glycine residue to a lysine ε-amino group of a target protein of the cell. This pathway proceeds through a cascade involving enzymes E1, E2 and ubiquitin-protein ligases (E3) 8; 9. Ubiquitin itself may form polymeric chains of various unit lengths and linkages involving distinct lysine residues of ubiquitin. Several lines of evidence have established that the number and connectivity of individual ubiquitin subunits in the attached chain determines the fate of the modified protein 10. For example, chains of four or longer units linked via K48 or K29 signal for proteasome-targeted proteolysis 11 while K63-linked chains lead to non-proteolytic pathways including DNA repair 12 and monoubiquitination has been shown to promote the internalization of plasma membrane proteins and other protein trafficking processes 13 (for a more complete description of ubiquitylation-dependent pathways see reference 5). UIMs directly participate in the formation, transfer and trafficking of poly ubiquitin chains attached to specific proteins of the cell by providing a ubiquitin recognition interface to a diverse range of ubiquitin receptor proteins.
The structure of UIMs and their interactions with ubiquitin have been in the focus of several investigations. Solution studies focusing on UIMs from the S5a, Vps27 and Hrs proteins report a common UIM binding surface on ubiquitin formed by key hydrophobic residues L8, I44 and V70 14; 15; 16; 17; 18. The three highly conserved residues form a continuous hydrophobic patch on the surface of ubiquitin and ubiquitin-like proteins that interacts with modest affinity (micromolar range) with a variety of proteins, such as the E2 and deubiquitinating enzymes 19. These studies provide the structural basis for sequence conservation patterns of the two interacting partners and previous results from alanine scanning mutagenesis experiments 20. The high resolution crystal structure of the Vps27 UIM, which is involved in endosomal sorting 21; 22, in the absence of ubiquitin revealed the presence of an ideal amphipathic α-helical conformation 14. In the later study the unit cell contained four individual UIMs arranged as a four-helix bundle suggesting the possibility of UIM oligomerization with a functional role in the cell. Nevertheless, this class of UIMs engages ubiquitin in its monomeric form, as exemplified in the solution structure of the Vps27 UIM-ubiquitin complex 23. This study has confirmed previous findings on UIM/ubiquitin interactions and further supported that the UIM adopts an extended α-helical conformation 23. Deviations from this ideal α-helical geometry have been previously reported in the solution structures of the S5a UIM in complex with a ubiquitin-like domain 24 and di-ubiquitin 25. Both of these studies report an N-terminal hairpin conformation that forms part of the interaction interface on the UIM. The structural diversity among UIM-ubiquitin interactions is also exemplified by the solution structures of the two tandem UIMs from the S5a proteasome subunit: a recent NMR study reported distinct ubiquitin engaging mechanisms among the two UIMs and further suggested a functional role for distinct binding specificities in the recognition of poly-ubiquitin chains by the proteasome 26. Taken together, these results suggest a plethora of UIM binding modes on a highly conserved ubiquitin surface that are dependent on the atomic details of each system.
While many structural aspects of UIM-ubiquitin recognition have been revealed in these past studies, a detailed characterization for the role of conformational dynamics in UIM recognition is lacking. Protein conformational dynamics are a natural consequence of the energetics of protein structure that are typically manifested as allosteric effects, loop motions, conformational changes and active-site rearrangements, with major implications for biological function 27; 28. In particular, conformational dynamics on the surface of proteins provide the structural diversity that can facilitate molecular recognition through conformational selection mechanisms 29; 30. Conformational dynamics also promote communication among distal parts of proteins, thus explaining conformational coupling effects that are not apparent in terms of average and/or static structures. These include effects on enzymatic activity due to site specific mutations in regions that are far from the active site of enzymes 31 or effects on antigen specificity of mutations in regions of antibodies that do not contact the antigen directly 32. The recent development of robust experimental methods for the evaluation of protein dynamics by nuclear magnetic resonance (NMR) analysis offers a powerful tool to characterize protein motions spanning a wide range of timescales 33; 34 that are relevant to biological function.
As part of an effort to characterize the structural, dynamic and thermodynamic details of the specificity of interactions of UIMs with ubiquitin, we have determined the solution ensemble and characterized conformational dynamics of a fusion protein between the N-terminal UIM from the yeast protein Vps27p and ubiquitin. Fusion proteins are frequently employed for the study of protein-protein interactions 35; 36 or to improve purification and crystallization of proteins of interest 37 and offer the advantage of increased solubility and apparent affinity that results in more efficient binding relative to a bimolecular system. Results from our structure determination process are complemented by backbone mobility measurements describing dynamics at the picoseconds to nanoseconds timescale. Comparative analysis of the results obtained for ubiquitin and the ubiquitin-UIM fusion protein indicate a high degree of plasticity throughout the interface. In agreement with our solution ensemble, inspection of the squared generalized order parameters (S2) suggests the presence of a bipartite UIM helical structure with a dynamic C-terminal region. Furthermore, we characterize the effects of UIM binding to the conformational dynamics of the system using Carr-Purcell-Meiboom-Gill (CPMG) 38; 39 relaxation dispersion experiments which are sensitive to atomic motions on the microseconds to milliseconds timescale. Analysis of CPMG data reveals the presence of conformational dynamics within the binding interface and further indicates long-range dynamical coupling with distal loops of ubiquitin. Notably, we observe changes in the dynamics of lysine residues at positions 6 and 48 which are known ubiquitylation sites involved in linkages with distinct functional profiles 40. These relaxation results complement the structural characterization of the system and reveal effects that could potentially have a functional role in interactions of ubiquitin with downstream components of the endocytosis pathway, or in distinguishing among different types of linkages in polyubiquitin chains. In addition to providing insight into a very important ubiquitin interacting system, the interplay between short range interactions among protein domains and long-range protein dynamics observed here may be widely representative of protein domain interactions.
Results
Structure calculations
The solution structure of the ubiquitin-UIM fusion protein was determined using standard nuclear magnetic resonance spectroscopy methods 41. In general, we obtained well-dispersed NMR spectra from samples free of precipitation, aggregation or degradation over the course of data acquisition, as confirmed with both 1H-15N and 1H-13C HSQC spectra. Near-complete resonance assignments were obtained using a semi-automated approach (BMRB accession number 16114), with a notable exception of H77, whose amide group was not observed in the spectra likely due to extensive line-broadening.
Automated NOE assignments were performed during preliminary structure calculations using the CYANA software suite42 and resulted in the production of 2252 structurally unique distance restraints of which 852 are long-range (Table 1). The final refined ensemble of 100 converged structures was calculated with the inclusion of 71 RDC and 49 hydrogen-bond restraints using a combined torsion angle dynamics/simulated annealing protocol in the program XPLOR 43. The 20 lowest energy conformers that do not show any significant violations in NOE, dihedral restraints and the ideality of bonds and angles are reported in the final ensemble (figure 1). RMSD values for backbone atoms of ubiquitin (residues 1-76) are 0.39Å, which indicates a highly converged structural ensemble. A strong correlation between regions of low NOE density and decreased amide heteronuclear NOE (hnNOE) values is observed, which indicates that regions of high RMSD values in the ensemble are likely structurally dynamic on the picosecond to nanosecond timescale (figure 1). In particular, residues within the C-terminus of the UIM and engineered inter-domain linker have reduced hnNOE values, low inter-residue NOE densities and also appear highly dynamic in the NMR ensemble.
Table 1.
Summary of conformational constraints and statistics for the 20 lowest energy conformations of the Ubiquitin/UIM fusion protein.
| Input Restraints | |
| NOE total | 2252 |
| Intraresidue | 452 |
| Sequential | 576 |
| medium-range | 394 |
| long-range | 830 |
| Dihedral | 158 |
| Φ | 79 |
| Ψ | 79 |
| hydrogen bonds | 49 |
| RDCs | 71 |
| Statistics for accepted structures: | |
| XPLOR energy components (kcal/mol) | |
| NOE | 5.71±1.9 |
| Dihedrals | 2.8±1.2 |
| RDC | 88.3±20.4 |
| Bonds | 8.39±1.9 |
| Angles | 102.4±23.3 |
| Impropers | 12.9±3.0 |
| van der Waals | -565.1 |
| Structural Precision | |
| atomistic RMSD values | |
| Backbone | |
| core residues (10-80) | 0.39 |
| core plus UIM helix (10-80+94-112) | 0.91 |
| heavy atoms | |
| core residues | 1.06 |
| core plus UIM helix | 1.37 |
| Ramachandran plot statistics | |
| most favored regions | 86.1% |
| additional allowed regions | 13.7% |
| generously allowed regions | 0.2% |
| disallowed regions | 0.0% |
Figure 1. Solution structure of the Ubiquitin/UIM fusion protein.

(a) An overlay of the 20 lowest energy conformations of the converged NMR ensemble (PDB ID 2kdi). A superposition of the x-ray structure of Ubiquitin (PDB ID iubq) is shown to illustrate perturbations in the structure induced by UIM binding. (b) Residue-specific structural statistics for the UIM/Ubiquitin complex. NOE constraint densities per residue (top) are compared to the 1H-induced 15N NOE (middle) and residue-specific RMSD values for the NMR ensemble. Under restrained regions in the ensemble (bottom) coincide with the regions of increased plasticity (middle) as illustrated by reduced 1H-induced 15N NOE and NOE density. Residue numbering includes an initial 9 residue-long Histidine-containing sequence.
A well defined ubiquitin domain spanning residues 2-76 is found in the structural models that is with few exceptions consistent with previously reported structures of ubiquitin 44; 45. The overall fold consists of a 5-stranded mixed β-sheet with a strand order 2-1-5-3-4 (two parallel inner strands at positions 1–7 and 64–72, and three other strands spanning residues 10–17, 40–45, and 48–50) an extended α-helix spanning residues 23-34 and a short 310 helix at residues 56-59. Notably, the UIM is found here in a mostly α-helical conformation with a well defined helical segment spanning residues 85-95 and a separate C-terminal helical segment at residues 96-105. This bipartite helical structure is promoted by a break from the ideal α-helical conformation at S95, as indicated by φ,ψ dihedral angles in the range -85 to -100, -20 to -10 among the NMR ensemble members. These structural observations for the UIM domain are supported by the analysis of NOE patterns and RDC values as well as protection patterns from hydrogen exchange with bulk solvent. Furthermore, we observe a type II′ β-turn at the N-terminus of the UIM with sequence ADEE, in agreement with previously reported structures of the S5a UIM bound to ubiquitin-like domains 15; 24, despite the lack of a consensus sequence pattern for this segment. The turn observed here is open, in the sense that there is no hydrogen bonding between the terminal residues, as further indicated by a broader distribution of dihedral angles among the ensemble. This feature is also supported by a distinct NOE density region spanning residues 82-85 (figure 1b). These deviations from an ideal helical structure have not been described in previous solution studies of the Vps27 UIM 23.
Structural characterization of the ubiquitin-UIM interface
The UIM domain docks into a complementary surface of ubiquitin in a parallel orientation to strands β3 and β5 and is engaged on each flanking side by the β-turns connecting strands β1 to β2 and β3 to β4 respectively. Interactions with the core are mediated mainly by two parts of the UIM helix, involving highly conserved residues among UIM sequences (figure 2):
Figure 2. Structure diagram of UIM-Ubiquitin interactions.

UIM binding is accommodated by a continuous hydrophobic patch on ubiquitin's solvent-exposed surface, formed by residues L8, V70, I44 and A56 shown in red from left to right in the structure. The UIM uses hydrophobic residues on two sides of the helical structure (shown as yellow and green sticks respectively) to engage the clamp-like surface on ubiquitin. Additional stabilizing electrostatic interactions are formed between the UIM n-terminal DEEE motif (red) and R42 and R72 on ubiquitin's β3 strand and C-terminus (blue surface).
A highly negatively charged segment at the N-terminus of the UIM (residues 83DEEE); this peptide motif forms electrostatic interactions with R42 and R72 located on strand β3 and the C-terminus of ubiquitin respectively.
An extensive hydrophobic surface of the UIM involving two faces of the helix. One part of this surface is formed by I88, I92 and L96, located on the side that engages the β3-β4 turn of ubiquitin. These residues are involved in an extensive interaction network with the hydrophobic patch of ubiquitin in which I44 plays a central role. The second side of the hydrophobic surface is formed by L87, A91 and L94, which are facing towards the β1-β2 turn of ubiquitin. The sidechains of these residues pack against a hydrophobic surface formed by L8 and V70. The small size of A91, which is located at the center of the UIM helix, is crucial for its placement in a confined hydrophobic environment formed by the side chains of surrounding UIM and ubiquitin residues. This position is universally conserved among UIM sequences.
The formation of intermolecular contacts and structural rearrangements involving ubiquitin residues that line strands β4 and β5, as well residues that compose the β1-β2 and β3-β4 turns are observed upon UIM binding. As shown in figure 1a, a superposition of the crystal structure of wild-type ubiquitin 44 highlights the presence of significant structural perturbations in these two turns as a consequence of UIM binding (figure 1). We observe key hydrophobic interactions of the UIM helix with L8 from the β1-β2 turn, the 42RLI45W segment at strand β3, 46AGK49Q at turn β3-β4 and the 68HLVLRL74R sequence that includes the C-terminal part of strand β5. These regions are highlighted in Figure 2. The UIM-contacting residues are highly conserved among the sequences of ubiquitin and ubiquitin-like domains, thus indicating that both hydrophobic and electrostatic contributions are essential for ubiquitin-UIM recognition (for a comprehensive review of UIM-ubiquitin complexes see reference 1).
The interaction surface and structural perturbations induced upon UIM binding were further characterized here by chemical shift mapping (shown on the left-side structure diagrams of figure 3). These results indicate that UIM in the free form binds ubiquitin at the same interaction surface as observed in the solution structure of the complex formed by the two domains expressed as a single polypeptide chain. The regions that are highlighted here are vey similar to results from a previous NMR mapping study on the interaction between a variety of ubiquitin-like protein domains and the UIM from S5a proteasome subunit 18. Patterns in binding-induced chemical shift changes support the role of residues within the β1-β2 and β3-β4 turns and β3 and β5 strands in binding the UIM helix. In particular, significant perturbations of the amide nitrogen and proton chemical shifts are observed for three groups including residues 8, 51-59, as well as the C-terminal residues 71-76 as shown in figure 3b. These results further highlight the role of conserved residue sites on the surface of ubiquitin for the molecular recognition of UIM domains.
Figure 3. Backbone dynamics of ubiquitin decrease within the UIM interaction surface upon binding.
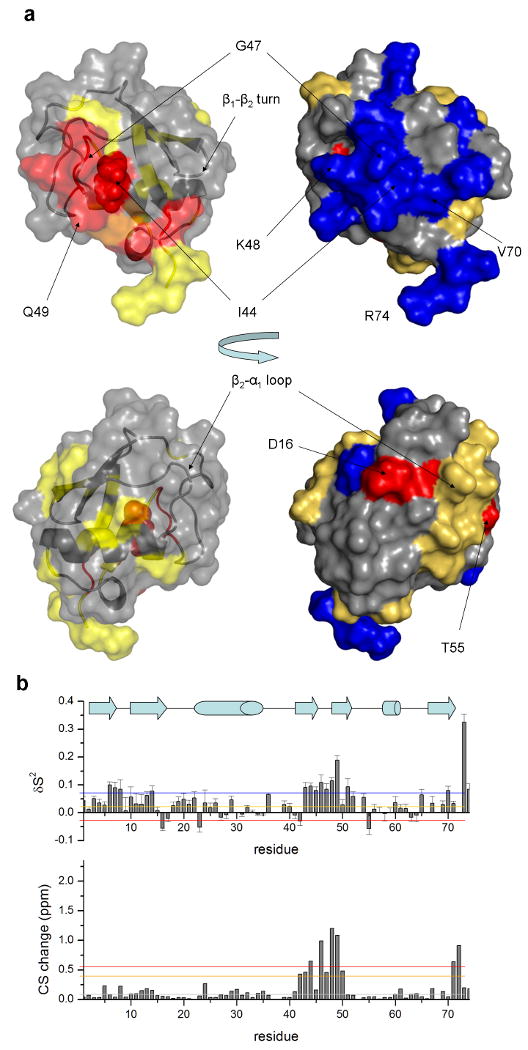
Changes in backbone chemical shifts and fast timescale motions are shown. (a) sites that undergo chemical shift perturbations closely correlate with those that show a decrease in the generalized order parameter. Two opposite views of the molecule are shown, the top containing the UIM-binding interface. Colors are based on grouping according to upper and lower bounds of ppm distances and order parameter changes respectively, as indicated by the color bars in the corresponding graphs (b).
The 6-residue linker peptide that connects the UIM to ubiquitin is found to be unstructured in the ensemble, with no elements of regular secondary structure. We observe no participation of any linker residues in the formation of the ubiquitin/UIM interface and no indication of any steric restrictions, thus concluding that the linker does not distort the structure of the complex.
Plasticity of the Ubiquitin/UIM interface evaluated by ps-ns dynamics
UIM induced changes in fast motions of the ubiquitin backbone were assessed here using model-free analysis of transverse (R2) and longitudinal (R1) 15N relaxation rate constants and hnNOE values at two magnetic field strengths (600 and 800 MHz). Relaxation data were acquired for ubiquitin in an unbound state and in complex with UIM as a fusion construct (see figures S1 and S2). Elevated R2 values were measured at both field strengths for D24 located in the C-terminal end of the first helix in ubiquitin due to the contribution of conformational exchange as reported previously 25. For the fusion protein, in addition to D24, relatively large R2 rate constants were observed for residues N60, D64, S65 and L66 located within the β3-β4 turn and the extended loop that connects β4 and β5. The experimental data were analyzed with the use of the model-free formalism to obtain generalized order parameters. These parameters describe the degree of spatial restriction of the motions of amide bond vectors in the molecular frame and are distributed between 0 and 1 46.
An inspection of the order parameter for the ubiquitin-UIM complex indicates the existence of relatively rigid parts of the molecule which correspond to the different elements of secondary structure as well as more dynamic regions, corresponding to turns and loops that connect these elements. In addition, S2 values below 0.6 were obtained for residues 73 to 83 within the peptide linker that joins the ubiquitin and UIM domains (figure S1), indicating that the linker peptide and D83 located at the N-terminal part of the UIM are highly mobile within the ps-ns timescale. A sharp decrease in all relaxation rates and the hnNOE at both fields is observed after L96 of the UIM that is reflected by a decrease in the order parameter. Taken together, these results support the presence of a relatively dynamic C-terminal segment of the UIM spanning residues 97-105 which retains helical characteristics as shown in the structural models presented here.
Comparison of the generalized order parameters obtained for ubiquitin in the unbound state and in complex with the UIM domain indicates significant changes in ps-ns backbone motions. As illustrated in the right-side structure diagrams of figure 3, these changes are largely found for residues that compose the UIM binding interface but also include sites within distal parts of the molecule. More specifically, a significant restriction in the backbone mobility of the region spanning residues 43-53, a region that includes strands β3, β4 and the extended loop. In this region, K48 and Q49 show the largest reduction in mobility, as indicated by a large increase in order parameter magnitude. Furthermore, residues V70, R74 and G75 of the interaction interface within the ubiquitin-UIM complex also become more restricted in the presence of the UIM. Small increase in the order parameter values is also observed for β1-β2 turn residues 7-9, and 10-14 on strand β2, a motif with extensive interactions with the UIM. In addition, the loop segment 18ESSD22T that connects strand β2 to the extended α-helix becomes less mobile upon UIM binding. However, our results also identify three residues (E16, I23 and T55) that become more dynamic within the ps-ns timescale in the presence of the UIM (>0.05 decrease in the order parameter). These residues are distal to the UIM interface, as they are located on the opposing side of ubiquitin at a distance of more that 20Å in the structure (figure 3), indicating that UIM binding induces long-range changes in fast backbone motions.
Effects of UIM binding on conformational exchange processes
Changes in ubiquitin conformational dynamics upon UIM binding that occur in the μsec-msec timescale range were evaluated using 15N CPMG relaxation dispersion analysis. Relaxation dispersion data were experimentally measured for ubiquitin in the unbound state and in complex with the UIM as a fusion construct at two static magnetic fields and over a range of pulse inversion frequencies, as described in methods. Conformational exchange processes that result in a change in chemical shift of the amide nitrogen can be rigorously characterized through the dependence of the effective relaxation rate on the CPMG frequency 47 (representative examples are shown in figure 5). The data can be analyzed in the context of a model involving states A and B with distinct chemical shifts that exchange stochastically with an intrinsic rate constant. By convention, state B is considered to be the less populated state. As a result, both the relative populations (pA,B) and exchange rate constants (kex = kAB + kBA) can be extracted from the data and the validity of various possible models can be assessed in terms of the resulting χ2 errors of the fit. In addition, calculated differences in chemical shift (Δω) can offer further insight into the structural basis for the conformational exchange process.
Figure 5. Representative CPMG relaxation dispersion data.

Effective transverse relaxation rates are shown as a function of the CPMG power, in units of Hz. For each site, independent fits of the modified form of the Carver-Richards equation (equation 1 in materials and methods) are also plotted. The fitted parameters for these sites can be found at table 2. Residues participating in similar conformational motions, in terms of the global parameters (kex and pA,B) are presented in the same row. From these sites, Lys6, Ile44, Val70 and Leu96 are located on the interaction interface while Leu50 and Leu56 on the extended loop. For the ubiquitin without the UIM the corresponding curves are flat, indicating induced dynamics at the μsec to msec timescale upon UIM docking.
The relaxation dispersion analysis for ubiquitin failed to identify the presence of any significant conformational exchange processes with the exception of residue D24 which also displayed severe line broadening in all spectra. In contrast, extensive conformational exchange was detected for 21 residues in the UIM-ubiquitin complex (Figure 4). The quantification of kinetic and thermodynamic parameters describing the exchange processes were obtained here by fitting relaxation dispersion curves to various forms of the Carver-Richards equation 47, according to the procedure described in methods. Under a two-state exchange model, data were analyzed in a residue specific manner to extract populations and chemical shift differences between the two states, along with the relevant exchange rate constants describing the process (Table 2). An inspection of the extracted parameters that are not sensitive to the chemical environment (kex and pA,B) suggests the presence of at least two distinct exchange processes which are selectively affecting distinct clusters of residues:
Figure 4. Summary of CPMG-derived exchange rates.

As an indication of conformational exchange, the exchange contribution to the transverse relaxation rate, Rex, is shown for amide nitrogen atoms along the sequence of ubiquitin and fusion proteins (a). These values are derived as the difference between peak intensities obtained at zero and high (938Hz) CPMG frequencies. For comparison with the location the UIM binding interface, 15N chemical shift changes in the presence of the UIM are shown (b). No significant exchange contribution to R2 was observed for most sites in ubiquitin (blue), while for the fusion protein the presence of conformational dynamics is manifested as high Rex values at several sites, for both the 600 and 800MHz data (green and red respectively). For Asp24, Rex values could not be accurately measured due to extended line broadening in both proteins, which indicates the presence of conformational exchange at the μsec-msec timescale.
Table 2.
Summary of CPMG-derived parameters that describe exchange processes are shown for 21 sites that participate in conformational motions.
| Residue | kex(sec−1) | pB | ΔωCPMG(ppm) | ΔωCSM(ppm) * |
|---|---|---|---|---|
| 3 | 1751±361 | 14%±2% | 0.5±0.2 | 0.06 |
| 6 | 1165±237 | <1% | 3.0±0.2 | 0.10 |
| 7 | 1812±522 | <1% | 1.9±0.9 | 0.07 |
| 13 | 1153±349 | <1% | 4.0±0.3 | 0.43 |
| 14 | 921±393 | <1% | 4.3±0.4 | 0.37 |
| 20 | 2371±662 | <1% | 2.8±0.9 | 0.07 |
| 43 | 2676±334 | 8%±3% | 1.2±0.2 | 1.12 |
| 44 | 1480±593 | <1% | 4.2±0.7 | 1.51 |
| 45 | 3358±1019 | 9%±7% | 1.3±0.4 | 0.40 |
| 48 | 939±634 | <1% | 2.9±0.7 | 2.92 |
| 49 | 2115±149 | 5%±3% | 1.4±0.4 | 2.63 |
| 50 | 2001±341 | 14%±3% | 1.1±0.7 | 1.16 |
| 51 | 1804±766 | 50%±3% | 0.7±0.6 | 0.06 |
| 56 | 1403±538 | 30%±10% | 0.3±0.4 | 0.04 |
| 60 | 1463±535 | 3%±0.1% | 6.7±0.5 | 0.21 |
| 64 | 1941±696 | 10%±3% | 1.5±0.2 | 0.23 |
| 65 | 2572±316 | 50%±10% | 1.2±0.1 | 0.02 |
| 66 | 2568±516 | 5%±2% | 1.1±0.2 | 0.03 |
| 70 | 1882±495 | 3%±1% | 2.5±0.7 | 0.06 |
| 92 | 957±454 | <1% | 2.5±0.6 | - |
| 96 | 1668±576 | 38%±9% | 0.6±0.3 | - |
ΔωCSM (ppm) : 15N Chemical shift change in the presence of the UIM from Chemical shift mapping. The error in this measurement is at the order of 0.04ppm.
A fast exchange process (exchange rate in the range 1,600-1,800 sec-1) with a small fraction of the population in state B (<10% of the ensemble) affecting 11 out of 21 sites. These sites were also shown by chemical shift mapping to have significant UIM-induced changes in the chemical shift of the nitrogen atoms.
A fast exchange process (exchange rate in the range 1,400-2,200 sec-1) with a significant fraction of the population in state B (>10% of the ensemble) that affects 7 sites (6 on ubiquitin and 1 on the UIM). In general, these sites do not show significant changes in chemical shift upon UIM binding.
In more detail, many of the residues that participate in exchange process (a) compose the interaction interface. I44, located at the center of the hydrophobic surface of the UIM recognition site, shows a clear CPMG frequency dependence that is consistent with this exchange process (shown in figure 5). In addition similar kex and pA,B parameters were obtained for the residues 48KQ in the β2-β3 turn. These residues interact directly with the UIM and also contact V70 on strand β5, a key residue within the hydrophobic patch on ubiquitin that is centered about I44. The values for the fit change in chemical shift due to exchange are in the range 0.5-6.6ppm. Although these values do not correlate quantitatively with the observed changes in the chemical shift of nitrogen atoms as obtained by chemical shift mapping, the location of the exchange sites coincides with sites showing strong chemical shift perturbations (Table 2 and figure 4). Furthermore K6 and T7, located at the C-terminus of strand β1, I13 and T14 at the center of strand β2 participate in this process. In addition this process appears to involve S20 and N60 which are found to form a stable tertiary interaction in the solution structure but are located at a distance of 22.8 and 16.5Å from UIM residues in the ubiquitin complex, respectively. However, these sites are connected to secondary structural elements that change conformation upon UIM binding and interact directly with the UIM (strands β2 and β5 respectively). Given the structural proximity and similarities in fitted parameters, this fast exchange process also likely involves UIM residue I92. Taken together, these results suggest the presence of a conformational exchange process that corresponds to a collective structural rearrangement of the hydrophobic docking interface that extends to structurally coupled sites in ubiquitin. This is further supported by the fact that several sites affected by this process also show significant perturbations of the nitrogen chemical shifts upon UIM binding (Table 2).
The second exchange process is identified for residues within the β3-β4 hairpin and turn and the extended loop that bridges strands β4 and β5. Since residues within the β3, β4, and β5 strands compose key elements of the UIM recognition site, a correlated conformational rearrangement of these structural elements appears to become active in the presence of the UIM domain. This conformational exchange process may be induced by perturbations in the network of interactions within ubiquitin's fold, as a result of UIM interactions. The presence of a significant fraction of the population in state B that is interconverting on the μsec timescale with state A (population 10-50%, kex >1,000Hz) indicates that both the activation barrier and free energy difference between the two conformations is small. The sites that are perturbed by this motion are L50 and E51 located at the end of strand β4 and following turn β3-β4, L56 which initiates the short 310-helix, and the two residues D64 and S65 at the beginning of strand β5 (figure 6). I3, contacting D64 in the structure is also part of this process. Also, the UIM residue L96 also participates in this second conformational exchange process since a similar population ratio (62:38) and exchange rate constant were obtained. As previously indicated, a break in the UIM helix continuity is observed at S95 about which the C-terminal helical segment appears to have rotational freedom (figure 1).
Figure 6. UIM-induced conformational dynamics in ubiquitin.

The sites that participate in conformational exchange processes are shown on the structure of the fusion protein. Interactions with the UIM induce conformational motions on the docking interface residues in addition to a second coordination layer of atoms and distal sites located in the extended loop that connects strands 3 and 4. Sites are color-coded according to the exchange process they participate in. Red: Process a – reorganization of the interface; Blue: Process b – induced loop motions; Magenta – undecided. The sidechains of Lysines at positions 6 and 48 that are confirmed ubiquitylation sites 40 are also shown. Raw relaxation data for a selection of these sites is shown in Figure 5 while fit parameters in table 2.
Ubiquitin residues L43, W45 and T66 were also observed to undergo conformational exchange process, however uncertainties in the extracted populations were too large to discriminate between the two aforementioned clusters or the presence of a third exchange process. The side chain of L43 is facing towards the hydrophobic core of ubiquitin, while the side chains of W45 and T66 are oriented towards the UIM but at a larger distance than the directly interacting residues (6-9Å) (figure 6). The amide of W45 is hydrogen-bonded with the carbonyl of N49 which interacts directly with the UIM, while T66 is located within the length of a solvent-separated contact from S95 of the UIM. In summary, these three residues appear to form a second coordination layer that, although does not interact directly with the UIM, is dynamically influenced by its presence.
Discussion
We have characterized by solution NMR the structure and dynamics of a fusion protein designed to investigate the interactions and molecular recognition between ubiquitin and ubiquitin-interacting motifs. Our results provide novel insight into structural details and dynamic features of the UIM interface and the structural basis for the observed sequence conservation patterns among UIMs. These findings should establish the foundation for further experiments to study structure-function relationships of the molecular recognition process in the ubiquitin-UIM system.
Overall, the features of the interaction interface observed in the solution ensemble are in agreement with previous studies of the Vps27 UIM-ubiquitin complex 23 and similar systems 15; 18; 24. However, we report a bipartite UIM domain with a well defined helical structure spanning residues 85-95 and a short more flexible helical segment after S95, which corresponds to 3 turns of a 36 α-helix, and a total equivalent helicity of ∼50%. Previous Circular Dichroism and NMR studies have shown that UIM peptides, in both the free and Ubiquitin-bound forms have an α-helical content in the range of 40-45% 14; 17. The presence of a bipartite helix is consistent with the mobility of the backbone, as evidenced by hnNOEs and order parameters (figures S1, S2), where enhanced mobility is observed after S95. To further validate the structure and relative orientation of the helix we performed comparisons of experimentally measured RDC values with those predicted by two different structural models of the ubiqutin-UIM complex: a) the ubiquitin-UIM NMR ensemble presented here and b) a hypothetical model of the structure containing the UIM in an idealized α-helical geometry. While close agreement among experimental and calculated values is found throughout the ubiquitin and N-terminal portion of the UIM domain, experimental RDC values for residues in the short C-terminal helical segment of the UIM (residues 97-107) correlate to a lesser extent with predicted data from the solution structures and the model with idealized helical geometry of the UIM (suppl. figure 3). These results suggest a trend where experimental determined values may be undergoing motional averaging thereby resulting in lower RDC values than those predicted from a given static structure. Such motions could be promoted by the break at S95 that is observed in the NMR ensemble, serving as a point to induce dynamics at the C-terminal UIM segment. In summary, the bipartite conformation of the UIM helix seen in this complex is supported by the NOE, dihedral and hydrogen-bond restraints, and is more consistent with the experimentally determined RDC values than the ideal helical conformation. Taken together, these results support the presence of a more dynamic C-terminus of the UIM helix. Such dynamics may be important in retaining conformational entropy required to enable the UIM to bind additional domains upon interaction with ubiquitin along the endocytosis pathway 1.
Results from the relaxation analysis presented here indicate that UIM binding reduces the amplitude of fast motional dynamics in the backbone throughout the binding interface and induces several conformational exchange processes that appear to involve concerted motions of ubiquitin residues. Notably, these processes involve coupling in sites that are far removed from the binding interface and appear to be free of dynamics on the μsec-msec timescale, as indicated by the CPMG data for ubiquitin alone. The presence of conformational dynamics in ubiquitin has been previously investigated with a variety of NMR relaxation experiments 48; 49; 50; 51. Although these studies identify several sites that undergo conformational exchange, the experimental techniques implemented were sensitive to different timescales and chemical details of the motion. In a recently published study, Massi and coworkers identify the presence of conformational dynamics for I23, N25, T55 and V70 by R1ρ relaxation analysis, a technique sensitive to motions at a shorter timescale relative to CPMG relaxation and longer to model-free analysis 48. Notably, these motions were shown to have a kinetic exchange rate of 25,000sec-1, while the processes reported in the present study occur with an exchange rate at the 1,000-2,000sec-1 regime. In addition, Bodenhausen and coworkers 49 identified exchange processes affecting the backbone amides of I23 and N25, by monitoring the relaxation of zero- and double-quantum coherences as a function of the CPMG pulse frequency. Although the exchange rate reported for these residues is in the 4,200sec-1 range, the dispersion data are sensitive to processes that result in slow, correlated modulations of both the nitrogen and proton chemical environment that cannot be identified with conventional 15N-based CPMG dispersion experiments 51. Taken together, these studies illustrate the importance of using a variety of experimental techniques to obtain a comprehensive view of protein conformational dynamics. In the present study we use results obtained for ubiquitin alone as a control to characterize the plasticity of the ubiquitin-UIM interface. To this extent, 15N T2 relaxation dispersion proves to be a powerful methodology for the study of ubiquitin-UIM interactions.
Analysis of our data indicates that the observed relaxation dispersion effects upon UIM binding arise from dynamics within the interacting UIM interface rather than involving a completely dissociated state. Several lines of evidence support our conclusion that, rather than sampling a completely unbound state, the UIM domain experiences displacements along the interaction interface which are accompanied with structural rearrangements within ubiquitin. These points can be summarized as follows. First, the chemical shifts that have been extracted by fitting to the relaxation dispersion data generally do not agree with the expected changes due to UIM binding, as measured by chemical shift mapping. The error in the fitted values, although relatively large (∼20%, as shown in table 2), are insufficient to account for this discrepancy. In addition, the UIM complex appears to contain widespread dynamics that are not necessarily limited within the bounds of the interaction interface, as identified by chemical shift mapping. Furthermore, the population ratio of the states undergoing exchange as extracted from the CPMG data does not agree with the UIM binding thermodynamics within the fusion protein, as measured using DSC experiments (unpublished data). Due to the highly favorable free energy of UIM binding in the fusion protein (15kJ/mol), a very minor population of the UIM-free state would be expected in the ensemble (<0.25%), and this is clearly not the case for several sites undergoing chemical exchange (Table 2). Finally, the expected exchange rate of UIM binding reaction is inconsistent with the derived rates from the CPMG data. Based on our analysis of published SPR data, we have estimated the kinetic off-rate (koff) to be in the 0.8 sec-1 range. Given the free energy of UIM-ubiquitin association within the fusion construct, we expect such a process to have minimal contribution to the relaxation dispersion measurements. Taken together, although our data do not exclude sampling of completely unbound state in the dynamics of the system, our results support the notion that the reported dynamics from the CPMG experiments reflect structural rearrangements within a bound ubiquitin-UIM complex.
We find that UIM binding induces conformational exchange on the μsec timescale for several remote residues from the interaction interface, including sites with important roles for the function of ubiquitin (Figures 4, 5 and 6). These sites are located mainly on the N-terminus of the β5 strand (sequence 64EST67L) but also on the extended loop (L50, D51, L56 and N60). Notably, with exception of L67, the side chains of these residues are oriented opposite from the UIM interface in the solution structure. Furthermore, our chemical shift mapping results show only minor perturbations in both the proton and amide nitrogen shifts upon UIM binding for these sites (Table 2). Taken together, these results suggest the influence of UIM binding on the motions of sites through mechanisms other than direct interactions. Such effects have previously reported to affect enzymatic function 31 and could be explained in the context of a perturbation/induction model, in which UIM binding induces a perturbation in the network of restraining intermolecular interactions that stabilize ubiquitin, thus promoting structural transitions in sites that are not in immediate proximity to the interface. Such a model of dynamical coupling among distal sites may have a functional significance in modulating interactions of ubiquitin with partners other than UIMs. To this extent, dynamics-mediated coupling among multiple binding sites may act to promote the integration of various signaling inputs, as previously shown for the effects of ligand binding in allosteric protein systems 27.
We report here that lysine residues at positions 6 and 48 have increased μsec-msec motilities in the ubiquitin-UIM complex relative to the unbound form. These two residues have side chains oriented towards the solvent and are well characterized ubiquitylation sites 40 that result in linkages with distinct biological roles 5; 8. For the Rap80 UIM, it has been proposed on a purely structural basis that recognition of K63-linked polyubiquitin chains occurs though a specific spatial arrangement of two tandem UIMs 52. Based on previously published chemical shift mapping observations on the Hrs UIM system, it has been proposed that UIM binding limits the accessibility of K48 to ubiquitylation enzymes by occluding their binding epitopes on ubiquitin 17. Our results suggest the presence of conformational exchange processes that affect the chemical environment of K48, and may also affect its accessibility to other ubiquitin-interacting proteins. In addition, chemical exchange was identified for K6, which is located 5.6Å away from the interface and is suggested to be involved in UIM-dependent ubiquitylation involving the Rap80 protein 53. Such unconventional lysine 6-linked polyubiquitin chains have been shown to be directly involved in non-proteolytic DNA damage response and double-strand (DSB) repair pathways 40; 54; 55. Conformational dynamics involving these residues could enhance their potential to interact with other molecules in spite of the presence of the UIM. Such interactions may stabilize the minor exchange conformations that are reveled by the CPMG experiments. Also, the dynamic coupling reported here provides a mechanism for connecting binding at the UIM interface with structural and dynamic changes at the K6 and K48 ubiquitylation sites. These findings may provide insights for the mechanisms UIMs use to discriminate among different types of linkages in polyubiquitin chains. Similar studies of the dynamics involved in UIM interactions with polyubiquitin chains are required to resolve the functional significance of these motions, as illustrated recently for a di-ubiquitin system 25.
In conclusion, we have conducted a detailed characterization of the structural details and conformational motions involved in the molecular recognition between a UIM domain derived from the Vps27 yeast protein and ubiquitin. Our results identify the presence of multiple conformational exchange processes which are induced upon UIM binding. These processes appear to involve a reorganization of the docking interface, as well as several long-range perturbations in loop motions of ubiquitin, rather that a complete dissociation of the UIM domain. A highly dynamic interaction interface may play a role in the biological function of the Vps27 protein, by facilitating the transfer of ubiquitylated cargo among different components of the endocytosis machinery on the membrane of the multivesicular endosome (reviewed in 1). Notably, the ubiquitin receptors involved in this system use a variety of ubiquitin-binding domains to engage a strikingly similar surface on ubiquitin. In addition, UIM binding induces dynamics in sites K6 and K48 which are directly involved in the interaction of ubiquitin with ubiquitinating enzymes and/or other components of the endocytosis machinery. We propose that dynamics induced by interactions with the UIM may provide a basis for the modulation of such downstream interactions, by perturbing restraining interactions within ubiquitin. Additional relaxation experiments that involve more complete sampling of the 15N spectral density, amide multiple-quantum coherence measurements 49 and relaxation analysis of nuclei within residue side-chains51 would be useful in further resolving the timescales and structural details of these motions, while molecular dynamics simulations of the system would complement this work and provide a model framework for structure-based interpretation of the conformational exchange processes that are accessible to these experimental methods.
Materials, experimental procedures and data analysis
Sample preparation
The expression vector for ubiquitin was generated by PCR step mutagenesis carried out on cDNA encoding ubiquitin (UBQ) to generate a V5A, F45W variant with an N-terminal 6xHis-tag 56; 57. This DNA sequence was further modified for the ubiquitin-UIM fusion construct by extending the C-terminus by multiple primer extension steps in order to fuse the coding sequence (corresponding to residues 258-279) of the n-terminal ubiquitin interacting motif from yeast Vps27 N (UIM), via an SMGG peptide linker segment. GGY residue segment was also added to the C-terminus of the UIM domain. The DNA encoding each construct were subcloned into a pGia expression vector system58 using standard molecular biology protocols.
Uniformly 15N and/or 13C labeled ubiquitin and ubiqutin-UIM samples were recombinately expressed in the E. coli strain BL21(DE3) pLysS. The cells were grown at 37 °C in MOPS minimal media 59 containing 15N-ammonium sulfate and/or 13C–glucose, (Cambridge Isotope Laboratories). Upon reaching mid-log phase, protein expression was induced by the addition of 1 mM of IPTG for 2-3 hours. Cells were harvested by centrifugation at 4,000 rpm and with the cell pellet from 1L of culture being resuspended in ∼35 ml of 8 M urea buffer, 0.1M sodium phosphate, 0.01M Tris and pH 8.5Cells were lysed by French press and proteins were purified from cell lysate using Ni-NTA resin (Novagen) under denaturing conditions, as recommended by the resin manufacturer. Size exclusion chromatography was employed in a second purification step using a Sephadex G-75 (Amersham Pharmacia) column (2.5×100 cm) in 5% acetic acid. As the final purification step, reverse-phase HPLC (Waters Corp.) on C18 column in 0.1% TFE and acetonitrile was employed.
NMR experiments, data processing and structure determination
NMR spectra were recorded at 27.85° C on Bruker AVANCE II spectrometers operating at 600 and 800 MHz and equipped with cryogenically cooled triple resonance probes with z-axis gradients using the Bruker software suite TopSpin2.1. Polypeptide backbone assignments were obtained using the HNCO, HN(CA)CO, HNCACB, and HN(CO)CACB triple-resonance experiments. Side-chain assignments were obtained through the analysis of 3D 13C-separated HCCH-TOCSY and COSY spectra. In addition the (H)CC(CO)NH, (H)CCH-TOCSY, HC(C)H-TOCSY, H(CCO)NH-TOCSY and (H)C(CO)NH straight-through experiments were used for side chain assignments. The pulse sequences for these experiments are described elsewhere 41. Resonance assignment was carried out in a semi-automated manner using the PINE online server 60 and manual processing/validation of the results, and resulted in a completeness of 81% for all backbone and side chain atoms (including the 6-residue polyhistidine-tag). Temperature calibrations were performed using a 4% methanol in d4-methanol sample.
Chemical shift mapping experiments were performed by both titrating free UIM peptide against a solution of purified ubiquitin protein, and by comparison with a cleaved protein without the UIM. The UIM peptide was synthesized as described previously 61. Backbone assignments for the cleaved protein (residues 1-77) were transferred from the full-length fusion protein and further confirmed by additional triple resonance experiments. We observe a linear shift of several peaks in the HSQC spectrum of ubiquitin that correspond to the positions of interface residues. Comparison of our results from 1H-15N HSQC experiments obtained by titrating free UIM peptide to ubiquitin versus the fusion protein supports that the chemical shifts sampled in the fusion protein are very similar to those for the bimolecular complex extrapolated at 100% occupancy (UIM solubility prevented full occupancy from being achieved), indicating a highly stable UIM-ubiquitin interface in the fusion protein. Combined changes in the proton and nitrogen chemical shifts were quantified according to the weighted distance metric: .
Structure determination and final model refinement was guided by a combination of NOE, dihedral angle, hydrogen-bond and RDC restraints that are summarized in the top section of table 1. Multiple NOESY spectra were recorded using mixing times of 80 and 120 ms in the aliphatic and amide regions. Automated assignment of NOE spectra was performed using the CANDID module within the suite CYANA 42; 62. For the derivation of backbone hydrogen-bond restraints we used the preliminary structural ensemble in interpreting hydrogen exchange protection from bulk solvent that was observed in HSQC spectra obtained at different time intervals after transferring 15N-labelled protein in deutarated solvent. This procedure resulted in the construction of 49 hydrogen-bond restraints. Furthermore, backbone dihedral angle restraints were obtained from an analysis of the N, H, C, Cα and Cβ chemical shifts using the program TALOS 63 for 79 residues. Residual dipolar coupling (RDC) values for most amides of the fusion protein were measured under partial alignment conditions in stretched acrylamide gels, prepared according to the procedure described in 64 using the inphase(IP)-antiphase(AP) HSQC experiment 65. Control experiments were performed in isotropic sample conditions with the same buffer. A grid-search using the PALES program 66 was carried out to determine the values of the alignment tensor magnitude (Dα) and rhombicity (R). The converged values of Dα and R were 7.98Hz and 0.249 respectively. The final structure calculation was performed using RDCs in addition to the aforementioned restraints. AQUA3.2 and PROCHECK-NMR 67 in addition to XPLOR energy and restraint violation statistics were used to asses the quality of the ensemble.
Analysis of Relaxation data and error estimation
15N spin-lattice and spin-spin relaxation rates were obtained by standard relaxation recovery and CPMG-type experiments respectively. Pulse sequences for the measurement of T1, T2 and 1H-15N heteronuclear NOE (hnNOE) have been described elsewhere 68; 69; 70. For better probing of the spectral density function, experimental data were recorded at 600.13 and 800.13 MHz. T1 measurements were performed using a series of 12 experiments with relaxation delays ranging from 1 to 3500msec at 600MHz and 1 to 3400msec at 800MHz. T2 measurements were performed using a series of 12 experiments with relaxation delays in the range 15.8-285msec at 600MHz and 15.8-284msec at 800MHz. Heteronuclear NOE spectra were collected with and without proton saturation at both fields.
Peak intensities and background noise levels were extracted with the program SPARKY, and subsequently used to obtain an estimate of the relaxation rates R1 and R2 by fitting to a two-parameter exponential decay model. The quality of the fit is indicated by low χ2 values. Uncertainties in the fitted parameters were estimated using 300 fictitious data sets that were generated by sampling Gaussian distributions centered on the raw data. This procedure, implemented in the program SPARKY, was found to produce a relative error less than 10% for most rates. We also performed fitting to the raw data using the program curvefit (Dr. Arthur Palmer, Columbia University) and background noise levels as an estimate of the uncertainties in the peak heights. This method propagates uncertainties in the fitted parameters using a Monte Carlo simulation of the input data. However, in this case we found that the estimated error was at the order of 1% of the fitted values, therefore we used the higher error values produced by the first method. Errors in the hnNOE values were obtained as described in 71. In total, we measured R1, R2 and hnNOE values for 75 residues from ubiquitin and 103 residues from the fusion protein.
Estimation of the axially symmetric diffusion tensor and model-free analysis
Diffusion tensor properties were obtained using an analysis of the T1/T2 ratio for both the core and fusion proteins 72. Residues corresponding to highly dynamic regions (indicated by hnNOE values less than <0.65), or regions that participate in chemical exchange processes (indicated by a T2 value shorter from the average by more than 1.5 standard deviations), were excluded 73. This led to the analysis of T1/T2 ratios from 64 and 74 residues for the core and fusion proteins respectively. Calculations of the T1/T2 ratio were carried out using a fixed S2 value of 0.9, an N-H bond length of 1.02Å and a 15N chemical shift anisotropy (CAS) of -160ppm. The angle between the amide bond vector and the PAS of the CSA was assumed to be 170 74. For the consideration of a fully anisotropic form of the diffusion tensor, we used the quadratic representation approach 75; 76 as implemented in the program quadric (Dr. Arthur Palmer, Columbia University). Statistical analysis of the predicted ratio for different diffusion models under the F-partial test indicated that an axially symmetric model reproduces the experimental data better than the isotropic or fully anisotropic models for both the core and fusion proteins. A ratio of 1.21 and 0.81 between the axial and perpendicular components of the diffusion tensor was used for subsequent analysis of ubiquitin and the fusion protein respectively. Optimization of model-free forms of the spectral density function 46; 77 to the relaxation data was performed with the program modelfree 4.1 78 using the Fast-model free interface for model selection 79. For these calculations we used a 15N magnetogyric ratio of -2.71, an N-H bond distance of 1.02Å and a value of -160 for the CSA of the 15N atom. Errors in the derived model-free parameters were propagated from the relaxation data using 300 Monte Carlo steps. Further optimization of the diffusion tensor properties during the model-free fitting process yielded values very closed to their initial estimates (1.32 and 0.85 respectively). Differences in the correlation time (equivalent to 1/6Diso) between ubiquitin and the fusion protein were also taken into account.
CPMG relaxation dispersion data acquisition and analysis
Relaxation dispersion data series were acquired at both external magnetic fields using the relaxation-compensated pulse sequences described in 80; 81 under standard Bruker implementations. We used the set of CPMG frequencies 31.25, 62.5, 93.75, 125, 156.25, 187.5, 250, 406.25, 500, 625, 812.5 and 937.5Hz and a total relaxation period of 64ms. In order to minimize off-resonance effects due to the high-power 180 CPMG pulses, data recorder at the high external field were acquired in two separate experiments that made use of different carrier frequencies to cover the desired spectral width. Effective relaxation rates were extracted by their two-point estimates:
where I0 and If are the peak intensities for zero and CPMG pulse inversion frequency respectively, as measured at the end of the τ = 64ms relaxation period. Duplicate points at 3 CPMG frequencies were used to estimate uncertainties in I0 and If, which were that propagated in to obtain experimental errors for the χ2 analysis of the fits.
In order to obtain thermodynamic (chemical shift changes, populations) and kinetic (exchange rates) parameters of the exchange process we used a numerical solution of the expression for a two-site chemical exchange process between states A and B, as initially proposed by Carver and Richards 47, presented here in a more compact form by Davis, Perlman and London 33:
| (1) |
In the above equations, tcp is the time interval between the successive inversion pulses, and are the intrinsic relaxation rates of states A and B respectively, pA and pB are the equilibrium populations of the two states, kex is the exchange rate describing the process and Δω is the chemical shift different between the two states. In the following analysis, we assumed that the intrinsic relaxation rates of the two spin states were identical, thus leading to the fitting of 5 parameters (Δω, pB = 1 − pA, kex and at two magnetic fields) to 24 experimental points (12 for each external magnetic field). We define kex and pB as site-independent parameters, in the sense that they should be equal among different amides participating in the same motion, regardless of the changes in the local chemical environment.
For subsequent analysis, we selected residues for which a) An F-test based on the observed χ2 reduction when going from a model with no exchange (flat curve) to a two-site exchange model is valid at a confidence level of 95%, b) The dispersion profiles obtained at the 800.13MHz experiments show a significant difference (larger than 1 standard deviation) between the effective relaxation rates obtained at zero and the highest pulse inversion frequency, indicating a dependence of on the CPMG inversion frequency and c) experimental errors are less than 20% and there is no significant overlap with other peaks in the HSQC spectrum. This selection process resulted in the identification of 21 sites (19 from ubiquitin and 2 from the UIM) that undergo exchange on the NMR chemical shift timescale.
Each site was fitted individually to the above two-state equation with 5 free parameters, using the procedure described in 82; 83 as implemented in a script generously provided by Dr. Lewis Kay. An attempt to use a three-state model did not lead to a statistically significant reduction in χ2 errors, as shown by an F-test comparison to the two-state model results. Groups of residues participating in similar processes were identified based on the site-independent parameters pB and kex. We performed an analysis of the static field dependence of the exchange rate according to Millet and coworkers to further investigate the timescale of the identified exchange processes 84. This method is based on the estimation of a scaling parameter:
The scaling parameter α was found to be greater than one within error for all sites considered, thus indicating fast exchange, in agreement with the results obtained from fitting to the Carver-Richards equation.
Global fits were also tested for all sites, in addition to those grouped by similarity in populations. However, insufficient decreases in the residual chi-square were obtained to account for the decrease in the degrees of freedom according to a partial F-test of significance. The structural implications of these processes are described in the main text.
Supplementary Material
Spin-lattice and spin-spin relaxation rates as measured by relaxation recovery and CPMG-type experiments respectively, are shown at two fields for both ubiquitin (left) and the fusion protein (right) along with the 1H-induced 15N NOE values. The final rates were fitted to the decay of peak intensities according to the procedures described in methods. High R2 values at Asn24 in ubiquitin and residues Asn24, Asn60, Asp64, Ser65 and Thr66 in the fusion protein are due to conformational exchange contributions, as confirmed by the CPMG experiments. Error bars were estimated from the background noise levels and 300 independent Monte Carlo simulations. Smooth lines drawn along the data are shown as a guide.
Extracted order parameters based on a Lipari-Szabo analysis of relaxation rates 46 and hnNOE data measured at two magnetic fields are shown along the primary sequences of the fusion protein (top). Model selection was performed in an automated way to include the effects of sub-picosecond motions and chemical exchange contributions. Order parameters for ubiquitin (bottom) were also extracted from similar experiments and used as a control to assess the effects of UIM binding to the ps-ns timescale plasticity of the interaction interface. Uncertainties in the order parameters were propagated from the relaxation data using 300 Monte Carlo simulations. Splines drawn along the data are shown to guide the eye.
Experimentally obtained RDCs are compared to predicted values for two different models of the ubiquitin-UIM fusion protein. Results are shown for a representative structure from the ubiquitin-UIM NMR ensemble presented in this study (green symbols) versus a hypothetical model of the solution structure that had been modified to contain the UIM in an idealized α-helical geometry (red symbols) based on the model proposed by Swanson and coworkers (PDB ID 1Q0W). Results are shown here for residues 82-107 belonging to the UIM domain and are further separated in two groups: residues 82-96, corresponding to the first, more ordered part of the UIM (circles) and resides 97-107, which are more dynamic. Similar values of the alignment tensor magnitude (Dα) and rhombicity (R) were obtained for both models, after a rigid-body optimization to the experimental data.
Acknowledgments
This work is funded by the NIH grant GM054537 awarded to G.I.M and NSF grant (MCB0543769) awarded to A.E.G.
Footnotes
Accession numbers: Atomic coordinates of the 20 lowest energy conformations have been deposited at the PDB (PDB ID 2KDI), while atom assignment data have been deposited at the BMRB data bank (accession code 16114).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005;6:610–21. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann K, Falquet L. A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem Sci. 2001;26:347–50. doi: 10.1016/s0968-0004(01)01835-7. [DOI] [PubMed] [Google Scholar]
- 3.Young P, Deveraux Q, Beal RE, Pickart CM, Rechsteiner M. Characterization of two polyubiquitin binding sites in the 26 S protease subunit 5a. J Biol Chem. 1998;273:5461–7. doi: 10.1074/jbc.273.10.5461. [DOI] [PubMed] [Google Scholar]
- 4.Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annual Review of Biochemistry. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 5.Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–6. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Polo S, Sigismund S, Faretta M, Guidi M, Capua MR, Bossi G, Chen H, De Camilli P, Di Fiore PP. A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature. 2002;416:451–455. doi: 10.1038/416451a. [DOI] [PubMed] [Google Scholar]
- 7.Miller SL, Malotky E, O'Bryan JP. Analysis of the role of ubiquitin-interacting motifs in ubiquitin binding and ubiquitylation. J Biol Chem. 2004;279:33528–37. doi: 10.1074/jbc.M313097200. [DOI] [PubMed] [Google Scholar]
- 8.Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–78. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- 9.Pickart CM. Mechanisms underlying ubiquitination. Annual Review of Biochemistry. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 10.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 11.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. Embo Journal. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spence J, Sadis S, Haas AL, Finley D. A Ubiquitin Mutant with Specific Defects in DNA-Repair and Multiubiquitination. Molecular and Cellular Biology. 1995;15:1265–1273. doi: 10.1128/mcb.15.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih SC, Sloper-Mould KE, Hicke L. Monoubiquitin carries a novel internalization signal that is appended to activated receptors. Embo Journal. 2000;19:187–198. doi: 10.1093/emboj/19.2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher RD, Wang B, Alam SL, Higginson DS, Robinson H, Sundquist WI, Hill CP. Structure and ubiquitin binding of the ubiquitin-interacting motif. J Biol Chem. 2003;278:28976–84. doi: 10.1074/jbc.M302596200. [DOI] [PubMed] [Google Scholar]
- 15.Fujiwara K, Tenno T, Sugasawa K, Jee JG, Ohki I, Kojima C, Tochio H, Hiroaki H, Hanaoka F, Shirakawa M. Structure of the ubiquitin-interacting motif of S5a bound to the ubiquitin-like domain of HR23B. Journal of Biological Chemistry. 2004;279:4760–4767. doi: 10.1074/jbc.M309448200. [DOI] [PubMed] [Google Scholar]
- 16.Hirano S, Kawasaki M, Ura H, Kato R, Raiborg C, Stenmark H, Wakatsuki S. Double-sided ubiquitin binding of Hrs-UIM in endosomal protein sorting. Nat Struct Mol Biol. 2006;13:272–7. doi: 10.1038/nsmb1051. [DOI] [PubMed] [Google Scholar]
- 17.Shekhtman A, Cowburn D. A ubiquitin-interacting motif from Hrs binds to and occludes the ubiquitin surface necessary for polyubiquitination in monoubiquitinated proteins. Biochem Biophys Res Commun. 2002;296:1222–7. doi: 10.1016/s0006-291x(02)02006-5. [DOI] [PubMed] [Google Scholar]
- 18.Walters KJ, Kleijnen MF, Goh AM, Wagner G, Howley PM. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry. 2002;41:1767–77. doi: 10.1021/bi011892y. [DOI] [PubMed] [Google Scholar]
- 19.Hamilton KS, Ellison MJ, Barber KR, Williams RS, Huzil JT, McKenna S, Ptak C, Glover M, Shaw GS. Structure of a conjugating enzyme-ubiquitin thiolester intermediate reveals a novel role for the ubiquitin tail. Structure. 2001;9:897–904. doi: 10.1016/s0969-2126(01)00657-8. [DOI] [PubMed] [Google Scholar]
- 20.Sloper-Mould KE, Jemc JC, Pickart CM, Hicke L. Distinct functional surface regions on ubiquitin. J Biol Chem. 2001;276:30483–9. doi: 10.1074/jbc.M103248200. [DOI] [PubMed] [Google Scholar]
- 21.Bilodeau PS, Urbanowski JL, Winistorfer SC, Piper RC. The Vps27p Hse1p complex binds ubiquitin and mediates endosomal protein sorting. Nat Cell Biol. 2002;4:534–9. doi: 10.1038/ncb815. [DOI] [PubMed] [Google Scholar]
- 22.Piper RC, Cooper AA, Yang H, Stevens TH. VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae. J Cell Biol. 1995;131:603–17. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swanson KA, Kang RS, Stamenova SD, Hicke L, Radhakrishnan I. Solution structure of Vps27 UIM-ubiquitin complex important for endosomal sorting and receptor downregulation. Embo J. 2003;22:4597–606. doi: 10.1093/emboj/cdg471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mueller TD, Feigon J. Structural determinants for the binding of ubiquitin-like domains to the proteasome. Embo Journal. 2003;22:4634–4645. doi: 10.1093/emboj/cdg467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haririnia A, Verma R, Purohit N, Twarog MZ, Deshaies RJ, Bolon D, Fushman D. Mutations in the hydrophobic core of ubiquitin differentially affect its recognition by receptor proteins. J Mol Biol. 2008;375:979–96. doi: 10.1016/j.jmb.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang QH, Young P, Walters KJ. Structure of S5a bound to monoubiquitin provides a model for polyubiquitin recognition. Journal of Molecular Biology. 2005;348:727–739. doi: 10.1016/j.jmb.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 27.Popovych N, Sun SJ, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nature Structural & Molecular Biology. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kraut DA, Carroll KS, Herschlag D. Challenges in enzyme mechanism and energetics. Annual Review of Biochemistry. 2003;72:517–571. doi: 10.1146/annurev.biochem.72.121801.161617. [DOI] [PubMed] [Google Scholar]
- 29.Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KF, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–5. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 30.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299:1362–7. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 31.Watt ED, Shimada H, Kovrigin EL, Loria JP. The mechanism of rate-limiting motions in enzyme function. Proc Natl Acad Sci U S A. 2007;104:11981–6. doi: 10.1073/pnas.0702551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boder ET, Midelfort KS, Wittrupt KD. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10701–10705. doi: 10.1073/pnas.170297297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis DG, Perlman ME, London RE. Direct measurements of the dissociation-rate constant for inhibitor-enzyme complexes via the T1 rho and T2 (CPMG) methods. J Magn Reson B. 1994;104:266–75. doi: 10.1006/jmrb.1994.1084. [DOI] [PubMed] [Google Scholar]
- 34.Palmer AG., 3rd NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–40. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 35.Johnsson N, Varshavsky A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci U S A. 1994;91:10340–4. doi: 10.1073/pnas.91.22.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwin B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–7. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 37.Graslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, Ming J, dhe-Paganon S, Park HW, Savchenko A, Yee A, Edwards A, Vincentelli R, Cambillau C, Kim R, Kim SH, Rao Z, Shi Y, Terwilliger TC, Kim CY, Hung LW, Waldo GS, Peleg Y, Albeck S, Unger T, Dym O, Prilusky J, Sussman JL, Stevens RC, Lesley SA, Wilson IA, Joachimiak A, Collart F, Dementieva I, Donnelly MI, Eschenfeldt WH, Kim Y, Stols L, Wu R, Zhou M, Burley SK, Emtage JS, Sauder JM, Thompson D, Bain K, Luz J, Gheyi T, Zhang F, Atwell S, Almo SC, Bonanno JB, Fiser A, Swaminathan S, Studier FW, Chance MR, Sali A, Acton TB, Xiao R, Zhao L, Ma LC, Hunt JF, Tong L, Cunningham K, Inouye M, Anderson S, Janjua H, Shastry R, Ho CK, Wang D, Wang H, Jiang M, Montelione GT, Stuart DI, Owens RJ, Daenke S, Schutz A, Heinemann U, Yokoyama S, Bussow K, Gunsalus KC. Protein production and purification. Nat Methods. 2008;5:135–46. doi: 10.1038/nmeth.f.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meiboom S, Gill D. Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Review of Scientific Instruments. 1958;29:688–691. [Google Scholar]
- 39.Carr HY, Purcell EM. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Physical Review. 1954;94:630–638. [Google Scholar]
- 40.Nishikawa H, Ooka S, Sato K, Arima K, Okamoto J, Klevit RE, Fukuda M, Ohta T. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J Biol Chem. 2004;279:3916–24. doi: 10.1074/jbc.M308540200. [DOI] [PubMed] [Google Scholar]
- 41.Rule GS, KT H. In: Fundamentals of Protein NMR spectroscopy FOCUS ON STRUCTURAL BIOLOGY. Kaptein R, editor. Vol. 5. Springer; 2006. [Google Scholar]
- 42.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–78. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 43.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 44.Vijay-Kumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 A resolution. J Mol Biol. 1987;194:531–44. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- 45.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. Journal of the American Chemical Society. 1998;120:6836–6837. [Google Scholar]
- 46.Lipari G, Szabo A. Model-Free Approach to the Interpretation of Nuclear Magnetic-Resonance Relaxation in Macromolecules .1. Theory and Range of Validity. Journal of the American Chemical Society. 1982;104:4546–4559. [Google Scholar]
- 47.Carver JP, Richards RE. General 2-Site Solution for Chemical Exchange Produced Dependence of T2 Upon Carr-Purcell Pulse Separation. Journal of Magnetic Resonance. 1972;6:89–&. [Google Scholar]
- 48.Massi F, Grey MJ, Palmer AG., 3rd Microsecond timescale backbone conformational dynamics in ubiquitin studied with NMR R1rho relaxation experiments. Protein Sci. 2005;14:735–42. doi: 10.1110/ps.041139505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dittmer J, Bodenhausen G. Evidence for slow motion in proteins by multiple refocusing of heteronuclear nitrogen/proton multiple quantum coherences in NMR. J Am Chem Soc. 2004;126:1314–5. doi: 10.1021/ja0386243. [DOI] [PubMed] [Google Scholar]
- 50.Mills JL, Szyperski T. Protein dynamics in supercooled water: the search for slow motional modes. J Biomol NMR. 2002;23:63–7. doi: 10.1023/a:1015397305148. [DOI] [PubMed] [Google Scholar]
- 51.Wist J, Frueh D, Tolman JR, Bodenhausen G. Triple quantum decoherence under multiple refocusing: slow correlated chemical shift modulations of C′ and N nuclei in proteins. J Biomol NMR. 2004;28:263–72. doi: 10.1023/B:JNMR.0000013699.48099.38. [DOI] [PubMed] [Google Scholar]
- 52.Sato Y, Yoshikawa A, Mimura H, Yamashita M, Yamagata A, Fukai S. Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by tandem UIMs of RAP80. Embo Journal. 2009;28:2461–2468. doi: 10.1038/emboj.2009.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan J, Kim YS, Yang XP, Li LP, Liao G, Xia F, Jetten AM. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 2007;67:6647–56. doi: 10.1158/0008-5472.CAN-07-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morris JR, Solomon E. BRCA1 : BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum Mol Genet. 2004;13:807–17. doi: 10.1093/hmg/ddh095. [DOI] [PubMed] [Google Scholar]
- 55.Wu-Baer F, Lagrazon K, Yuan W, Baer R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J Biol Chem. 2003;278:34743–6. doi: 10.1074/jbc.C300249200. [DOI] [PubMed] [Google Scholar]
- 56.Ermolenko DN, Richardson JM, Makhatadze GI. Noncharged amino acid residues at the solvent-exposed positions in the middle and at the C terminus of the alpha-helix have the same helical propensity. Protein Sci. 2003;12:1169–76. doi: 10.1110/ps.0304303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khorasanizadeh S, Peters ID, Butt TR, Roder H. Folding and stability of a tryptophan-containing mutant of ubiquitin. Biochemistry. 1993;32:7054–63. doi: 10.1021/bi00078a034. [DOI] [PubMed] [Google Scholar]
- 58.Gribenko AV, Hopper JE, Makhatadze GI. Molecular characterization and tissue distribution of a novel member of the S100 family of EF-hand proteins. Biochemistry. 2001;40:15538–48. doi: 10.1021/bi0114731. [DOI] [PubMed] [Google Scholar]
- 59.Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–47. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eghbalnia HR, Bahrami A, Wang L, Assadi A, Markley JL. Probabilistic Identification of Spin Systems and their Assignments including Coil-Helix Inference as Output (PISTACHIO) J Biomol NMR. 2005;32:219–33. doi: 10.1007/s10858-005-7944-6. [DOI] [PubMed] [Google Scholar]
- 61.Richardson JM, Lopez MM, Makhatadze GI. Enthalpy of helix-coil transition: Missing link in rationalizing the thermodynamics of helix-forming propensities of the amino acid residues. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1413–1418. doi: 10.1073/pnas.0408004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. Journal of Biomolecular Nmr. 2002;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- 63.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 64.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. Journal of Biomolecular Nmr. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 65.Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. Journal of Magnetic Resonance. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 66.Zweckstetter M. NMR: prediction of molecular alignment from structure using the PALES software. Nature Protocols. 2008;3:679–690. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- 67.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 68.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–9. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 69.Palmer AG, Skelton NJ, Chazin WJ, Wright PE, Rance M. Suppression of the Effects of Cross-Correlation between Dipolar and Anisotropic Chemical-Shift Relaxation Mechanisms in the Measurement of Spin Spin Relaxation Rates. Molecular Physics. 1992;75:699–711. [Google Scholar]
- 70.Boyd J, Hommel U, Campbell ID. Influence of Cross-Correlation between Dipolar and Anisotropic Chemical-Shift Relaxation Mechanisms Upon Longitudinal Relaxation Rates of N-15 in Macromolecules. Chemical Physics Letters. 1990;175:477–482. [Google Scholar]
- 71.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Formankay JD, Kay LE. Backbone Dynamics of a Free and a Phosphopeptide-Complexed Src Homology-2 Domain Studied by N-15 Nmr Relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 72.Tjandra N, Feller SE, Pastor RW, Bax A. Rotational diffusion anisotropy of human ubiquitin from N-15 NMR relaxation. Journal of the American Chemical Society. 1995;117:12562–12566. [Google Scholar]
- 73.Pawley NH, Wang CY, Koide S, Nicholson LK. An improved method for distinguishing between anisotropic tumbling and chemical exchange in analysis of N-15 relaxation parameters. Journal of Biomolecular Nmr. 2001;20:149–165. doi: 10.1023/a:1011249816560. [DOI] [PubMed] [Google Scholar]
- 74.Fushman D, Cowburn D. The effect of noncollinearity of N-15-H-1 dipolar and N-15 CSA tensors and rotational anisotropy on N-15 relaxation, CSA/dipolar cross correlation, and TROSY. Journal of Biomolecular Nmr. 1999;13:139–147. doi: 10.1023/a:1008349331773. [DOI] [PubMed] [Google Scholar]
- 75.Bruschweiler R, Liao XB, Wright PE. Long-Range Motional Restrictions in a Multidomain Zinc-Finger Protein from Anisotropic Tumbling. Science. 1995;268:886–889. doi: 10.1126/science.7754375. [DOI] [PubMed] [Google Scholar]
- 76.Lee LK, Rance M, Chazin WJ, Palmer AG. Rotational diffusion anisotropy of proteins from simultaneous analysis of N-15 and C-13(alpha) nuclear spin relaxation. Journal of Biomolecular Nmr. 1997;9:287–298. doi: 10.1023/a:1018631009583. [DOI] [PubMed] [Google Scholar]
- 77.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. Deviations from the Simple 2-Parameter Model-Free Approach to the Interpretation of N-15 Nuclear Magnetic-Relaxation of Proteins. Journal of the American Chemical Society. 1990;112:4989–4991. [Google Scholar]
- 78.Mandel AM, Akke M, Palmer AG. Backbone Dynamics of Escherichia-Coli Ribonuclease-H1 - Correlations with Structure and Function in an Active Enzyme. Journal of Cellular Biochemistry. 1995:29–29. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 79.Cole R, Loria JP. FAST-Modelfree: A program for rapid automated analysis of solution NMR spin-relaxation data. Journal of Biomolecular Nmr. 2003;26:203–213. doi: 10.1023/a:1023808801134. [DOI] [PubMed] [Google Scholar]
- 80.Loria JP, Rance M, Palmer AG. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. Journal of the American Chemical Society. 1999;121:2331–2332. [Google Scholar]
- 81.Tollinger M, Skrynnikov NR, Mulder FAA, Forman-Kay JD, Kay LE. Slow dynamics in folded and unfolded states of an SH3 domain. Journal of the American Chemical Society. 2001;123:11341–11352. doi: 10.1021/ja011300z. [DOI] [PubMed] [Google Scholar]
- 82.Korzhnev DM, Bezsonova I, Lee S, Chalikian TV, Kay LE. Alternate Binding Modes for a Ubiquitin-SH3 Domain Interaction Studied by NMR Spectroscopy. Journal of Molecular Biology. 2009;386:391–405. doi: 10.1016/j.jmb.2008.11.055. [DOI] [PubMed] [Google Scholar]
- 83.Korzhnev DM, Karlsson BG, Orekhov VY, Billeter M. NMR detection of multiple transitions to low-populated states in azurin. Protein Science. 2003;12:56–65. doi: 10.1110/ps.0225403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Millet O, Loria JP, Kroenke CD, Pons M, Palmer AG. The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. Journal of the American Chemical Society. 2000;122:2867–2877. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spin-lattice and spin-spin relaxation rates as measured by relaxation recovery and CPMG-type experiments respectively, are shown at two fields for both ubiquitin (left) and the fusion protein (right) along with the 1H-induced 15N NOE values. The final rates were fitted to the decay of peak intensities according to the procedures described in methods. High R2 values at Asn24 in ubiquitin and residues Asn24, Asn60, Asp64, Ser65 and Thr66 in the fusion protein are due to conformational exchange contributions, as confirmed by the CPMG experiments. Error bars were estimated from the background noise levels and 300 independent Monte Carlo simulations. Smooth lines drawn along the data are shown as a guide.
Extracted order parameters based on a Lipari-Szabo analysis of relaxation rates 46 and hnNOE data measured at two magnetic fields are shown along the primary sequences of the fusion protein (top). Model selection was performed in an automated way to include the effects of sub-picosecond motions and chemical exchange contributions. Order parameters for ubiquitin (bottom) were also extracted from similar experiments and used as a control to assess the effects of UIM binding to the ps-ns timescale plasticity of the interaction interface. Uncertainties in the order parameters were propagated from the relaxation data using 300 Monte Carlo simulations. Splines drawn along the data are shown to guide the eye.
Experimentally obtained RDCs are compared to predicted values for two different models of the ubiquitin-UIM fusion protein. Results are shown for a representative structure from the ubiquitin-UIM NMR ensemble presented in this study (green symbols) versus a hypothetical model of the solution structure that had been modified to contain the UIM in an idealized α-helical geometry (red symbols) based on the model proposed by Swanson and coworkers (PDB ID 1Q0W). Results are shown here for residues 82-107 belonging to the UIM domain and are further separated in two groups: residues 82-96, corresponding to the first, more ordered part of the UIM (circles) and resides 97-107, which are more dynamic. Similar values of the alignment tensor magnitude (Dα) and rhombicity (R) were obtained for both models, after a rigid-body optimization to the experimental data.