

Abstract
Hippocampal mossy fiber (MF) synapses on area CA3 lacunosum-moleculare (L-M) interneurons are capable of undergoing a Hebbian form of NMDA receptor (NMDAR)-independent long-term potentiation (LTP) induced by the same type of high-frequency stimulation (HFS) that induces LTP at MF synapses on pyramidal cells. LTP of MF input to L-M interneurons occurs only at synapses containing mostly calcium-impermeable (CI)-AMPA receptors (AMPARs). Here, we demonstrate that HFS-induced LTP at these MF-interneuron synapses requires postsynaptic activation of protein kinase A (PKA) and protein kinase C (PKC). Brief extracellular stimulation of PKA with forskolin (FSK) alone or in combination with 1-Methyl-3-isobutylxanthine (IBMX) induced a long-lasting synaptic enhancement at MF synapses predominantly containing CI-AMPARs. However, the FSK/IBMX-induced potentiation in cells loaded with the specific PKA inhibitor peptide PKI6-22 failed to be maintained. Consistent with these data, delivery of HFS to MFs synapsing onto L-M interneurons loaded with PKI6-22 induced posttetanic potentiation (PTP) but not LTP. Hippocampal sections stained for the catalytic subunit of PKA revealed abundant immunoreactivity in interneurons located in strata radiatum and L-M of area CA3. We also found that extracellular activation of PKC with phorbol 12,13-diacetate induced a pharmacological potentiation of the isolated CI-AMPAR component of the MF EPSP. However, HFS delivered to MF synapses on cells loaded with the PKC inhibitor chelerythrine exhibited PTP followed by a significant depression. Together, our data indicate that MF LTP in L-M interneurons at synapses containing primarily CI-AMPARs requires some of the same signaling cascades as does LTP of glutamatergic input to CA3 or CA1 pyramidal cells.
Introduction
The involvement of protein kinase A (PKA) and protein kinase C (PKC) in the induction and maintenance of long-term potentiation (LTP) in hippocampal pyramidal cells has been studied intensively (Nicoll and Malenka, 1995; Soderling and Derkach, 2000). In area CA3, PKA plays a fundamental role in the induction and maintenance of the NMDA receptor (NMDAR)-independent form of LTP at mossy fiber (MF)-to-pyramidal cell synapses (Hopkins and Johnston, 1988; Huang et al., 1994; Weisskopf et al., 1994; Villacres et al., 1998; Yeckel et al., 1999; Calixto et al., 2003). Furthermore, it is well established that PKA modulates the activation of the L-type voltage-gated Ca2+ channels (VGCCs) required for the induction of NMDAR-independent LTP at Schaffer collateral-to-pyramidal cell synapses (Grover and Teyler, 1990) as well as MF-to-pyramidal cell synapses (Kapur et al., 2001). PKC also was found to play a role in LTP at MF-to-pyramidal cell synapses. Early evidence showed that presynaptic PKC activation was required for MF LTP induction (Son and Carpenter, 1996; Son et al., 1997; Henze et al., 2000), and more recently, postsynaptic PKC was shown to be necessary for NMDAR-LTP at MF synapses on pyramidal cells (Kwon and Castillo, 2008).
Numerous studies have revealed the extensive heterogeneity in interneuron long-term synaptic plasticity, especially with respect to the mechanisms controlling the induction and polarity of synaptic change (Kullmann and Lamsa, 2007). However, little is known about the intracellular signaling cascades subserving synaptic plasticity at excitatory inputs to hippocampal interneurons. Previous work has shown that postsynaptic activation of the Ca2+-calmodulin signaling pathway upregulates glutamatergic transmission at inputs to interneurons residing in stratum pyramidale of area CA1 (Wang and Kelly, 2001). Potentiation of dendritic Ca2+ signaling at inputs to CA1 alveus/oriens interneurons was found to require the coactivation of mGluR5s and PKC (Topolnik et al., 2009). Other studies have focused on the role of protein kinases in activity-dependent plasticity at MF synapses on CA3 interneurons. For example, the presynaptic induction of LTD at the MF input to CA3 interneurons in stratum lucidum is mediated by activation of mGluR7 and PKC at MF terminals (Pelkey et al., 2005). Furthermore, the internalization of mGLuR7s enables a presynaptic form of MF LTP that requires PKA activation (Pelkey et al., 2008). In the dentate gyrus, the postsynaptic induction of MF LTP in basket cells was reported to be blocked by the extracellular application of an inhibitor of PKC (Alle et al., 2001). Therefore, it remains an open question as to whether PKC signaling exerts its action on MF LTP within the presynaptic or postsynaptic compartment of the MF-to-basket cell synapse.
We previously demonstrated that HFS delivered to MF synapses on CA3 lacunosum-moleculare (L-M) interneurons containing predominantly Ca2+-impermeable (CI)-AMPA receptors (AMPARs) causes a robust PTP and a NMDAR-independent form of Hebbian LTP at these synapses (Galván et al., 2008). Here we provide compelling evidence that induction of this form of LTP at the MF-to-L-M interneuron synapse requires postsynaptic action of PKA and PKC, signaling molecules typically associated with several forms of LTP at glutamatergic synapses on hippocampal pyramidal cells.
Materials and Methods
Electrophysiological recordings.
Animal use was in accordance with the University Institutional Animal Care and Use Committee. Male Sprague Dawley rats (25 ± 5 d old; Zivic Miller) were deeply anesthetized (Nembutal, 50 mg/kg, i.p.) and perfused intracardially with a modified artificial CSF ACSF in which sucrose was substituted for NaCl (in mm): 230 sucrose, 1.9 KCl, 1.2 Na2PO4*7H2O, 25 NaHCO3, 10 glucose, 1 CaCl2, 4 MgCl2, at 4°C. Following 1–2 min of perfusion, animals were decapitated and the brains removed. Blocks of hippocampus were glued to the stage of a Leica VT1000S and cut in 385 μm thick sections. Slices were maintained for at least 120 min in an incubation solution of the following composition (in mm): 125 NaCl, 2 KCl, 1.2 NaH2PO4, 25 NaHCO3, 10 glucose, 1 CaCl2, and 6 MgCl2. The solution was maintained at pH 7.3 with bubbled with O2 (95%)/CO2 (5%) at room temperature. The slices were then transferred to a submersion recording chamber and superfused at a constant flow rate (2.5 ml/min; 24 s exchange rate including the volume inside the tubing) via a peristaltic pump (Watson Marlow 400) using the following solution (in mm): 125 NaCl, 3 KCl, 1.25 Na2HPO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 10 glucose, 0.01 (−)-bicuculline methobromide, and 0.05 d-AP5 (d-2-amino-5-phosphonopentanoic acid). The temperature of the solution in the recording chamber was maintained at 33 ± 1°C. Interneurons were identified visually with infrared video microscopy and DIC optics. Patch pipettes were pulled from borosilicate glass and had resistances of 3–6 MΩ when filled with a solution containing (in mm) 120 K-methylsulfate, 10 NaCl, 10 KCl, 10 HEPES, 0.5 EGTA, 4.5 Mg · ATP, 0.3 Na2 · GTP, and 14 phosphocreatine. Biocytin (0.1%) was routinely added to the pipette solution to allow subsequent morphological identification and reconstruction of interneurons (Fig. 1A). In experiments aimed at assessing the current–voltage relationship (I–V) of MF AMPA EPSCs, QX-314 (5 mm) was added to the internal solution. Osmolarity was adjusted to 295–300 mOsm with a pH 7.2–7.3. Access resistance was monitored throughout the duration of the experiments using 10 ms, 5 mV voltage steps. Interneurons were accepted for analysis only when the seal resistance was >1 GΩ and the series resistance (4–9 MΩ) did not change >15% during the experiment.
Figure 1.
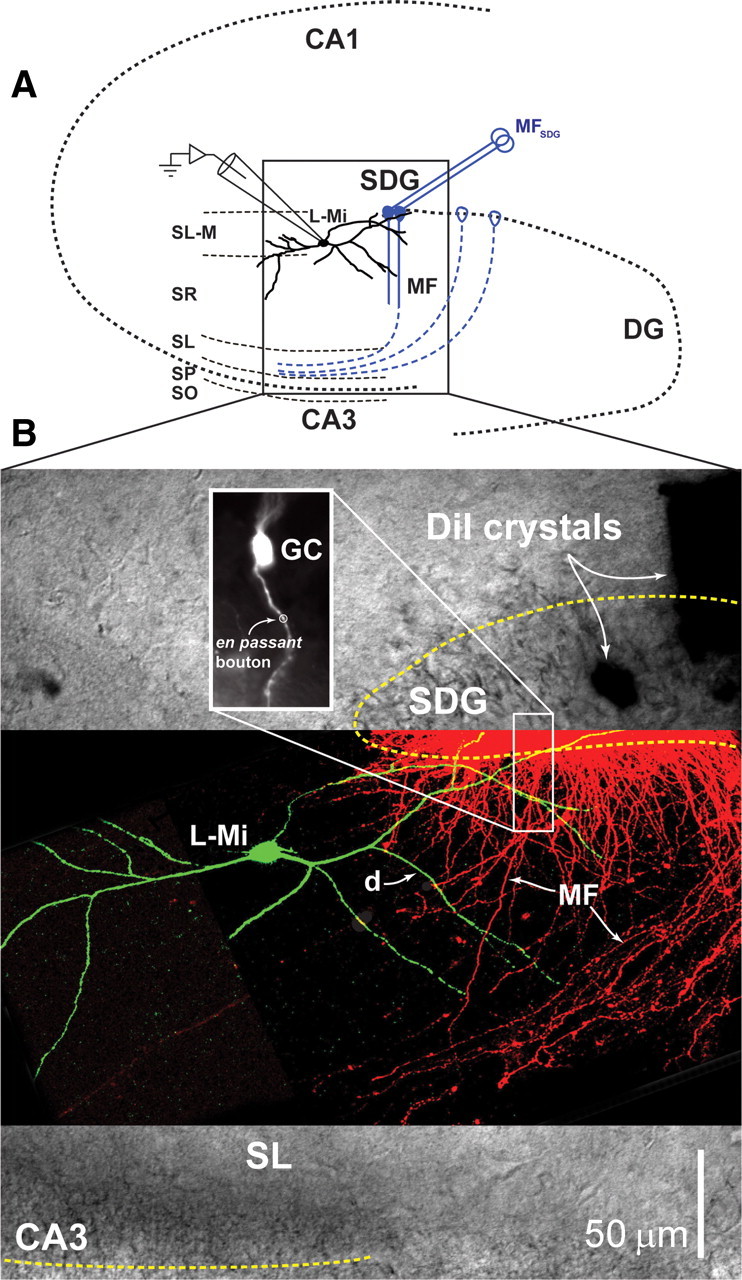
Selective activation of MF synapses onto the spineless dendrite of L-M interneurons. A, Schematic representation of the hippocampus slice preparation showing the position of the bipolar stimulating electrode (MFSDG) placed on the tip of the SDG and the whole-cell recording pipette in a L-M interneuron (L-Mi). The laminar diagram of the hippocampus identifies the CA1 and CA3 regions, the dentate gyrus (DG), the SDG, L-M, stratum radiatum (SR), stratum lucidum (SL), stratum pyramidale (SP), and stratum oriens (SO). B, Microscopic image of the cellular elements. A DIC image of the region identified by the box in A is overlaid with a confocal fluorescent image of a Lucifer yellow-filled L-M interneuron (L-Mi) and DiI-labeled granule cells projecting MFs from the SDG. The inset in the upper left shows an individual granule cell (GC) labeled with DiI, to highlight the presence of several putative en passant boutons along the mossy fiber projection (one of which is identified with a circle) as it courses through SL-M and SR layers of the hippocampus. The inset in the lower right is an enlargement of one region of crossing between mossy fiber axons (MF) and dendrites (d) of the L-M interneuron. In fact, the L-M interneuron dendrites extend across the MF projection at several positions (ranging from close to the SDG to near the SL).
MF synaptic responses were evoked by extracellular stimulation using concentric bipolar electrodes (12.5 μm inner pole diameter, 125 μm outer pole diameter; FHC Inc.) positioned on the suprapyramidal blade of the dentate gyrus (SDG) (Galván et al., 2008) (Fig. 1A). Stimulation provided by an A.M.P.I. Master 8 connected to a stimulus isolation unit (WPI stimulus isolator A365) consisted of single monopolar pulses (100–300 μA; 50–100 μs duration) at ranges of 0.25 Hz to 0.16 Hz. To reduce the probability of antidromic activation of CA3 pyramidal cells via stimulation of their axon collaterals, we used low current intensities that resulted in composite EPSPs with amplitudes <30% of the threshold amplitude required to evoke action potentials in the recorded interneurons. Current- and voltage-clamp recordings were obtained with an Axoclamp-1D amplifier (Molecular Devices) in the presence of (−)-bicuculline methiodide (10 μm) and d-AP5 (50 μm), to block GABAAR- and NMDAR-mediated responses, respectively. Paired-pulse facilitation (PPF) was expressed as the paired-pulse ratio (PPR) between the average amplitude of the second EPSP to that of the first EPSP in the pair. Loading of the peptides (PKI6-22, Rp-cAMPs or chelerythrine) was performed at least 20–25 min before the recording of control MF EPSPs. Signals were low-pass filtered at 5 kHz, digitized at 10 kHz, and stored on disk for off-line analysis. Data acquisition and analysis were performed using customized LabView programs (National Instruments). The group II metabotropic glutamate receptor agonist 2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl) glycine (DCG-IV; 1 or 5 μm) was applied at the end of every experiment to confirm the MF origin of the EPSP. Although inhibition of MF transmission by DGC-IV is consistently complete at synapses on pyramidal cells (≥90%); (Kamiya and Yamamoto, 1997), it is variable at MF synapses on interneurons (Alle et al., 2001; Lawrence et al., 2004). Therefore, synaptic responses were classified as of MF origin only if DCG-IV application resulted in inhibition >70% (Lawrence et al., 2004; Galván et al., 2008). LTP was induced by HFS consisting of 3 trains of 100 pulses delivered at 100 Hz with an intertrain interval of 10 s. Delivery of each stimulation train to MFs was paired with a simultaneous postsynaptic depolarizing current step (30 ± 0.6 pA; 1 s). This postsynaptic depolarizing pulse was strong enough to evoke a robust AP burst.
Drugs.
d-AP5, (−)-bicuculline methobromide, and DCG-IV were purchased from Tocris Bioscience. H-89 was from BioMol. Otherwise drugs were purchased from Sigma Chemical. Forskolin, IBMX, H89, bisindolymaleimide-1, and PDA were dissolved in DMSO at concentrations of 100, 100, 10, 2, and 25 mm, respectively, and then added to the bath solution. The concentration of DMSO in the final bath solution was 0.1%; otherwise, drugs were dissolved in double distillated H2O.
Confocal imaging.
L-M interneurons were filled with Lucifer yellow (1 mg/ml) using visually guided whole-cell patch clamp. After filling the cell, the slice was loaded with DiI crystals in the SDG (location matched typical placement of stimulating electrode described above) to visualize axons from the dentate gyrus granule cells. In a separate set of experiments, DiI crystals were localized in the stratum radiatum to label granule cell soma in a retrograde manner. Slices were returned to the incubation solution for at least 2 h before being fixed and mounted on slides using Fluoromount-G mounting medium. DiI was excited at 514 nm and Lucifer yellow was excited at 488 nm. Images were collected using an inverted Olympus Fluoview 1000 at the Center for Biologic Imaging at the University of Pittsburgh, Pittsburgh, PA.
Immunoperoxidase localizations.
Rats were anesthetized with an intraperitoneal injection of chloral hydrate (300 mg/kg; dissolved in 0.9% saline) and rapidly perfused transcardially with 0.9% saline, followed by 4% paraformaldehyde in 0.1 m phosphate buffer. The brains were removed following perfusion and placed in a 20% sucrose solution until they sank. The brains were frozen and sectioned at a thickness of 40 μm on a cryostat at −16°C and stored in cryopreservative at −20°C (Watson et al., 1986) until staining. To probe the sections for PKA immunoreactivity, sections were brought to room temperature over a 30 min period, washed with sequential rinses in 50 mm Tris-buffered saline (TBS) on a rocker table, pretreated with an antigen retrieval citrate buffer at 80°C for 20 min, and rinsed again several times in 50 mm TBS. The tissue was treated to permeabilize and prevent unwanted nonspecific staining with 50 mm TBS containing 0.4% Triton X-100 and 10% normal goat serum at room temperature for 1 h. Sections then were incubated with an antibody that selectively recognizes the α isoform of the catalytic subunit of PKA (1:1000; Cell Signaling Technology) in 50 mm TBS containing 0.4% Triton X-100 and 10% normal goat serum at 4°C for 48–72 h. To confirm specificity of the immunocytochemical localization, alternate sections were incubated with primary antibody (PKA C-α) that had been preincubated with a blocking peptide (Cell Signaling Technology) following the manufacturer's specifications. Briefly, the antibody was incubated in a solution containing the blocking peptide at 2× concentration of the primary antibody for 1 h at room temperature before incubation of sections. After washing in 50 mm TBS over 45 min, sections were incubated in biotinylated goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories) in 50 mm TBS containing 0.4% Triton X-100 for 90 min, followed by incubation in ABC complex in 50 mm TBS containing 0.4% Triton X-100 (Vectastain Elite Kit) for 90 min at room temperature on a rotator. After washing the sections in 50 mm TBS, immunostaining was visualized with a substrate solution containing DAB 0.6 mg/ml + 0.03% H2O2 + 4 mm NiCl2 in TBS. Sections were washed several times and then mounted on gelatin-coated slides, dried at room temperature, dehydrated in ethanol, cleared in xylene, and coverslipped with Protocol xylene-based medium. Immunocytochemical staining was photographed using an Olympus BX51 photomicroscope equipped with DIC optics. All images were captured using Simple PCI software (C-Imaging Systems). Adobe Illustrator and Adobe PhotoShop software were used to assemble illustrations. The brightness and contrast of images was adjusted using Adobe PhotoShop, but the color balance was not altered.
Immunofluorescence localizations.
To probe the sections for both glutamate decarboxylase (GAD), an interneuron-specific marker, and PKA immunoreactivity, sections were brought to room temperature over a 30 min period, washed with sequential rinses in 0.1 m PBS on a rocker table, pretreated with an antigen retrieval Tris buffer at 80°C for 20 min, and rinsed again several times in 0.1 m PBS. The tissue was pretreated with 0.15 m glycine in 0.1 m PBS to prevent autofluorescence, and then permeabilized and blocked with 0.1 m PBS containing 0.4% Triton X-100 and 10% normal goat serum at room temperature for 1 h to prevent unwanted nonspecific staining. Sections then were incubated with the antibody that selectively recognizes PKA C-α (1:500; Cell Signaling Technology) and an antibody that selectively recognizes the 67 isoform of GAD (1:500; Millipore) in 0.1 m PBS containing 0.4% Triton X-100 and 10% normal goat serum at 4°C for 48–72 h. After washing in 0.1 m PBS over 45 min, sections were incubated in biotinylated goat anti-rabbit secondary antibody (Vector Laboratories) in 0.1 m PBS containing 0.4% Triton X-100 for 90 min, followed by sequential washes and incubation in Cy3-Streptavidine and Cy2-anti-mouse secondary antibody in 0.1 m PBS containing 0.4% Triton X-100 for 90 min at room temperature on a rotator. After washing the sections in 0.1 m PBS, they were mounted on gelatin-coated slides, dried at room temperature, and coverslipped with Fluoromount. Immunofluorescent staining was visualized and captured using a Nikon Diaphot 2000 confocal microscope equipped with a Bio-Rad MRC1024 acquisition system. Improvision Openlab software was used to adjust the brightness and contrast of the images, without altering color balance, and merge the adjusted images for detection of overlap. Adobe Illustrator and Adobe PhotoShop software were used to assemble illustrations.
Statistical analysis.
Group measures are expressed as means ± SEM. Normality of sample distributions was tested with Kolmogorov–Smirnov test, followed by one-way ANOVA and Student–Newman–Keuls pairwise comparisons. In all cases differences were considered significant if P was less than α = 0.05. In figures, statistical significance is denoted as follows: *p < 0.05, **p < 0.01, and ***p < 0.001 (or higher).
Results
Interneurons were identified by visual localization of their soma within L-M (200–300 μm from the boundary between stratum pyramidale and stratum lucidum, 50–180 μm from tip of dentate gyrus) (Ascoli et al., 2009). Recordings were obtained using visually guided whole-cell patch-clamp techniques, as previously described (Calixto et al., 2008; Galván et al., 2008). CA3 interneurons receive synaptic input from MF small en passant varicosities emerging from the parent axon, and from filopodial extensions of the giant MF bouton on CA3 pyramidal cells (Acsády et al., 1998). Figure 1A shows a schematic representation of the hippocampal slice preparation detailing the typical location of L-M interneurons and the stimulating electrode used to evoke responses from MF synapses. Post hoc reconstruction of biocytin-labeled L-M interneurons revealed wide-reaching dendritic branches, some of which course near the SDG (Ascoli et al., 2009), where they are coextensive with MF traveling to stratum lucidum or with MF collateral plexuses in the hilus of the dentate gyrus (Claiborne et al., 1986; Acsády et al., 1998) (Fig. 1B). We have previously shown that focal applications of 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) restricted to the stratum lacunosum-moleculare of area CA3 selectively abolished MF responses in L-M interneurons evoked by stimulation applied to SDG (Cosgrove et al., 2009). We surmise from these data that the results presented here were obtained from MF responses that were selectively evoked by synapses near SDG.
Adenylyl cyclase stimulation induces MF potentiation of naive CI-AMPAR synapses on L-M interneurons
MF synapses on hippocampal interneurons have both CI-AMPARs and CP-AMPARs (Dingledine et al., 1999; Toth et al., 2000; Bischofberger and Jonas, 2002). We previously demonstrated that NMDAR-independent LTP at MF inputs to L-M interneurons is induced exclusively at synapses containing predominantly CI-AMPARs (Galván et al., 2008). CI-AMPAR EPSCs exhibited a nearly linear current–voltage relationship (I–V) whereas CP-AMPAR EPSCs showed strong inward rectification (Tóth and McBain, 1998; Laezza et al., 1999; Galván et al., 2008). Despite that the large and complex dendritic arbors of L-M interneurons (Ascoli et al., 2009) (Fig. 1B) are likely to distort the somatically recorded EPSC waveform due to inadequate space clamp (Williams and Mitchell, 2008), MF synaptic responses can be easily grouped by their nonrectifying or rectifying properties (Fig. 2A).
Figure 2.
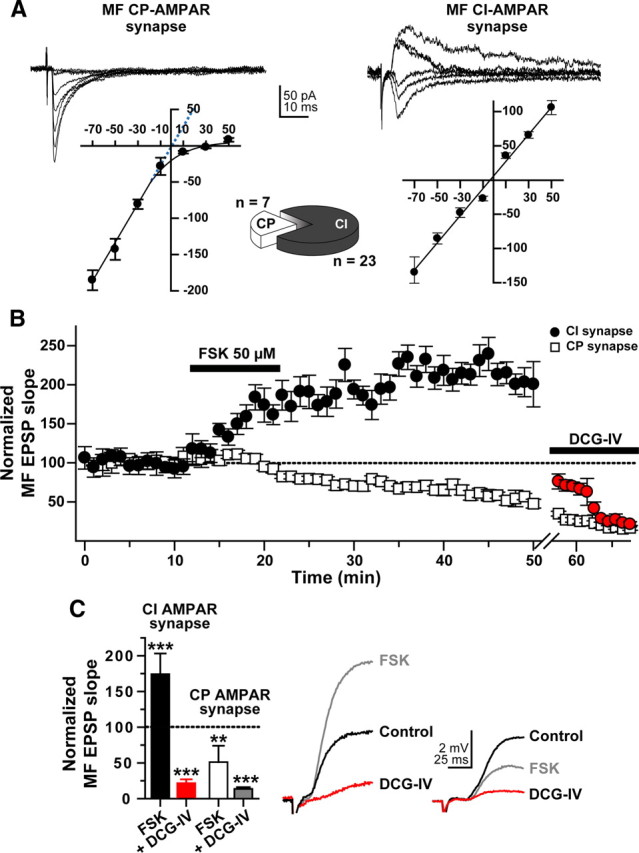
Forskolin induces MF potentiation only at predominantly CI-AMPAR synapses on L-M interneurons. All experiments were performed in the presence of bicuculline (10 μm) and d-AP5 (50 μm). A, Representative current–voltage (I–V) relationship for rectifying CP-AMPAR MF synapses (left traces) and nonrectifying CI-AMPAR MF synapses (right traces) found in L-M interneurons of area CA3. The scatter plot in the left panel shows the rectification characteristic of CP-AMPAR-mediated responses. The dotted blue line in this plot is an extrapolation of the linear regression drawn through the data points between −70 mV and −10 mV to demonstrate that in the absence of rectification, the currents would reverse near 0 mV. The scatter plot in the right panel demonstrates the linearity characteristic of CI-AMPAR-mediated responses, and a reversal near 0 mV. Five EPSCs were evoked at each potential, from −70 mV to +50 mV (20 mV increments) and averaged. Pie chart shows the proportion of CP- and CI-AMPAR responses identified with either AMPAR I–V curves or Philanthotoxin (5 μm). B, Time course profile of normalized slopes of MF-evoked L-M interneuron EPSPs for CP- (open squares) and CI-AMPAR responses (filled circles) to bath application of FSK (50 μm; bar represents the period of drug application, 10 min). Response reduction upon bath application of DCG-IV (5 μm) near the end of the recording period confirms the MF origin of the recorded responses. C, Bar graph summarizing the effect of FSK on the slope of the MF-evoked L-M interneuron EPSP mediated by either CI-AMPARs (filled bar) or CP-AMPARs (open bar) recorded 30 min after FSK wash-out. The 10 min application of FSK induced MF potentiation at all CI-AMPAR synapses (n = 6), whereas synapses containing CP-AMPAR underwent MF depression (n = 3). The effect of DCG-IV on the respective responses is shown as well (red and gray bar, respectively). **p < 0.01; ***p < 0.001 or higher statistical significance. Error bars indicate SEM. Right panel shows representative traces from a CI-AMPAR synapse and a CP-AMPAR synapse.
Activation of adenylyl cyclase with forskolin (FSK) was found to induce NMDAR-independent long-lasting enhancement of MF synaptic transmission to CA3 pyramidal cells (Hopkins and Johnston, 1988; Huang et al., 1994; Weisskopf et al., 1994; Villacres et al., 1998; Calixto et al., 2003) and dentate gyrus basket cells (Alle et al., 2001). Both types of potentiation were shown to be blocked by inhibition of PKA (Huang et al., 1995). Here we induced plasticity at MF inputs to L-M interneurons by bath application of FSK (50 μm for 10 min) and used the differential current–voltage characteristic of CI- versus CP-AMPAR EPSCs to determine the predominant AMPAR type mediating FSK-induced MF synaptic plasticity.
Consistent with our previous report (Galván et al., 2008), L-M interneurons with MF synapses expressing mostly nonrectifying AMPARs constituted the majority of the recorded cells (∼80%; pie chart in Fig. 2). The slope of the MF EPSP evoked from this significant subpopulation of interneurons exhibited a sustained increase in the EPSP slope after wash-out of FSK (174 ± 29% of baseline 30 min after FSK; p < 0.0001, n = 5). In contrast, the slope of MF EPSPs from interneurons with predominantly rectifying AMPARs showed a persistent depression after FSK treatment (51 ± 23% of baseline 30 min after FSK; p < 0.01, n = 3) (Fig. 2B,C), in agreement with previous observations by others (Maccaferri et al., 1998; McBain et al., 1999). Bath application of the group II mGluR agonist DCG-IV (1 μm) confirmed the MF origin of both CI- and CP-AMPAR-mediated responses (86 ± 2% and 78 ± 5% inhibition 20 min after onset of DCG-IV application, respectively; p < 0.001) (Fig. 2B,C). Unlike FSK, 1,9-dideoxyforskolin (50 μm for 10 min, supplemental Fig. 1, available at www.jneurosci.org as supplemental material), an inactive analog of FSK, did not affect either the slope of the MF EPSP (104 ± 5% of control at 10 min postapplication, p > 0.5; DCG-IV sensitivity, 77 ± 29% inhibition, p < 0.001; n = 6) or the paired-pulse ratio (PPR; control PPR: 1.52 ± 0.20; PPR at the end of the 10 min application of 1,9-dideoxyforskolin: 1.51 ± 0.15, p > 0.4, n = 6). Together these data show that transient activation of adenylyl cyclase is sufficient to trigger potentiation of MF EPSPs produced by naive CI-AMPA synapses on L-M interneurons. The data also demonstrate that nonrectifying MF synapses on L-M interneurons are highly responsive to FSK, strongly suggesting that CI-AMPARs may selectively be linked to the activation of the cAMP/PKA cascade.
We also examined the effect of FSK (50 μm) in combination with the cyclic nucleotide phosphodiesterase inhibitor, 1-Methyl-3-isobutylxanthine (IBMX, 50 μm) (Huang et al., 1994; Duffy and Nguyen, 2003), on the isolated MF CI-AMPAR component. Bath perfusion with the polyamine toxin philanthotoxin (PhTx, 5 μm), a selective blocker of CP-AMPARs, minimally decreased the amplitude of MF EPSCs (Vh = −70 mV; PhTx reduction: 9 ± 3% of baseline; p > 0.2; n = 4) (Fig. 3A). At the end of PhTx application, the recording was switched to current-clamp conditions (−70 mV), a baseline was recorded for 8 min, and FSK (50 μm) + IBMX (50 μm) were added to the bath perfusion for 10 min. Compared to the effect of application of FSK, coapplication of FSK/IBMX induced a fast and sustained potentiation of the slope of the MF EPSP that was larger than the potentiation caused by FSK alone (334 ± 54% of control 10 min after onset of FSK/IBMX; p < 0.0001) (Fig. 3A). After wash-out, the slope of the MF EPSP gradually decayed to a plateau level that remained potentiated above control level for at least 30 min after wash-out (170 ± 15.6% of baseline; p < 0.001) (Fig. 3A). DCG-IV (5 μm) was applied at the end of the experiments to confirm the MF nature of the responses (84 ± 3% inhibition; p < 0.001) (Fig. 3A). We use a higher concentration of DCG-IV because the stimulation of the adenylyl cyclase by FSK produces a PKA-dependent phosphorylation of the mGluR2 receptor, which counteracts the selective inhibitory action of group II mGluR activation on MF boutons (Kamiya and Yamamoto, 1997; Schaffhauser et al., 2000).
Figure 3.

The simultaneous inhibition of phosphodiesterases and stimulation of adenylyl cyclase induces potentiation of predominantly CI-AMPAR MF synapses on L-M interneurons. A, Representative experiment showing the time course of the amplitude of MF-evoked EPSCs before PhTx (1), during PhTx (2), after PhTx during baseline recording in current-clamp mode (3), during the combined application of FSK + IBMX (4), 30 min after drug wash-out (4), and during DCG-IV (5). Each circle represents a single EPSC (0.3 Hz) or EPSP (0.2 Hz). First break in the time axis represents the switch from voltage-clamp to current-clamp mode. MF EPSCs were weakly (<8%) sensitive to PhTx (5 μm), and MF EPSPs remained potentiated at least 30 min after the removal of FSK + IBMX. B, Paired-pulse facilitation (paired-pulse ratio, PPR index) was monitored throughout the experiment. PhTx did not alter the PPR in voltage-clamp mode. However, the PPR index decreased during bath-application of FSK + IBMX and partially recovered after wash-out of the drugs. Bars in the panel on the right summarize the PPR results at the time points indicated by the letters in the line graph on the left. C, Summary bar graph of MF-evoked responses during PhTx (n = 4), FSK + IBMX, wash-out, and DCG-IV (n = 11). Insets show representative traces (average of 10 sweeps) of MF-evoked EPSCs (center traces) and the initial phase of MF-evoked EPSPs (right traces) at the times indicated by the numbers in A. ***p < 0.001.
The FSK/IBMX-induced potentiation of the MF EPSP was associated with a decrease in paired-pulse facilitation. A decrease in PPR generally is regarded as indicative of a presynaptic effect due to increase in transmitter release (Weisskopf et al., 1994). Figure 3B shows the time course of the PPR in the presence of FSK/IBMX. Bath-application of PhTx (5 μm) did not alter the PPR (control PPR: 1.44 ± 0.1; PPR during PhTx application: 1.42 ± 0.12, p > 0.3; n = 4). However, PPR decreased significantly during application of FSK/IBMX (control PPR: 1.45 ± 0.07; FSK/IBMX PPR: 0.97 ± 0.08; p < 0.01; n = 4), and recovered only partially during wash-out (1.31 ± 0.06, at 30 min after wash-out).
A comparable MF potentiation as described above also was observed in experiments in which the CI-AMPAR component was not isolated (slope of MF EPSP during the FSK/IBMX application, 397 ± 31% of baseline, p < 0.0001; 30 min after wash-out, 185 ± 30% of baseline; p < 0.001; n = 7; DCG-IV sensitivity, 77 ± 29% inhibition; n = 7) (Fig. 3C). Given that these cells exhibited a level of potentiation upon FSK/IBMX application similar to that observed after isolation of CI-AMPAR-mediated responses, it seems reasonable to assume that the MF-evoked EPSPs recorded here also were mediated predominantly by CI-AMPARs. Because the magnitude of the FSK/IBMX effect did not differ significantly between the two groups of recorded interneurons, the data were pooled for the analysis shown in Figure 3C. Recordings from cells for which the MF EPSP was contaminated by a polysynaptic component that evoked action potentials during FSK/IBMX application were excluded from all analyses.
The demonstration of FSK-mediated potentiation of MF responses in L-M interneurons is indicative that FSK binding sites are present as previously shown for the giant MF boutons on CA3 pyramidal cells (Worley et al., 1986). Therefore, one possible interpretation of these results is that MF potentiation during FSK application is due to a presynaptic effect (Weisskopf et al., 1994; Nicoll and Malenka, 1995; López-García et al., 1996; Villacres et al., 1998). However, there is also strong evidence for postsynaptic regulation of MF synaptic transmission by cAMP-dependent mechanisms (Yeckel et al., 1999; Sivakumaran et al., 2009). Indeed, our data demonstrating that FSK-mediated potentiation is selective for MF synapses containing predominantly CI-AMPARs are consistent with a postsynaptic locus of PKA activation. To obtain direct evidence on the postsynaptic contribution to the MF potentiation induced by stimulation of the cAMP/PKA pathway, L-M interneurons were loaded with the specific cell-impermeant PKA inhibitor, peptide PKI6-22 (20 μm) (Duffy and Nguyen, 2003; Skeberdis et al., 2006; Sivakumaran et al., 2009) before the application of FSK/IBMX. Bath-application of FSK (50 μm) + IBMX (50 μm) resulted in a similar level of potentiation of the slope of MF EPSPs from control versus PKI6-22-loaded cells (maximum slope value in the presence of FSK/IBMX in control cells, 397.4 ± 31% of baseline, n = 10, Fig. 4A, filled circles, B, filled bars; in cells loaded with PKI6-22, 394.7 ± 35.3% of baseline; n = 6) (Fig. 4A, open circles, B, open bars; p > 0.5). The effect of FSK/IBMX on PPR also was similar in control versus PKI6-22-loaded cells (PPR at 10 min of application of FSK/IBMX in control cells, 0.97 ± 0.08; in PKI6-22-loaded cells, 0.96 ± 0.1; p > 0.5; data not shown). However, in contrast to the persistence of the FSK/IBMX-induced potentiation in the absence of the inhibitor (Fig. 4A, filled circles; Fig. 4B, filled bars), the FSK/IBMX-induced potentiation in cells loaded with PKI6-22 was not maintained but decayed rapidly such that the slope of the MF EPSP no longer differed significantly from baseline values 30 min later (116 ± 35% of baseline, p > 0.1; Fig. 4A, open circles; Fig. 4B, open bars). Together, these data suggest that PKA activation in presynaptic elements is sufficient to produce a robust but transient potentiation of transmission at MF synapses on L-M interneurons. However, increased PKA activation in postsynaptic elements is required for the maintenance of FSK/IBMX-induced MF potentiation at synapses on L-M interneurons, similar to the scenario that was described for FSK/IBMX-induced potentiation at synapses on pyramidal cells (Frey et al., 1993; Huang et al., 1994; Duffy and Nguyen, 2003).
Figure 4.
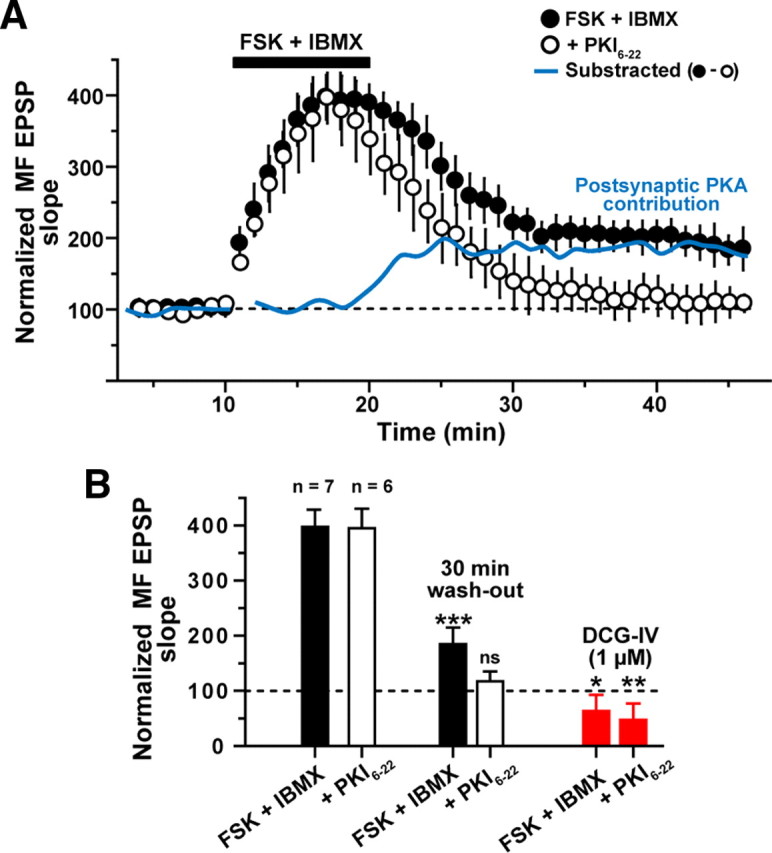
Temporal sequence of projected presynaptic and postsynaptic PKA contributions to MF potentiation. A, Time course of the normalized slope of MF-evoked EPSPs before, during, and after a 10 min application of IBMX + FSK (indicated by filled bar), as recorded either without (filled circles, n = 7) or with the PKA inhibitor PKI6-22 (20 μm; open circles, n = 6) in the patch-pipette. Application of FSK + IBMX induced an enhancement of the MF EPSP that in the absence of the PKA inhibitor persisted for at least 30 min after wash-out of the drugs. In cells loaded with PKI6-22, application of FSK + IBMX induced a similar enhancement that, however, failed to persist; the evoked response decayed to control levels within 15 min after wash-out of the drugs. The subtraction of the normalized values observed in the presence of FSK + IBMX without PKI6-22 minus FSK + IBMX with PKI6-22 in the patch-pipette (blue line) suggests a hypothetical time course for the requirement of postsynaptic PKA activity. The requirement for postsynaptic PKA activity appears to begin before the wash-out of drugs. B, Summary bar graph of MF-evoked responses during and 30 min after FSK + IBMX either without the PKA inhibitor (filled bars) or with PKI6-22 in the patch-pipette (open bars). MF origin of the EPSPs was confirmed by DCG-IV application at the end of the experiment (red bars). *p < 0.05, **p < 0.01, ***p < 0.001.
To determine the time course of the isolated postsynaptic component of the FSK-mediated MF potentiation, we performed a mathematical subtraction between the values observed in the presence of FSK/IBMX and those observed in the presence of FSK/IBMX + PKI6-22. The plot of the residual values suggests that shortly before the beginning of the wash-out, PKA activity in postsynaptic compartments emerges as the critical contributor to the FSK/IBMX-induced MF potentiation at synapses on L-M interneurons (Fig. 4A, blue line). A critical contribution of postsynaptic PKA signaling also was found to apply to the maintenance of the late phase of LTP at hippocampal pyramidal cell synapses (Frey et al., 1993; Huang et al., 1994; Bolshakov et al., 1997; Woo et al., 2002).
Postsynaptic inhibition of PKA prevents HFS-induced LTP at MF synapses on L-M interneurons
The previous results show that postsynaptic PKA activation is necessary for the maintenance of FSK-induced MF potentiation in L-M interneurons. Therefore, we hypothesized that HFS (100 pulses at 100 Hz, 10 s interpulse interval) that triggers LTP at MF inputs to L-M interneurons is sensitive to inhibition of PKA. In agreement with previous work (Galván et al., 2008), HFS delivered to the MF input innervating predominately CI-AMPAR synapses on L-M interneurons (confirmed by only a slight PhTx effect: 6 ± 2% of baseline; n = 11; p > 0.11, supplemental Fig. 2A, available at www.jneurosci.org as supplemental material) caused robust PTP of the MF EPSP (203 ± 6% of baseline) followed by sustained LTP of the slope of the MF EPSP (163 ± 17% of baseline 30 min post-HFS; p < 0.001; DCG-IV inhibition, 67 ± 11% of baseline; n = 11; p < 0.01; supplemental Fig. 2A,B). To test the effect of PKA inhibition on MF LTP, hippocampal slices were incubated in ASCF containing the general kinase inhibitor H-89 (10 μm) for 30–45 min before recording. After obtaining a stable baseline, delivery of HFS induced a robust PTP (181 ± 22% of baseline), but a prolonged potentiation was not observed. Instead, the evoked response returned to baseline level within 30 min (89 ± 16% 30 min post-HFS; n = 7; p > 0.11; DCG-IV (1 μm) inhibition, 70 ± 5%; p < 0.01) (Fig. 5A). To determine whether inhibition of PKA may have contributed to this effect, we included in the patch pipette Rp-cAMP (50 μm), a membrane-permeable inhibitor of PKA activation. Baseline MF EPSPs were obtained for at least 10 min to allow Rp-cAMP to diffuse into the cells. Delivery of HFS to the MF input induced PTP (185 ± 14% of baseline) but failed to induce LTP (87 ± 10% at 30 min post-HFS; n = 8; p > 0.1; DCG-IV sensitivity, 82 ± 10% inhibition; p < 0.001) (Fig. 5A).
Figure 5.

Postsynaptic blockade of PKA prevents LTP at MF synapses on L-M interneurons. A, Bar graphs summarizing MF-evoked responses after delivery of HFS (3 trains with 100 pulses at 100 Hz, delivered with a 10 s intertrain interval) to the MFs. LTP was induced successfully at CI-AMPAR synapses on L-M interneurons (left panel). When slices were incubated (45 min to 60 min) with the PKA inhibitor H-89, LTP was abolished. In cells loaded with either Rp-cAMPs (50 μm), a membrane-permeable inhibitor of PKA activation, or PKI6-22 (20 μm), a membrane-impermeable inhibitor of PKA, a normal level of PTP was preserved but MF LTP failed to develop. For the case of cells loaded with PKI6-22, the CI AMPAR component of the response was isolated using PhTx (5 μm). All the responses recorded were sensitive to DCG-IV (1 μm). **p < 0.01, ***p < 0.001. B, Recordings from a representative experiment in which the patch-pipette was loaded with PKI6-22 (20 μm). Time course of the amplitude of MF-evoked EPSCs or EPSPs before PhTx (1), during PhTx (2), during baseline in current-clamp mode (3), 30 min after HFS (4), and during DCG-IV (5). Each circle represents the amplitude value from a single EPSC (0.3 Hz) or EPSP (0.2 Hz). First break in the time axis represents the switch from voltage-clamp to current-clamp mode. MF-evoked EPSCs were weakly (<5%) sensitive to PhTx (5 μm), and LTP was blocked as a result of loading of the postsynaptic cell with PKI6-22. Traces (average of 10 sweeps) show representative responses at the times indicated by the numbers in the line graph.
Because Rp-cAMP was shown to affect other targets in addition to PKA, its use for determining the role of PKA in LTP has been put into question (Otmakhov and Lisman, 2002; Duffy and Nguyen, 2003). Therefore, to determine the role of postsynaptic PKA in HFS-induced LTP at MF synapses on interneurons, we loaded L-M interneurons with PKI6-22 (20 μm), as described above. In a first group of experiments (n = 4), we assessed the effect of PKI6-22 on the isolated CI-AMPAR component of the MF-evoked response by including PhTx (5 μm) in the perfusion bath before delivery of HFS to the MF. In voltage-clamp conditions (Vh = −70 mV), PhTx moderately reduced the amplitude of MF EPSCs (6 ± 1% of baseline; p > 0.12) (Fig. 5A,B). At the end of PhTx application, recording was switched to current-clamp conditions (−70 mV) and HFS was delivered to the MF input. Following HFS, the isolated CI-AMPAR component of the MF-evoked EPSP exhibited robust PTP (188 ± 30% of baseline), but the potentiation did not persist and the response declined to control levels over the course of the subsequent 10–20 min (102 ± 4% of control level 30 min post-HFS; p > 0.1; DCG-IV (1 μm) sensitivity, 75 ± 4% inhibition; p < 0.0001, Fig. 5A,B). With a second group of experiments (n = 9), we omitted PhTx from the perfusion. The effect of postsynaptic PKA inhibition, however, was similar in that we observed robust PTP (182 ± 19% of baseline; p < 0.001), but the response potentiation failed to be maintained (97 ± 11% at 30 min post-HFS; p > 0.11; DCG-IV sensitivity, 76 ± 7% inhibition; p < 0.001).
Our observations here and in the foregoing section similarly point to PKA in the postsynaptic elements as a critical player in plasticity at MF synapses. Whereas there is ample evidence for localization of PKA in hippocampal pyramidal cells (Gundlach and Urosevic, 1989; Mucignat-Caretta and Caretta, 2002; Havekes et al., 2007), no evidence exists about the presence of PKA in hippocampal interneurons. We therefore immunostained hippocampal sections for the catalytic subunit of PKA and observed abundant immunoreactivity not only in CA1 and CA3 pyramidal cells, dentate granule cells, and MF (Fig. 6A,B), but also in interneurons located in strata radiatum and L-M of area CA3 (Fig. 6B). To confirm that PKA-immunopositive cells in the dendritic strata of CA3 are indeed interneurons, we performed double-immunofluorescence staining hippocampal sections for both the catalytic subunit of PKA (Fig. 6C) and the interneuron-specific marker glutamate decarboxylase (GAD) (Fig. 6D). Merger of the confocal images revealed that most of the cells in strata radiatum and L-M staining immunopositive for GAD also were immunopositive for PKA (Fig. 6E). These findings provide strong evidence for the presence of PKA in CA3 interneurons, including L-M interneurons. Importantly, these findings are in agreement with the idea that PKA signaling within L-M interneurons contributes to plasticity at MF synapses on these interneurons.
Figure 6.
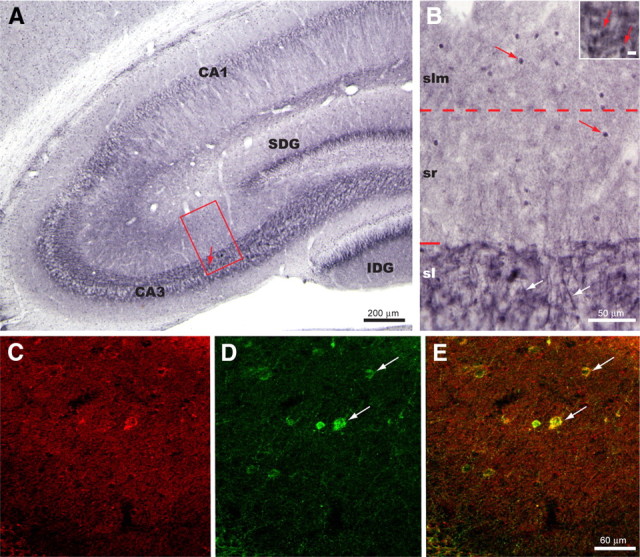
Localization of PKA immunoreactivity within the dorsal hippocampus is illustrated in the coronal plane. A, Immunoreactivity was observed consistently within the apical dendrites of pyramidal cells in areas CA3 and CA1, within the MF pathway of the stratum lucidum (sl), and within interneurons present within strata radiatum (sr) and lacunosum moleculare (slm). The arrow in boxed area identifies the location of the high-magnification inset of B. SDG = suprapyramidal blade of dentate gyrus; IDG = infrapyramidal layer of dentate gyrus. B, Boxed area in A, presented at a higher magnification to illustrate to the best advantage the distribution pattern of PKA immunoreactivity shown in A. Immunopositive apical dendrites of pyramidal neurons (white arrows) can be seen passing through stratum lucidum. Densely stained large varicosities with a distribution and size similar to that observed for mossy fiber boutons were also evident in stratum lucidum (red arrows of inset; marker bar = 10 μm). Immunopositive interneurons are present within both sr and slm but are most prevalent within slm. C–E, Immunofluorescence localizations revealed that interneurons within sr and slm colocalized PKA and GAD. C and D illustrate immunofluorescence localization of PKA (C) and GAD (D) in the same field. Merger of the confocal images shown in E reveals that PKA and GAD immunoreactivity are colocalized in a subset of these sr and slm interneurons (white arrows). Whereas the PKA signal was detected primarily in the nuclear compartment in our immunohistochemical experiments, its appearance was stronger in the cytosol in our immunofluorescent studies. This difference in subcellular detection is likely related to methodological differences between the two types of study.
PKC activation induces potentiation and PKC inhibition prevents HFS-induced LTP at MF synapses on L-M interneurons
Previous work revealed that PKC is present in MF terminals (Worley et al., 1986; Terrian et al., 1991) and is required for induction of LTP at MF synapses on both pyramidal cells (Son and Carpenter, 1996) and basket cell interneurons of the dentate gyrus (Alle et al., 2001). We therefore tested whether direct activation of PKC with phorbol 12,13-diacetate (PDA; 2 μm) affects MF-evoked EPSPs recorded from L-M interneurons. Bath-perfusion of PDA for at least 30 min resulted in a persistent increase in the amplitude of MF EPSPs (200 ± 19% of baseline 20 min after onset of PDA application; n = 6; p < 0.001) (Fig. 7A). The enhancement was associated with a decrease in the PPR (PPR control, 1.56 ± 0.1; PPR during PDA application, 1.03 ± 0.04, p < 0.0001; supplemental Fig. 4, available at www.jneurosci.org as supplemental material) indicating a presynaptic expression. We also examined the effect of PDA on the isolated CI-AMPAR component of MF EPSPs by applying PhTx (5 μm) to the perfusion bath before delivery of HFS to the MF. In voltage-clamp conditions (Vh = −70 mV), PhTx only slightly reduced the amplitude of MF EPSCs (96 ± 2% of baseline; p > 0.10; Fig. 7A,B). At the end of PhTx application, recordings were switched to current-clamp conditions (−70 mV) before bath-application of PDA. Similar to our previous observations, PDA induced a pronounced increase in the amplitude of the isolated CI-AMPAR synaptic component MF-evoked EPSPs (200 ± 21% of baseline 30 min after onset of PDA application; p < 0.001; n = 4), and this potentiation effect was associated with a decrease in the PPR (control PPR: 1.57 ± 0.2; PDA PPR: 1.01 ± 0.01; p < 0.0001; n = 4; supplemental Fig. 3A,B). Because activation of PKC with PDA reduces the ability of group II mGluRs to suppress MF synaptic transmission (Kamiya and Yamamoto, 1997; Macek et al., 1998), we applied an increased concentration of DCG-IV (5 μm) to confirm the MF nature of the EPSPs (DCG-IV sensitivity: 68 ± 2% inhibition; p < 0.001, Fig. 7A,B).
Figure 7.
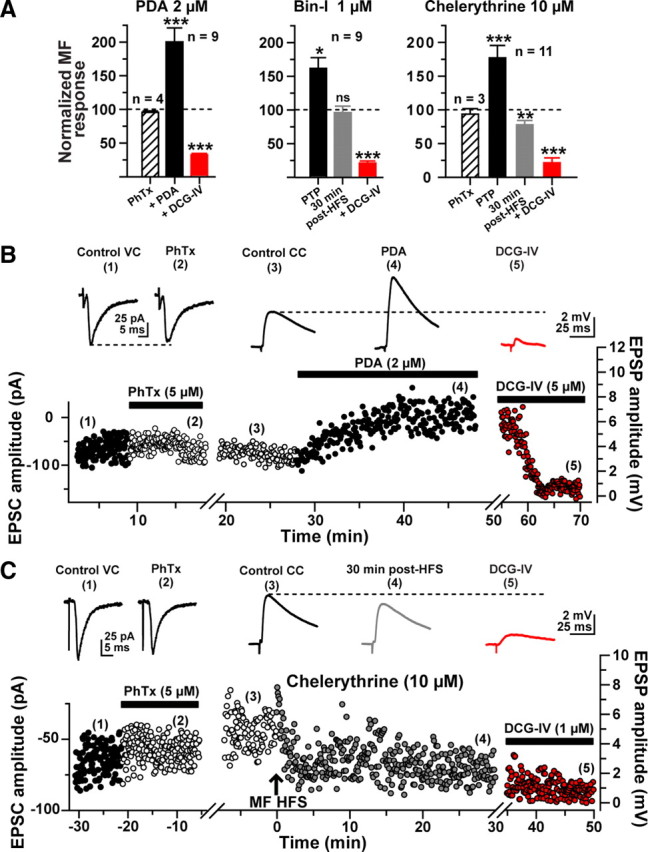
Postsynaptic PKC activity is required for LTP at MF synapses on L-M interneurons. A, Bar graphs summarizing the effect of the phorbol ester phorbol 12,13-diacetate (PDA; 2 μm) on the slope of MF-evoked EPSPs (left); the effect of preincubation (45 min to 1 h) with the PKC inhibitor bisindolylmaleimide I (Bin-I, 1 μm) on LTP induced by delivery of HFS to the MF (center); and the effect of loading of the postsynaptic cell with the PKC inhibitor chelerythrine (10 μm) on MF-induced LTP of the CI-AMPAR component (right). B, Representative experiment showing the time course of the amplitude of MF-evoked EPSCs or EPSPs before PhTx (1), during PhTx (2), during baseline in current-clamp mode (3), during PDA (4), and during DCG-IV (5). Each circle represents a single EPSC (0.3 Hz) or EPSP (0.2 Hz). First break in the time axis represents the switch from voltage-clamp to current-clamp mode. C, Representative experiment showing the CI-AMPAR-mediated component of the EPSC recorded from a typical L-M interneuron. MF-evoked EPSCs were weakly (<5%) sensitive to PhTx (5 μm; n = 3), and LTP was blocked as a result of loading of the postsynaptic cell with the PKC inhibitor chelerythrine. The effect of bath application of DCG-IV (1 μm) confirmed the MF-evoked nature of the recorded responses. Insets are representative traces obtained at the time indicated by the numbers.
Next, we examined whether PKC activity is necessary for HFS-induced LTP at MF synapses on L-M interneurons. We incubated hippocampal slices in the presence of the PKC inhibitor bisindolylmaleimide (Bin-1, 1 μm) for 45 min to 60 min before the start of recordings. As previously reported (Alle et al., 2001), slices pretreated with Bin-1 showed diminished but significant PTP after delivery of HFS (149 ± 9% of baseline; p < 0.05; n = 9), and this blunted potentiation failed to persist, as indicated by a return of the EPSP amplitude to baseline level in the subsequent minutes (96 ± 10% of control 30 min post-HFS, p > 0.1) (Fig. 7A). DCG-IV (1 μm) confirmed the MF origin of these responses (82 ± 5% inhibition; n = 9).
Finally, we tested whether postsynaptic PKC activity is required for MF LTP at synapses on L-M interneurons. To that end, we included the PKC inhibitor chelerythrine (10 μm) in the patch pipette (Kwon and Castillo, 2008). Although chelerythrine had little effect on PTP (183 ± 10% of baseline; n = 3), HFS resulted in a weak but significant depression of the isolated CI-AMPAR component of the MF EPSP (87 ± 4% of baseline 30 min post-HFS, n = 3, p < 0.05; DCG-IV sensitivity: 89 ± 10% inhibition; p < 0.001; Figs. 7A,C). Recordings from cells in which the CI-AMPAR component was not isolated revealed a similar failure to induce LTP, and the development of a slight depression of the MF EPSP when chelerythrine was included in the patch pipette (PTP: 178 ± 9% of baseline; EPSP amplitude at 30 min post-HFS: 78 ± 6%; n = 8; p < 0.01; DCG-IV sensitivity: 82 ± 5% inhibition, p < 0.001, Fig. 7A). Together, our results showing that stimulation of PKC activity with phorbol esters causes an increase in MF-evoked EPSPs of L-M interneurons suggest that PKC can modulate transmission at MF inputs to these interneurons. Our findings that LTP fails to develop in the presence of Bin-1 or chelerythrine indicate that LTP of the CI-AMPAR-mediated component of MF-evoked EPSPs requires PKC. Finally, our observations that postsynaptic application of the PKC inhibitor chelerythrine prevented the development of LTP, despite minimal effect on HFS-evoked PTP, suggest that postsynaptic PKC activity is required for LTP of the isolated CI-AMPAR component of MF synapses on L-M interneurons.
Discussion
Although much is known about the signaling events involved in plasticity at glutamatergic synapses on pyramidal cells, there is considerably paucity of information about the identity and regulation of critical molecular players in plasticity at excitatory synapses on interneurons. The purpose of the present experiments was to assess the role of PKA and PKC in LTP of synaptic transmission at MF synapses on interneurons with soma residing in the stratum lacunosum moleculare of hippocampal area CA3. We previously showed that MF synapses on L-M interneurons that contain mainly CI-AMPARs are capable of undergoing LTP induced by the same HFS protocol that induces LTP at MF synapses on pyramidal cells (Galván et al., 2008). The present results demonstrate for the first time that LTP-inducing activity at naive MF-to-interneuron synapses leads to the downstream activation of postsynaptic signaling cascades that involve the activation of both PKA and PKC. Specifically, we showed that bath application of the adenylyl cyclase activator FSK causes a lasting potentiation at CI-AMPAR-containing MF synapses on L-M interneurons and that this potentiation, as well as HFS-induced LTP at these synapses, is disrupted by introduction of a membrane-impermeable inhibitor of PKA into the postsynaptic cell. We also showed that bath application of the PKC activator PDA similarly causes a lasting potentiation of transmission at CI-AMPAR-containing MF synapses on L-M interneurons. Postsynaptic injection of a membrane-impermeable inhibitor of PKC interfered with LTP induced by HFS of the MF input to L-M interneurons. Collectively, these findings demonstrate that postsynaptic signaling events shown to play a critical role at glutamatergic synapses on pyramidal cells in areas CA3 and CA1 also are crucial participants in the signaling sequelae underlying long-term modification of transmission at naive MF synapses that contain primarily CI-AMPARs on L-M interneurons.
Our observations of PKA expression in L-M interneurons are consistent with findings by others showing that the induction of MF LTP in CA3 pyramidal cells requires postsynaptic PKA (Yeckel et al., 1999; Sivakumaran et al., 2009). On the other hand, FSK binding sites are highly expressed at the giant MF bouton on CA3 pyramidal cells (Worley et al., 1986), and previous work demonstrated that presynaptic PKA activity is required for the induction of MF LTP in pyramidal cells (Lonart and Südhof, 1998; Castillo et al., 2002; Pelkey et al., 2008) and stratum lucidum interneurons (Pelkey et al., 2008) as well at Schaffer collateral synapses on CA1 pyramidal cells (Blitzer et al., 1995, 1998). Indeed, we found that FSK application produced a robust, albeit transient, presynaptic potentiation in L-M interneurons loaded with the specific PKA inhibitor peptide PKI6-22. These data would indicate that FSK bindings sites are also present in MF boutons on L-M interneurons. Presynaptic activation of PKA could result in an accelerated vesicle cycling of the readily releasable pool of neurotransmitter (Chavis et al., 1996). Nevertheless, we also show that postsynaptic accumulation of cAMP and PKA activity is necessary for the maintenance of both the chemically induced potentiation and the electrical-induced MF LTP in L-M interneurons. These findings are consistent with our previous observations indicating that MF LTP in L-M interneurons is postsynaptically induced by a rise in cytosolic [Ca2+]I via activation of L-type VGCCs and mGluR1α, which leads to Ca2+ mobilization via IP3 receptors (IP3R), and ryanodine receptors (RyR)-mediated Ca2+-induced Ca2+ release (CICR) from internal stores (IP3R); (Galván et al., 2008). We hypothesize that LTP-inducing activities at predominantly CI-AMPAR containing MF synapses lead to PKA activation, most likely via the increase in cAMP production triggered by the Ca2+ activation of adenylyl cyclase 1 (Xia et al., 1991; Villacres et al., 1998). In turn, activation of PKA is known to phosphorylate L-type VGCCs (Davare et al., 1999), and enhance the activity of these calcium channels in hippocampal neurons (Kavalali et al., 1997). Furthermore, PKA activation also stimulates Ca2+ release from internal stores (Riegel and Williams, 2008). Therefore, the blockade of MF LTP produced by the intracellular injection of PKI6-22 may be explained by the complete disruption of postsynaptic Ca2+ dynamics that follows HFS. Although postsynaptic action of PKA signaling molecules was also shown to be critical for LTP of glutamatergic synapses on hippocampal CA1 pyramidal cells (Duffy and Nguyen, 2003; Esteban et al., 2003; Yang et al., 2008), a postsynaptic role for PKA in hippocampal interneurons does not appear to be a universal principle. For example, examination of LTP at MF synapses on dentate gyrus basket cells revealed that extracellular application of an inhibitor of PKA did not prevent the electrically induced postsynaptic form of LTP at these synapses, even though bath application of FSK was shown to produce a sustained increase of transmission at these same MF-basket cell synapses (Alle et al., 2001). Furthermore, examination of transmission at naive MF synapses on interneurons in stratum lucidum of area CA3 was revealed to be insensitive to bath application of FSK despite a pronounced potentiating effect of the adenylyl cyclase activator on the MF input to CA3 pyramidal cells (Maccaferri et al., 1998). It was only after L-AP4-mediated internalization of mGLuR7s that MF terminals in the stratum lucidum of area CA3 expressed a cAMP/PKA/-mediated presynaptic form of LTP (Pelkey et al., 2008).
Previous work has also shown that the presence of PKC activity in presynaptic MF terminals is necessary for the induction of LTP at MF synapses on pyramidal cells (Terrian et al., 1991; Son and Carpenter, 1996). Furthermore, phorbol ester activation of PKC induces a transient enhancement of glutamate release from the giant MF bouton on CA3 pyramidal cells (Son and Carpenter, 1996; Kamiya and Yamamoto, 1997; Macek et al., 1998; Honda et al., 2000). Here, we show that PDA application also induced a pronounced increase in the amplitude of MF responses from synapses on L-M interneurons. This PDA-induced potentiation was accompanied by a decrease in PPR indicating that an increase in glutamate release due to presynaptic activation of PKC is partly responsible for the synaptic enhancement. This interpretation is consistent with the observations that MF PTP is reduced during bath application of the PKC inhibitor Bin-1. However, postsynaptic PKC blockade prevented the induction of MF LTP while leaving MF PTP intact. A postsynaptic requirement for PKC also was observed for NMDAR-LTP at MF synapses on CA3 pyramidal cells (Kwon and Castillo, 2008), and for NMDAR-dependent LTP in CA1 pyramidal cells (Ling et al., 2002). Similar to the present findings, inhibition of PKC with bath-applied Bin-1 resulted in decreased PTP and blockade of LTP at MF-to-dentate gyrus basket cell synapses (Alle et al., 2001). The observations that PKC activation is involved in LTP at MF synapses on L-M and dentate gyrus basket cell interneurons stand in stark contrast to the findings that PKC activation underlies the HFS-induced LTD at naive MF synapses on stratum lucidum interneurons (Pelkey et al., 2005).
Blockade of L-type VGCCs prevents MF long-term plasticity in L-M interneurons but interference with IP3R or RyR results in MF LTD induced by the same HFS that induces MF LTP in naive slices (Galván et al., 2008). This strongly suggests that the rise in postsynaptic [Ca2+]i produced by activation of L-type VGCCs is supplemented by the concomitant activation of IP3R and RyR-mediated CICR from the internal stores (Nakamura et al., 2000). This interpretation would be consistent with experimental observations suggesting that the mechanisms that regulate the polarity of synaptic efficacy are controlled by the levels of postsynaptic [Ca2+]i (Bear et al., 1987; Artola and Singer, 1993; Hansel et al., 1997; Cormier et al., 2001). Therefore, it is tempting to speculate that the small but persistent post-HFS MF depression in chelerythrine-loaded cells is due to the loss of PKC-mediated facilitatory effects on calcium release from the internal stores (Petersen et al., 1994; Dermitzaki et al., 2004). Presumably this reduction in postsynaptic [Ca2+]i was partially counteracted by the parallel stimulatory actions of PKA activation on L-type VGCCs and CICR, which could have prevented the induction of a full-blown LTD in the absence of PKC activation.
Conclusions
Our current findings provide additional evidence supporting the general notion that the mechanisms underlying MF short-term (Scott et al., 2008) and long-term plasticity (Pelkey et al., 2006) are target-cell-specific and thus, likely to vary across the diversity of interneuron phenotypes receiving input from the dentate granule cells. This diversity shows that any attempt to provide a unified account of the mechanisms underlying interneuron synaptic plasticity from studies of a particular interneuron subtype is problematic. Our data also reveal that the molecular cascades involved in LTP at glutamatergic synapses on some interneuron subtypes may be more similar to those underlying LTP at glutamatergic synapses on pyramidal cells than previously appreciated. Therefore, an intriguing possibility is that other known properties of synaptic plasticity at pyramidal cell synapses, such as MAP kinase signaling (Roberson et al., 1999), de novo synthesis of proteins (Huang et al., 1994; Calixto et al., 2003), cAMP-mediated recruitment of new sites of synaptic transmission (Bolshakov et al., 1997), and synaptic tagging (Frey and Morris, 1998), may also be found to apply to plasticity at MF synapses on L-M interneurons.
Footnotes
This work was supported by National Institutes of Health Grants NS24288 (G.B.) and NS046423 (E.T.). We thank Dr. Spencer D. Watts for his invaluable assistance in the creation of the confocal images.
References
- Acsády L, Kamondi A, Sík A, Freund T, Buzsáki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 1998;18:3386–3403. doi: 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alle H, Jonas P, Geiger JR. PTP and LTP at a hippocampal mossy fiber-interneuron synapse. Proc Natl Acad Sci U S A. 2001;98:14708–14713. doi: 10.1073/pnas.251610898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artola A, Singer W. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- Ascoli GA, Brown KM, Calixto E, Card JP, Galván EJ, Perez-Rosello T, Barrionuevo G. Quantitative morphometry of electrophysiologically identified CA3b interneurons reveals robust local geometry and distinct cell classes. J Comp Neurol. 2009;515:677–695. doi: 10.1002/cne.22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synapse modification. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Jonas P. TwoB or not twoB: differential transmission at glutamatergic mossy fiber-interneuron synapses in the hippocampus. Trends Neurosci. 2002;25:600–603. doi: 10.1016/s0166-2236(02)02259-2. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998;280:1940–1942. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- Calixto E, Thiels E, Klann E, Barrionuevo G. Early maintenance of hippocampal mossy fiber-long-term potentiation depends on protein and RNA synthesis and presynaptic granule cell integrity. J Neurosci. 2003;23:4842–4849. doi: 10.1523/JNEUROSCI.23-12-04842.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calixto E, Galván EJ, Card JP, Barrionuevo G. Coincidence detection of convergent perforant path and mossy fibre inputs by CA3 interneurons. J Physiol. 2008;586:2695–2712. doi: 10.1113/jphysiol.2008.152751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Claiborne BJ, Amaral DG, Cowan WM. A light and electron microscopic analysis of the mossy fibers of the rat dentate gyrus. J Comp Neurol. 1986;246:435–458. doi: 10.1002/cne.902460403. [DOI] [PubMed] [Google Scholar]
- Cormier RJ, Greenwood AC, Connor JA. Bidirectional synaptic plasticity correlated with the magnitude of dendritic calcium transients above a threshold. J Neurophysiol. 2001;85:399–406. doi: 10.1152/jn.2001.85.1.399. [DOI] [PubMed] [Google Scholar]
- Cosgrove KE, Galván EJ, Meriney SD, Barrionuevo G. Area CA3 interneurons receive two spatially segregated mossy fiber inputs. Hippocampus. 2009 doi: 10.1002/hippo.20713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Dong F, Rubin CS, Hell JW. The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J Biol Chem. 1999;274:30280–30287. doi: 10.1074/jbc.274.42.30280. [DOI] [PubMed] [Google Scholar]
- Dermitzaki I, Tsatsanis C, Alexaki VI, Castanas E, Margioris AN. Roles of protein kinase A (PKA) and PKC on corticotropin-releasing hormone (CRH)-induced elevation of cytosolic calcium from extra-and intra-cellular sources. Hormones (Athens) 2004;3:252–258. doi: 10.14310/horm.2002.11134. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Duffy SN, Nguyen PV. Postsynaptic application of a peptide inhibitor of cAMP-dependent protein kinase blocks expression of long-lasting synaptic potentiation in hippocampal neurons. J Neurosci. 2003;23:1142–1150. doi: 10.1523/JNEUROSCI.23-04-01142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 1998;21:181–188. doi: 10.1016/s0166-2236(97)01189-2. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Galván EJ, Calixto E, Barrionuevo G. Bidirectional Hebbian plasticity at hippocampal mossy fiber synapses on CA3 interneurons. J Neurosci. 2008;28:14042–14055. doi: 10.1523/JNEUROSCI.4848-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Gundlach AL, Urosevic A. Autoradiographic localization of particulate cyclic AMP-dependent protein kinase in mammalian brain using [3H]cyclic AMP: implications for organization of second messenger systems. Neuroscience. 1989;29:695–714. doi: 10.1016/0306-4522(89)90142-5. [DOI] [PubMed] [Google Scholar]
- Hansel C, Artola A, Singer W. Relation between dendritic Ca2+ levels and the polarity of synaptic long-term modifications in rat visual cortex neurons. Eur J Neurosci. 1997;9:2309–2322. doi: 10.1111/j.1460-9568.1997.tb01648.x. [DOI] [PubMed] [Google Scholar]
- Havekes R, Timmer M, Van der Zee EA. Regional differences in hippocampal PKA immunoreactivity after training and reversal training in a spatial Y-maze task. Hippocampus. 2007;17:338–348. doi: 10.1002/hipo.20272. [DOI] [PubMed] [Google Scholar]
- Henze DA, Urban NN, Barrionuevo G. The multifarious hippocampal mossy fiber pathway: a review. Neuroscience. 2000;98:407–427. doi: 10.1016/s0306-4522(00)00146-9. [DOI] [PubMed] [Google Scholar]
- Honda I, Kamiya H, Yawo H. Re-evaluation of phorbol ester-induced potentiation of transmitter release from mossy fibre terminals of the mouse hippocampus. J Physiol. 2000;529:763–776. doi: 10.1111/j.1469-7793.2000.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins WF, Johnston D. Noradrenergic enhancement of long-term potentiation at mossy fiber synapses in the hippocampus. J Neurophysiol. 1988;59:667–687. doi: 10.1152/jn.1988.59.2.667. [DOI] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER, Varshavsky L, Brandon EP, Qi M, Idzerda RL, McKnight GS, Bourtchouladze R. A genetic test of the effects of mutations in PKA on mossy fiber LTP and its relation to spatial and contextual learning. Cell. 1995;83:1211–1222. doi: 10.1016/0092-8674(95)90146-9. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Yamamoto C. Phorbol ester and forskolin suppress the presynaptic inhibitory action of group-II metabotropic glutamate receptor at rat hippocampal mossy fibre synapse. Neuroscience. 1997;80:89–94. doi: 10.1016/s0306-4522(97)00098-5. [DOI] [PubMed] [Google Scholar]
- Kapur A, Yeckel M, Johnston D. Hippocampal mossy fiber activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway. Neuroscience. 2001;107:59–69. doi: 10.1016/s0306-4522(01)00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Hwang KS, Plummer MR. cAMP-dependent enhancement of dihydropyridine-sensitive calcium channel availability in hippocampal neurons. J Neurosci. 1997;17:5334–5348. doi: 10.1523/JNEUROSCI.17-14-05334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Lamsa KP. Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci. 2007;8:687–699. doi: 10.1038/nrn2207. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron. 2008;57:108–120. doi: 10.1016/j.neuron.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laezza F, Doherty JJ, Dingledine R. Long-term depression in hippocampal interneurons: joint requirement for pre- and postsynaptic events. Science. 1999;285:1411–1414. doi: 10.1126/science.285.5432.1411. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Grinspan ZM, McBain CJ. Quantal transmission at mossy fibre targets in the CA3 region of the rat hippocampus. J Physiol. 2004;554:175–193. doi: 10.1113/jphysiol.2003.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- Lonart G, Südhof TC. Region-specific phosphorylation of rabphilin in mossy fiber nerve terminals of the hippocampus. J Neurosci. 1998;18:634–640. doi: 10.1523/JNEUROSCI.18-02-00634.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-García JC, Arancio O, Kandel ER, Baranes D. A presynaptic locus for long-term potentiation of elementary synaptic transmission at mossy fiber synapses in culture. Proc Natl Acad Sci U S A. 1996;93:4712–4717. doi: 10.1073/pnas.93.10.4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Tóth K, McBain CJ. Target-specific expression of presynaptic mossy fiber plasticity. Science. 1998;279:1368–1370. doi: 10.1126/science.279.5355.1368. [DOI] [PubMed] [Google Scholar]
- Macek TA, Schaffhauser H, Conn PJ. Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. J Neurosci. 1998;18:6138–6146. doi: 10.1523/JNEUROSCI.18-16-06138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, Freund TF, Mody I. Glutamatergic synapses onto hippocampal interneurons: precision timing without lasting plasticity. Trends Neurosci. 1999;22:228–235. doi: 10.1016/s0166-2236(98)01347-2. [DOI] [PubMed] [Google Scholar]
- Mucignat-Caretta C, Caretta A. Clustered distribution of cAMP-dependent protein kinase regulatory isoform RI alpha during the development of the rat brain. J Comp Neurol. 2002;451:324–333. doi: 10.1002/cne.10352. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Lisman JE. Postsynaptic application of a cAMP analogue reverses long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2002;87:3018–3032. doi: 10.1152/jn.2002.87.6.3018. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ. mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron. 2005;46:89–102. doi: 10.1016/j.neuron.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Lacaille JC, McBain CJ. Compartmentalized Ca(2+) channel regulation at divergent mossy-fiber release sites underlies target cell-dependent plasticity. Neuron. 2006;52:497–510. doi: 10.1016/j.neuron.2006.08.032. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Yuan XQ, Lacaille JC, McBain CJ. State-dependent cAMP sensitivity of presynaptic function underlies metaplasticity in a hippocampal feedforward inhibitory circuit. Neuron. 2008;60:980–987. doi: 10.1016/j.neuron.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OH, Petersen CC, Kasai H. Calcium and hormone action. Annu Rev Physiol. 1994;56:297–319. doi: 10.1146/annurev.ph.56.030194.001501. [DOI] [PubMed] [Google Scholar]
- Riegel AC, Williams JT. CRF facilitates calcium release from intracellular stores in midbrain dopamine neurons. Neuron. 2008;57:559–570. doi: 10.1016/j.neuron.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffhauser H, Cai Z, Hubalek F, Macek TA, Pohl J, Murphy TJ, Conn PJ. cAMP-dependent protein kinase inhibits mGluR2 coupling to G-proteins by direct receptor phosphorylation. J Neurosci. 2000;20:5663–5670. doi: 10.1523/JNEUROSCI.20-15-05663.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R, Lalic T, Kullmann DM, Capogna M, Rusakov DA. Target-cell specificity of kainate autoreceptor and Ca2+-store-dependent short-term plasticity at hippocampal mossy fiber synapses. J Neurosci. 2008;28:13139–13149. doi: 10.1523/JNEUROSCI.2932-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumaran S, Mohajerani MH, Cherubini E. At immature mossy-fiber-CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J Neurosci. 2009;29:2637–2647. doi: 10.1523/JNEUROSCI.5019-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- Son H, Carpenter DO. Protein kinase C activation is necessary but not sufficient for induction of long-term potentiation at the synapse of mossy fiber-CA3 in the rat hippocampus. Neuroscience. 1996;72:1–13. doi: 10.1016/0306-4522(95)00532-3. [DOI] [PubMed] [Google Scholar]
- Son H, Davis PJ, Carpenter DO. Time course and involvement of protein kinase C-mediated phosphorylation of F1/GAP-43 in area CA3 after mossy fiber stimulation. Cell Mol Neurobiol. 1997;17:171–194. doi: 10.1023/A:1026361711588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrian DM, Ways DK, Gannon RL. A presynaptic role for protein kinase C in hippocampal mossy fiber synaptic transmission. Hippocampus. 1991;1:303–314. doi: 10.1002/hipo.450010321. [DOI] [PubMed] [Google Scholar]
- Topolnik L, Chamberland S, Pelletier JG, Ran I, Lacaille JC. Activity-dependent compartmentalized regulation of dendritic Ca2+ signaling in hippocampal interneurons. J Neurosci. 2009;29:4658–4663. doi: 10.1523/JNEUROSCI.0493-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth K, McBain CJ. Afferent-specific innervation of two distinct AMPA receptor subtypes on single hippocampal interneurons. Nat Neurosci. 1998;1:572–578. doi: 10.1038/2807. [DOI] [PubMed] [Google Scholar]
- Toth K, Suares G, Lawrence JJ, Philips-Tansey E, McBain CJ. Differential mechanisms of transmission at three types of mossy fiber synapse. J Neurosci. 2000;20:8279–8289. doi: 10.1523/JNEUROSCI.20-22-08279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villacres EC, Wong ST, Chavkin C, Storm DR. Type I adenylyl cyclase mutant mice have impaired mossy fiber long-term potentiation. J Neurosci. 1998;18:3186–3194. doi: 10.1523/JNEUROSCI.18-09-03186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Kelly P. Calcium-calmodulin signalling pathway up-regulates glutamatergic synaptic function in non-pyramidal, fast spiking rat hippocampal CA1 neurons. J Physiol. 2001;533:407–422. doi: 10.1111/j.1469-7793.2001.0407a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RE, Jr, Wiegand SJ, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–159. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ. Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci. 2008;11:790–798. doi: 10.1038/nn.2137. [DOI] [PubMed] [Google Scholar]
- Woo NH, Abel T, Nguyen PV. Genetic and pharmacological demonstration of a role for cyclic AMP-dependent protein kinase-mediated suppression of protein phosphatases in gating the expression of late LTP. Eur J Neurosci. 2002;16:1871–1876. doi: 10.1046/j.1460-9568.2002.02260.x. [DOI] [PubMed] [Google Scholar]
- Worley PF, Baraban JM, De Souza EB, Snyder SH. Mapping second messenger systems in the brain: differential localizations of adenylate cyclase and protein kinase C. Proc Natl Acad Sci U S A. 1986;83:4053–4057. doi: 10.1073/pnas.83.11.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia ZG, Refsdal CD, Merchant KM, Dorsa DM, Storm DR. Distribution of mRNA for the calmodulin-sensitive adenylate cyclase in rat brain: expression in areas associated with learning and memory. Neuron. 1991;6:431–443. doi: 10.1016/0896-6273(91)90251-t. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, Zhou Q. Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc Natl Acad Sci U S A. 2008;105:11388–11393. doi: 10.1073/pnas.0802978105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]