

Abstract

The mono-triflate salts of some chiral non-racemic 1,2-diamines react with α-ketoenones in a stoichiometric reaction to form products of the Nazarov cyclization in high enantiomeric ratios. The mechanism appears to involve rearrangement of an enamine – iminium ion.
Recent years have seen a resurgence of interest in the Nazarov cyclization.1,2 An attractive approach to the asymmetric Nazarov cyclization is through iminium ion catalysis.3 For example, treatment of divinyl ketone 1 (Scheme 1) with an asymmetric secondary amine 2 can be expected to lead to equilibrium with iminium ion 3. Nazarov cyclization of the pentadienyl carbocation (cf. 4) might lead to cyclic species 5 through a conrotatory process. Through appropriate choice of amine 2 and reaction conditions, asymmetric induction might be observed during this process. Although this general approach appears to be sound, in fact the pentadienyl cation (cf. 4) is rendered more stable than the cyclic allylic cation 5 by conjugation with the non-bonding electron pair on the nitrogen atom.4 This generally results in an equilibrium favoring the acyclic iminium ion. This obstacle may be overcome by incorporating a structural feature into 1 that raises the energy of 3/4 compared to 5.
Scheme 1.

To the best of our knowledge the Nazarov cyclization of an iminium ion has been reported only twice. The first example is summarized in Scheme 2.5 Addition of lithioallenyl ether 7 to E-α-methyl cinnamonitrile 6 leads to N-lithio imine 8. Workup with mild aqueous acid protonates the imine that then undergoes Nazarov cyclization to 9. N-Acetylation in a separate step gave acetamide 10 in 73% overall yield from 6. The success of this reaction is probably due to release of allene strain (10.76 kcal/mol)3g as well as the irreversible loss of methoxymethyl cation during the cyclization. In the second example of an imino Nazarov cyclization the stability of the iminium ion was attenuated by N-tosylation.6
Scheme 2.

We first screened a small set of monoamines to determine whether they would catalyze the asymmetric conversion of 11 to 15 (Scheme 3). Stoichiometric L-proline, (S)-α-methylbenzylamine or (1R,2S)-(−)-ephedrine in the presence of a Brønsted acid led to small amounts (<10%) of racemic 15 in a very slow reaction. These negative results suggested a solution to the problem through a cooperative mechanism (vide infra).
Scheme 3.

We conceived of an alternative approach to the ones described above that would allow us to use iminium ion catalysis for a Nazarov cyclization. Scheme 3 summarizes the approach. Exposure of an α-ketoenone such as 11 to a diamine salt 12 is expected to generate enamine-iminium ion 13 under Brønsted acid catalysis. The carbonyl group of the methyl ketone is likely to be the more reactive of the two, and will form an enamine. Subsequent reaction of the free amino group with the enone carbonyl group leads to enamine-iminium ion 13. Intermediate 13 should be an excellent substrate for the Nazarov reaction since the enamine is polarized so as to favor cyclization.7 Critically, this Nazarov cyclization converts enamine-iminium ion 13 to enamine-iminium ion 14. Consequently, both 13 and 14 are stabilized by a non-bonding electron pair on a nitrogen atom therefore there is no reason to expect the equilibrium to favor 13. Hydrolysis of 14 would regenerate diamine salt 12, liberating the α-hydroxyenone product 15. We thought that this approach had some merit since we knew from our earlier work that efficient cyclization of 11 to 15 can be accomplished under a variety of reaction conditions.
We subsequently screened the series of diamines that are listed in Figure 1, all based on the (1R,2R)-(−)-1,2-diaminocyclohexane motif that has been widely used in asymmetric catalysis.8 All diamines were evaluated as their mono-triflate salts in the stoichiometric reaction with 11. We were surprised that neither primary-primary diamine 16, nor secondary-secondary diamines 17 and 18 led to cyclic product 15 under a variety of conditions. Primary-tertiary diamines 24–27 were also ineffective. On the other hand, primary-secondary diamines 19–23 were all effective in converting 11 to 15 with varying degrees of asymmetric induction in a 4 M solution in DMSO at room temperature. Diamines 19 and 20 led to 15 in 85/15 and 86/14 er’s, respectively, whereas diamines 21 and 22 gave enantiomeric ratios of 79/21 and 77.5/22.5, respectively. Diamine 23 led to nearly racemic product (er 53/47). Under optimized conditions of solvent and concentration (0.1 M in MeCN containing 25 mol % water), the mono-triflate salt of 19 led to 15 in 60% yield and 97/3 er after 7.5 d at room temperature (Figure 2). We were unable to find conditions that would allow 19 to function catalytically, either through inclusion of additives9 or by varying the reaction conditions. Acid catalyzed liberation of the diamine from its bound form is required, suggesting that product inhibition takes place.
Figure 1.

Diamines screened.
Figure 2.

ORTEP diagram of 51, the ester derivative of 15 with (−)-camphanic acid chloride. Ellipsoids are shown at the 50% level. Selected bond lengths (Å): C(1)-C(2) 1.472(2), C(2)-C(3) 1.329(2), O(1)-C(1) 1.2139(19). Selected bond angles (°): C(3)-C(2)-C(1) 113.11(14), C(2)-C(3)-C(4) 110.42(14), C(3)-C(4)-C(5) 104.28(12), C(1)-C(5)-C(4) 105.46(13).
A catalytic process was observed with diamine 28, however, this led to an unexpected product (Scheme 4). Exposure of 11 to 20 mol % of the mono-triflate salt of 28 in MeCN at room temperature overnight led to α-ketoacid 29 in ca. 45% yield (unoptimized) along with another unidentified dimeric product. α-Ketoacid 29 was formed in variable yields along with α-ketoaldehyde 30 from the reaction of 11 with some secondary amines, suggesting that aldehyde 30 was the primary product and that air oxidation to 29 had taken place during the reaction. The β-vinylic methyl group in 29 must have its origin as the acetyl methyl group in 11. It is difficult to imagine a means of forming the carbon-carbon bond between the β-vinyl carbon atom and the acetyl methyl group of 11 unless the two are first joined in a ring. Scheme 5 summarizes our postulated mechanism. Alternative mechanisms involving the initial formation of an enamine-iminium species similar to 13 that rationalize the formation of 30 can also be imagined. Protonated keto enamine 31 can undergo Nazarov cyclization to 32. A series of proton transfer steps converts 32 to 34. The cleavage step that converts 34 to 35 can be thought of as a retro-Nazarov reaction.10 Enol-keto conversion leads from 35 to 36. Hydrolysis of 36 gives 30 and regenerates the catalyst 28. The origins of the differences in reactivity between 28 and 19 are difficult to rationalize.
Scheme 4.

Scheme 5.

Table 1 summarizes the results of the stoichiometric reaction between the mono-triflate salt of 19 and a series of diketones. The reactions are in all cases slow, requiring approximately one week for completion. In the case of products bearing α gem-dimethyl substitution (38, 40, 42 and 44) the minor enantiomer was undetectable by chiral HPLC. The cyclization is, however, delicately balanced as indicated by the results with substrates 45, 47 and 49 that led to cyclic products of lower er in poor yield. These reactions also led to the generation of numerous byproducts, rendering the purification of the cyclopentenones difficult.
Table 1.
α-Hydroxycyclopentenones from the Nazarov reaction
| entry | diketone | cyclopentenone | yield | era | rxn time |
|---|---|---|---|---|---|
| 1 |
 11 |
 15 |
60(63)%b | 97/3 | 7.5 d |
| 2 |
 37 |
 38 |
66(73)% | >99/1 | 7.5 d |
| 3 |
 39 |
 40 |
49%c | >99/1 | 6.5 d |
| 4 |
 41 |
 42 |
65% | >99/1 | 7.5 d |
| 5 |
 43 |
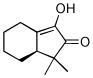 44 |
62% | >99/1d | 7.5 d |
| 6 |
 45 |
 46 |
11% | 90/10 | 5.5 d |
| 7 |
 47 |
 48 |
20% | 91/9 | 6 d |
| 8 |
 49 |
 50 |
24% | 81/19d | 5 d |
All reactions were performed in acetonitrile at 0.1 M with 25 mol % water and 1.05 equiv of the mono-triflate salt of 19.
Enantiomer ratios were determined by chiral HPLC with a Chiralcel AD-H column.
Yields in parentheses are based on recovered diketone.
Volatile solid.
The sequence of elution of major and minor enantiomers was inverted.
The absolute stereochemistry of 15 was determined crystallographically from the (1S)-(−)-camphanic acid chloride derivative 51 (Figure 2). Ester 51 crystallizes in space group P21, which is compatible with chiral crystal structures.11 The absolute stereochemistry of the other α-hydroxycyclopentenones shown in Table 1 has been assigned by analogy with 15. The inversion in chromatographic mobility that was observed for the enantiomers of 44 and 50 may indicate an inversion of the stereochemical preference in these cases, or it may be due to a unique interaction of these two cyclopentenones with the chiral stationary phase.1
In summary, we have described an iminium ion mediated asymmetric Nazarov cyclization of α-diketones through a mechanism requiring the formation of an enamine-iminium ion.2 To the best of our knowledge this represents only the third example of an imino Nazarov cyclization. Although the process is not catalytic at its present level of development, the results suggest a number of ways to overcome the problem of product inhibition. The excellent enantioselectivities provide an impetus to do so.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM57873) for generous support. We thank Mr. Paolo Larini (Università degli Studi di Torino) for his contribution to this work.
Footnotes
Supporting Information Available. Experimental procedures for 15 and 19; 1H and 13C NMR, HRMS and IR data for 19, 40, 42, 48 and 50; HPLC and optical rotations for 15, 19, 38, 42, 44, 46, 48, 50; reproductions of 1H and 13C NMR spectra for 15, 19, 38, 40, 42, 44, 46, 48 and 50. Crystallographic data for 51 and for the (−)-camphanic acid derivative of 38. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews of the Nazarov reaction see: Harmata M. Chemtracts. 2004;17:416–435.Frontier AJ, Collison C. Tetrahedron. 2005;61:7577–7606.Pellissier H. Tetrahedron. 2005;61:6479–6517.Tius MA. Eur J Org Chem. 2005:2193–2206.Habermas KL, Denmark S, Jones TK. In: Organic Reactions. Paquette LA, editor. Vol. 45. John Wiley & Sons, Inc; New York: 1994. pp. 1–158.
- 2.Recent examples of the Nazarov cyclization: Malona JA, Cariou K, Frontier AJ. J Am Chem Soc. 2009;131:7560–7561. doi: 10.1021/ja9029736.Rieder CJ, Winberg KJ, West FG. J Am Chem Soc. 2009;131:7504–7505. doi: 10.1021/ja9023226.Marx VM, Burnell DJ. Org Lett. 2009;11:1229–1231. doi: 10.1021/ol900029d.Bitar AY, Frontier AJ. Org Lett. 2009;11:49–52. doi: 10.1021/ol802329y.Basak AK, Tius MA. Org Lett. 2008;10:4073–4076. doi: 10.1021/ol801587u.He W, Huang J, Sun X, Frontier AJ. J Am Chem Soc. 2008;130:300–308. doi: 10.1021/ja0761986.Song D, Rostami A, West FG. J Am Chem Soc. 2007;129:12019–12022. doi: 10.1021/ja071041z.Grant TN, West FG. Org Lett. 2007;9:3789–3792. doi: 10.1021/ol7015669.Gao S, Wang Q, Chen C. J Am Chem Soc. 2009;131:1410–1412. doi: 10.1021/ja808110d.Nie J, Zhu HW, Cui HF, Hua MQ, Ma JA. Org Lett. 2007;9:3053–3056. doi: 10.1021/ol071114j.
- 3.Examples of the catalytic asymmetric Nazarov cyclization: Rueping M, Ieawsuwan W. Adv Synth Catal. 2009;351:78–84.Walz I, Togni A. Chem Comm. 2008:4315–4317. doi: 10.1039/b806870d.Rueping M, Ieawsuwan W, Antonchick AP, Nachtsheim BJ. Ang Chem Int Ed. 2007;46:2097–2100. doi: 10.1002/anie.200604809.Liang G, Trauner D. J Am Chem Soc. 2004;126:9544–9545. doi: 10.1021/ja0476664.Aggarwal VK, Belfield AJ. Org Lett. 2003;5:5075–5078. doi: 10.1021/ol036133h.Liang G, Gradl SN, Trauner D. Org Lett. 2003;5:4931–4934. doi: 10.1021/ol036019z.Examples of the use of chiral auxiliaries for the Nazarov: Banaag AR, Tius MA. J Org Chem. 2008;73:8133–8141. doi: 10.1021/jo801503c.Dhoro F, Kristensen TE, Stockmann V, Yap GPA, Tius MA. J Am Chem Soc. 2007;129:7256–7257. doi: 10.1021/ja0718873.de los Santos DB, Banaag AR, Tius MA. Org Lett. 2006;8:2579–2582. doi: 10.1021/ol060816q.Pridgen LN, Huang K, Shilcrat S, Tickner-Eldridge A, DeBrosse C, Haltiwanger RC. Synlett. 1999:1612–1614.
- 4.Smith DA, Ulmer CW., II J Org Chem. 1997;62:5110–5115. [Google Scholar]
- 5.Tius MA, Chu CC, Nieves-Colberg R. Tetrahedron Lett. 2001;42:2419–2422. [Google Scholar]
- 6.Suarez-Pantiga S, Rubio E, Alvarez-Rua C, Gonzalez JM. Org Lett. 2009;11:13–16. doi: 10.1021/ol8025523. [DOI] [PubMed] [Google Scholar]
- 7.(a) Tius MA, Kwok C-K, Gu X-q, Zhao C. Synthetic Commun. 1994;24:871–885. [Google Scholar]; (b) He W, Herrick IR, Atesin TA, Caruana PA, Kellenberger CA, Frontier AJ. J Am Chem Soc. 2008;130:1003–1011. doi: 10.1021/ja077162g. [DOI] [PubMed] [Google Scholar]
- 8.(a) Luo S, Xu H, Chen L, Cheng JP. Org Lett. 2008;10:1775–1778. doi: 10.1021/ol800471b. [DOI] [PubMed] [Google Scholar]; (b) Stead D, O’Brien P, Sanderson A. Org Lett. 2008;10:1409–1412. doi: 10.1021/ol800109s. [DOI] [PubMed] [Google Scholar]; (c) Uyeda C, Jacobsen EN. J Am Chem Soc. 2008;130:9228–9229. doi: 10.1021/ja803370x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Singh A, Johnston JN. J Am Chem Soc. 2008;130:5866–5867. doi: 10.1021/ja8011808. [DOI] [PubMed] [Google Scholar]; (e) Zhang W, Yamamoto H. J Am Chem Soc. 2007;129:286–287. doi: 10.1021/ja067495y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The following additives were evaluated: silica gel, alumina (neutral, basic, acidic), CuCl2, LiCl, LiClO4, aq NH4OH.
- 10.(a) Harmata M, Lee DR, Barnes CL. Org Lett. 2005;7:1881–1883. doi: 10.1021/ol050657v. [DOI] [PubMed] [Google Scholar]; (b) Harmata M, Schreiner PR, Lee DR, Kirchhoefer PL. J Am Chem Soc. 2004;126:10954–10957. doi: 10.1021/ja048942h. [DOI] [PubMed] [Google Scholar]; (c) Harmata M, Lee DR. J Am Chem Soc. 2002;124:14328–14329. doi: 10.1021/ja028729q. [DOI] [PubMed] [Google Scholar]
- 11.Flack HD. Helv Chim Acta. 2003;86:905–921. [Google Scholar]
- 12.Crystallographic data for the (−)-camphanic acid derivative of 38 show that the absolute stereochemistry of 38 is the same as 15. See Supporting Information.
- 13.A referee has suggested that the mechanism that we have proposed may in fact be better described as an “intramolecular enamine-Michael addition to enone”. We think that this is unlikely for two reasons. First, such a process would correspond to a 5-endo-trig reaction that is disfavored by Baldwin’s Rules. Second, if the reasonable assumption is made that the rate-limiting step is the ring-forming step, then one would have expected a very large difference in rate for the cyclizations leading to 15 and to 38. Since the enamine derived from 37 is β,β-disubstituted it is a much poorer Michael donor than the unsubstituted enamine derived from 11 and would be expected to react at a significantly slower rate. No difference in reaction rate was evident for the cyclizations of 11 and 37.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.