

Abstract
With the availability of genetic pathways or networks and accumulating knowledge on genes with variants predisposing to diseases (disease genes), we propose a disease-gene-centric support vector machine (DGC-SVM) that directly incorporates these two sources of prior information into building microarray-based classifiers for binary classification problems. DGC-SVM aims to detect the genes clustering together and around some key disease genes in a gene network. To achieve this goal, we propose a penalty over suitably defined groups of genes. A hierarchy is imposed on an undirected gene network to facilitate the definition of such gene groups. Our proposed DGC-SVM utilizes the hinge loss penalized by a sum of the L∞-norm being applied to each group. The simulation studies show that DGC-SVM not only detects more disease genes along pathways than the existing standard SVM and SVM with an L1-penalty (L1-SVM), but also captures disease genes that potentially affect the outcome only weakly. Two real data applications demonstrate that DGC-SVM improves gene selection with predictive performance comparable to the standard-SVM and L1-SVM. The proposed method has the potential to be an effective classification tool that encourages gene selection along paths to or clustering around known disease genes for microarray data.
Keywords: DAG, Gene expression, Gene network, Grouped penalty, Hierarchy, Penalization
1 Introduction
Genes interact with each other through their RNA and protein expression products. For example, the rate at which transcription factor genes are transcribed into RNA molecules may govern the transcriptional rate of their regulatory target genes, which as a result become either up- or down- regulated. A gene network is a collection of effective interactions, describing the multiple ways through which one gene affects all the others to which it is connected. A gene network reveals genetic dynamics underlying the aggregate function that the network maintains. High-throughput genomic advances have generated various databases providing gene network information, such as the Biomolecular Interaction Network Database (BIND) (Alfarano et al 2005), the Human Protein Reference Database (HPRD) (Peri et al 2004), and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al 2004). In recent years, genetic studies have uncovered hundreds of genes with variants that predispose to common diseases, such as cancer, Parkinson’s disease, and diabetes. For example, gene TP53 is among the most famous ones, which, as a tumor suppressor, is central to many anti-cancer mechanisms. Gene TP53 encodes tumor protein p53, the so-called ”the guardian of the genome”, which mediates cellular response to DNA damage and is involved in other important biological processes, e.g., cell cycle. Among its other functions, p53 activates other genes to fix the damage if p53 determines that the DNA can be repaired. Otherwise, p53 prevents the cell from dividing and signals its death. Most mutations that deactivate TP53 destroy protein p53’s ability to regulate other genes properly and thus leads to increasing risk of tumor development (Soussi and Béroud 2003; Børresen-Dale 2003). Hence, not just a single gene, but a subnetwork of TP53 and its interacting partners, are involved in the disease process.
With the availability of various repositories of gene networks and the accumulating knowledge on genes linked to diseases, one question naturally arises: how to integrate the two sources of prior information into a model to detect genes involved in disease-related biological processes. A network-based approach takes such a coherent view and makes use of the network information in building statistical models. Employing a network-based perspective not only sheds insight within the network modules (Calvano et al 2005; Benson 2006; Chuang et al 2007; Liu et al 2007) but also allows the possibility to identify disease genes that have only weak effects. Such genes often play a central role in discriminative subnetworks by interconnecting groups of genes involved in various biological processes. Chuang et al (2007) pointed out that several well-known cancer genes, such as TP53, KRAS, and HRAS, were ignored by gene-expression-alone analysis but successfully detected by using network information. However, their network-based approach involves a random search over subnetworks, leading to possibly instable and suboptimal final results.
Since its invention (Vapnik 1995; Cortes and Vapnik 1995), the support vector machine (SVM) has been acclaimed as a useful regularization method due to its excellent empirical performance, especially with high dimensional data (Brown et al 2000; Furey et al 2000), its possible extensions to accommodate various penalty functions, and resulting model sparsity if a suitable penalty (e.g. L1-norm) is employed. For binary classification, the standard L2-norm SVM (STD-SVM) has good predictive performance, but is incapable of performing variable selection. The L1-SVM (Zhu et al 2003; Wang and Shen 2007) produces sparse models for data with p ≫ n. Zou and Yuan (2008) developed a grouped variable selection scheme for factors by the use of an F∞-norm SVM such that all features derived from the same factor (i.e. categorical predictor) are included or excluded simultaneously. Note that their grouping scheme was based on non-overlapping groups. Zhao et al (to appear) generalized grouped variable selection and introduced the composite absolute penalties (CAP) family. CAP achieves both grouped selection for non-overlapping groups and hierarchical selection for overlapping groups. Extending the idea of grouping to gene networks, Zhu et al (2009) proposed a network-based SVM (NG-SVM), treating any two neighboring genes in a network as one group, and explicitly incorporating the network information into building classifiers. Both the simulation studies and real data applications showed that NG-SVM enjoyed advantages in gene selection and predictive performance compared with the popular STD-SVM and L1-SVM. However, a potential problem of NG-SVM lies in its tendency of selecting isolated genes or gene pairs, i.e., genes largely disconnected to each other in the network, which is not desirable given that some disease genes cluster together and form subnetworks.
In this paper, we embed the information of both a gene network and some crucial disease genes into the SVM framework by emploiting two ways of grouping genes to construct penalties. By considering an undirected network to be anchored on certain crucial disease gene(s), i.e., genes known to be central to a disease, a hierarchical structure is imposed on the network (with the anchoring crucial genes at the top) to facilitate the definition of various gene groups. By summing up an L∞-norm over each group, we obtain the penalty for DGC-SVM. Ideally, by DGC-SVM, identification of one gene triggers the inclusion of the disease genes along the connected paths towards the top crucial gene(s). In particular, we intend to capture disease genes, even if their direct effects on the outcome are weak, which are important in regulating functional activities of other genes along the pathways or within the subnetworks involved in the disease.
2 Methods
2.1 Orienting an undirected network
Starting from an undirected network G, we convert it into a directed acyclic graph (DAG) G̃. Suppose that G originates from only one disease gene g and consists of p genes in total. Genes (including g) in network G are indexed by {1, 2, …, p}. We have the expression levels of the p genes and a binary outcome for N samples, with xi ∈ ℝp and yi ∈ {1, −1}. The expression of each gene is normalized to have mean 0 and standard deviation 1 across samples. We define a directed edge by an ordered pair of ends (a, b) indicating that a is upstream to b, or equivalently, b is downstream to a. Since genetic interrelationships occur only between pairs of distinct genes, network G contains no loop, defined as a directed edge with identical ends. In addition, no two directed edges adjoin the same pair of genes. Gene g is the top (center) gene of network G. The distance between two genes a and b is the minimum number of directed edges traversed from a to b. Genes closer to the network origin, gene g, are said to be at an upper level than those farther apart. Genes with the same distance from the origin are at the same level. For example, the distance between gene g and any of its direct neighbors is 1. The distance between any two genes at the same level is 0. Thus, DAG G̃ is defined from the undirected network G. G̃ assigns directions from upper-level to lower-level genes but ignores edges connecting genes at the same level. Upper-level genes are called nodes whereas genes with no downstream genes are named as leaves. DAG G̃ captures the upper-lower interrelationships but ignoring the lateral ones.
If we have more than one center genes g1…gL, DAG G̃ can be defined as follows: (1) Derive DAGs G̃1…G̃L, each corresponding to one center gene in g1…gL; (2) if G̃1…G̃L share no common nodes; (3) if the DAGs have common nodes, pick up any of them, align all the associated DAGs at the level where that common node is seated, treat that node in each associated DAG as being located at the same level (named as levelv), and merge the associated DAGs by recognizing only the upper-lower interrelationships but ignoring the lateral ones. Then, identify the common nodes of the merged DAG and the remaining untouched DAGs, repeat step (3) until no common nodes exist. Note that each node in the merged DAG has the same downstream genes no matter which center the node is derived from. The above process may result in different DAGs if the combination of the associated DAGs occurs at different common nodes, introducing certain arbitrariness.
2.2 Pathway grouping
To achieve our goal of detecting collectives of genes involved in disease along pathways or within subnetworks, we form a penalty on suitably defined groups of genes. We experiment two ways of grouping: (linear) pathway (PW) grouping and partial tree (PT) grouping. We first describe the PW grouping. It forms groups along linear paths as an attempt to encourage (linear) pathway selection.
A path in G̃ is a connected sequence of directed edges and the length of the path is the number of directed edges traversed. Note that a path connects genes from upper-to lower-levels without any two consecutive genes from the same level. Since a path can be determined by the sequence of the nodes the path, a path is simply specified by its node sequence. We define a single node as a trivial path. Define a complete path of leaf k in G̃, ℰk (k = 1, …, K), as
Suppose ℰk contains a total of nk genes, including leaf k and gene g. Then we have nk groups with a hierarchical structure
(t = 1,…, nk) by grouping the genes in ℰk under the ”lower nested within upper” rule, that is, node/leaf at a lower level must appear in all the groups that contain any node at an upper-level. For example, in the network displayed in Figure 1, if gene 1 is considered to be at the top, then genes 1,…, 12 are nodes and genes 13, …, 26 are leaves. The complete path of leaf gene 16, ℰ16, is {1, 2, 6, 16}. Hierarchical groups derived from ℰ16 or leaf gene 16 are {2, 6, 16}, {6, 16}, {16}, and ℰ16 itself. Note that multiple distinct complete paths may exist between leaf k and gene g, for example, {g, a, c, k} and {g, b, c, k}. In this case, group {c, k} and group {k} are defined twice respectively. When forming groups, we count each distinct group only once. Therefore, groups formed from {g, a, c, k} and {g, b, c, k} include 6 groups: {g, a, c, k}, {g, b, c, k}, {a, c, k}, {b, c, k}, {c, k}, and {k}. Thus, we impose a grouping structure 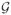 containing distinct groups on G̃, that is, every group in appears only once:
containing distinct groups on G̃, that is, every group in appears only once:
while a gene may appear in multiple groups, which causes no problem in the below formulation and computation.
Figure 1.

Left: Simple network originating from gene 1. Right: DAG derived from the simple network.
Corresponding to , we construct our penalty as
| (1) |
The hinge loss penalized by (1) leads to our proposed DGC-SVM with PW grouping (DGC-SVM-PW), which is developed as an attempt to encourage selecting genes along the pathway (pathway selection).
| (2) |
where the subscript ”+” denotes the positive part, i.e., z+ = max{z, 0}, λ is the tuning parameter, and with ws as a weight for gene s. For example, we can define with ds as the number of direct neighbors of gene s, or ws = ds, or simply ws = 1 for all genes. The solution to (2) can be found as a linear programming problem:
| (3) |
subject to
| (4) |
In the above parametrization, we have and , in which and denote the positive and negative parts of βs. Note that, by our construction, some genes fall within multiple groups , which however causes no computational problem with the above linear programming.
2.3 Partial tree grouping
The PT grouping is devised to achieve hierarchical selection, that is, the selection of a lower-level gene ensures the selection of its upper-level gene(s). In addition, selecting any gene in the DAG guarantees the inclusion of at least one center gene, which is desirable in view of the biological importance of any center gene. The DGC-SVM with PT grouping (DGC-SVM-PT) groups each node/leaf with all its downstream genes. Since a leaf has no downstream genes, the group derived from the leaf contains only one element, the leaf itself. For the above G̃, we have p groups in total, K of which contain only single elements derived from K leaves, and the rest p−K of which are formed as
For example, the simple network in Figure 1 derives 26 groups, including (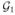 ) that contains all the 26 genes, and 14 single-leaf groups. Here we impose the grouping structure as = (, …,
) that contains all the 26 genes, and 14 single-leaf groups. Here we impose the grouping structure as = (, …, 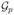 ). The formulation of DGC-SVM-PT is the same as its PW grouping counterpart (2)-(4).
). The formulation of DGC-SVM-PT is the same as its PW grouping counterpart (2)-(4).
The DGC-SVM-PT is a direct application of the CAP family of Zhao et al (to appear) in the context of SVM. It enjoys the hierarchical property that if any node/leaf at a lower-level is included in the model, the nodes at any upper-level in the group will be almost surely included. This property is important to our goal of capturing disease genes along pathways or within subnetworks, which offers the possibility of detecting genes that may have weak effects but play a central role in regulating multiple biological processes through connecting various functional groups of disease-relevant genes. In addition, this property guarantees the identification of a center gene of the network if any gene in the network is selected.
2.4 Choice of weight
DGC-SVM involves a weight function w. The choice of the weight depends on the goal of shrinkage and governs variable selection and predictive performance.
A main motivation behind the proposed penalties is the grouping effect of the L∞ norm. Because of the singularity of the penalty max(|a|, |b|) at |a| = |b|, by Fan and Li (2001), the penalty encourages the shrinkage to |a| = |b|, which can be achieved if the penalization parameter λ is large enough. For linear regression, this so-called grouping effect has been theoretically established by Bondell and Reich (2008) and Pan et al (2009) for two-gene groups, and by Wu et al (2008) for a more general case with more than two genes in a group. Now consider network G and its grouping structure derived from G̃. For simplicity, we assume that contains only two-gene and one-gene groups. For these two-gene groups, the weighted penalty encourages |βj1|/wj1= |βj2|/wj2 where βj1 and βj2 belong to the same group. Here we examine three weight functions specifically: ws = 1,
, and ws = ds, where ds is the degree of gene s, i.e., the number of direct neighbors of gene s. The new method encourages |βj1| = |βj2| if ws = 1,
if
, and
if ws = ds. The same reasoning also applies to groups with more than two genes. Therefore, larger weights (from ws = 1,
, to ws = ds) favor genes with more direct neighbors to have larger coefficient estimates; in other words, larger weights relax the shrinkage effect for those ”hub” genes that are connected to many genes and are known to be biologically more important. Due to this property, the choice of a large weight, as a simple strategy, enables us to alleviate the bias in the coefficient estimates from penalization and possibly improve predictive performance. The weight can be considered as a tuning parameter and determined by cross-validation or an independent tuning data set, though we will not pursue it here.
Since the proposed penalty is linear, linear programming can be used to solve the resulting optimization problem. We implemented the method by R package lpsolve.
3 Simulation
We numerically evaluated the new methods, DGC-SVM-PW and DGC-SVM-PT, in two simulation studies over a simple network and a more complex one. The DAG for a simple network is essentially a hierarchical tree where any two genes are connected by a unique path. In contrast, there exist multiple paths adjoining the same pair of genes in the complicated network. The grouping structure in either case is unique. We compare the performance of DGC-SVMs with that of STD-SVM, L1-SVM, and NG-SVM. The R package e1071 (with linear kernel) was used to obtain the solutions of the STD-SVM, while the other ones were implemented by the R package lpsolve.
3.1 A simple network
We applied the DGC-SVM to the simple network depicted in Figure 1. Any two genes in its DAG are connected by a unique path. The simulation data sets were generated following the set-ups of Li and Li (2008).
Generate the expression level of center gene 1, X1 ~ N (0, 1).
Assume node s and each of its downstream genes follow a bivariate normal distribution with means 0 and unit variances with correlation 0.7. Thus, the expression level of each downstream gene is distributed as N(0.7Xs, 0.51).
Generate outcome Y from a logistic regression model: Logit (Pr(Y = 1|X)) = XTβ + β0, β0 = 2, where X is a vector of the expression levels of all the genes, and β is the corresponding coefficient vector.
We considered three sets of informative genes. The effect of each informative gene on the outcome was equal to that of its upstream node divided by the square-root of the upstream node’s degree. All the other genes were noninformative, which had no effect on the outcome. Three sets of true coefficients, β = (β1, β2, …, βi, …, β26), were specified in three scenarios:
-
PT setting: one of the tree branches of the hierarchical tree or DAG (gene 1, 2, 5, 6, 14, 15, and 16) was informative.
-
PW setting: pathway {1, 3, 7, 17} was informative.
-
PW setting: pathway {1, 2, 5, 14} was informative.
In each scenario, we simulated 50, 50 and 10,000 observations for each training, tuning and test dataset. For each of tuning parameter values, we obtained a classifier from the training data, applied it to the tuning data, and identified λ̂ that yielded the minimal classification error over the tuning set. Then we used the classifier corresponding to λ̂ to compute the classification error on the test data. The entire process was repeated 100 times (i.e., 100 independent runs). The means of the test classification errors, false negatives (the number of informative genes whose coefficients were estimated to be zero), model sizes (the number of genes whose coefficients were estimated to be nonzero), and their corresponding standard errors ( ) are reported in Table 1.
Table 1.
Simulation results (averaged over 100 runs) for a simple network with p = 26 genes. There were 7, 4 and 4 informative genes in the three scenarios respectively.
| Scenario | Method | Test Error (SE) | # False Negative (SE) | Model Size (SE) |
|---|---|---|---|---|
| 1 | STD | 0.129 (0.003) | 0.00 (0.00) | 26.00 (0.00) |
| L1 | 0.122 (0.003) | 2.81 (0.14) | 7.19 (0.36) | |
| NG (w = 1) | 0.145 (0.004) | 0.26 (0.05) | 14.73 (0.47) | |
|
|
0.123 (0.004) | 0.10 (0.04) | 15.32 (0.50) | |
| NG (w = d) | 0.108 (0.003) | 0.12 (0.05) | 14.47 (0.56) | |
| PW (w = 1) | 0.152 (0.003) | 0.84 (0.07) | 15.57 (0.65) | |
|
|
0.136 (0.003) | 0.93 (0.08) | 16.90 (0.61) | |
| PW (w = d) | 0.126 (0.003) | 1.60 (0.13) | 14.44 (0.61) | |
| PT (w = 1) | 0.107 (0.003) | 1.94 (0.14) | 9.42 (0.53) | |
|
|
0.107 (0.004) | 2.51 (0.13) | 8.65 (0.53) | |
| PT (w = d) | 0.110 (0.004) | 3.08 (0.13) | 7.39 (0.44) | |
| 2 | STD | 0.147 (0.003) | 0.00 (0.00) | 26.00 (0.00) |
| L1 | 0.118 (0.004) | 0.99 (0.07) | 6.56 (0.33) | |
| NG (w = 1) | 0.162 (0.004) | 0.16 (0.05) | 17.83 (0.52) | |
|
|
0.138 (0.003) | 0.01 (0.01) | 17.33 (0.52) | |
| NG (w = d) | 0.125 (0.003) | 0.04 (0.02) | 16.09 (0.57) | |
| PW (w = 1) | 0.174 (0.003) | 0.32 (0.05) | 18.93 (0.48) | |
|
|
0.163 (0.003) | 0.42 (0.06) | 16.31 (0.48) | |
| PW (w = d) | 0.144 (0.003) | 0.43 (0.06) | 14.94 (0.47) | |
| PT (w = 1) | 0.111 (0.003) | 0.72 (0.08) | 7.74 (0.49) | |
|
|
0.109 (0.003) | 1.05 (0.08) | 6.64 (0.52) | |
| PT (w = d) | 0.113 (0.003) | 1.32 (0.07) | 6.07 (0.40) | |
| 3 | STD | 0.143 (0.002) | 0.00 (0.00) | 26.00 (0.00) |
| L1 | 0.120 (0.003) | 0.96 (0.07) | 6.67 (0.34) | |
| NG (w = 1) | 0.149 (0.003) | 0.09 (0.04) | 16.04 (0.53) | |
|
|
0.127 (0.003) | 0.02 (0.01) | 15.50 (0.51) | |
| NG (w = d) | 0.117 (0.003) | 0.06 (0.03) | 13.25 (0.52) | |
| PW (w = 1) | 0.165 (0.002) | 0.33 (0.05) | 17.98 (0.55) | |
|
|
0.147 (0.003) | 0.34 (0.05) | 16.96 (0.49) | |
| PW (w = d) | 0.141 (0.003) | 0.41 (0.06) | 15.39 (0.51) | |
| PT (w = 1) | 0.106 (0.003) | 0.59 (0.07) | 8.07 (0.53) | |
|
|
0.110 (0.003) | 1.10 (0.08) | 5.84 (0.34) | |
| PT (w = d) | 0.113 (0.003) | 1.28 (0.07) | 5.71 (0.27) | |
Evidently, DGC-SVM-PT generated models as sparse as that obtained from L1-SVM, and gave the most accurate predictions among all the other methods. In addition, the center gene, gene 1, was detected in each run by DGC-SVM-PT. NG-SVM and DGC-SVM-PW yielded fewer false negatives due to the larger models produced by each method. The weight w = d improved the classification accuracy, slightly shrank the model size, and kept almost the same false negatives for NG-SVM and DGC-SVM-PW compared with the other two weight functions. In contrast, w = 1 worked better for DGC-SVM-PT. It reduced the false negatives while produced models of comparable predictive performance to that with or w = 1. Therefore, DGC-SVM-PT with w = 1 was the winner. In addition, it also improved reproducibility. The most frequently-recovered pathways from each method (L1-SVM, NG-SVM , DGC-SVM-PW , and DGC-SVM-PT ) are displayed in Figure 2. Both DGC-SVM-PT and L1-SVM missed the leaves under each scenario. However, both identified the majority parts of the true pathways. Compared with all the other methods, DGC-SVM-PT detected the same pathway in a much higher frequency. Therefore, the identified pathways by this method were more reproducible.
Figure 2.

Most frequently recovered pathways. Rows from top to bottom: L1-SVM, NG-SVM, DGC-SVM-PW, and DGC-SVM-PT. Columns from left to right: scenario 1 (S1) {1, 2, 5, 6, 14, 15, 16}, scenario 2 (S2) {1, 3, 7, 17}, and scenario 3 (S3) {1, 2, 5, 14}. Frequencies of the recovered pathways are included in parentheses.
3.2 A complicated network
Next, we explored the complicated network originating from gene 1 as displayed in Figure 3. For this network, there exists one pair of genes that is connected by more than one path. Therefore, the DAG derived from the complicated network is not a tree. For example, gene 32 has both gene 23 and gene 3 at its upstream. In addition, genes at the same level are connected, such as gene 22 and gene 23, and gene 33 and gene 34. By definition, genes with no downstream genes are considered as leaves. Even though gene 22 is connected with gene 23, gene 22 is considered as a leaf because gene 23 is at the same level as gene 22. Likewise, genes 33 and 34 are both treated as leaves. Therefore, differing from the simple network, the DAG defined by the complicated network contains directed edges characterizing upper-lower relationships but ignores undirected edges describing lateral connections. Genes 1, 2, and 3 are assumed to be disease genes that affect the outcome weakly but importantly. Our goal is to identify disease genes, inlcuding those that play a critical role in mediating other genes in multiple biological processes even if their direct effects on the outcome are weak.
Figure 3.

Left: A complicated network originating from gene 1. Right: DAG defined from the network.
The simulated data were generated similarly as for the simple network. Here, we considered two scenarios: (1) genes 1, 2, and 3 had weak effects (β1 = β2 = β3 = 0.1), and leaf gene 32 had strong effect (β32 = 10); (2) the three disease genes had the same effect as in scenario (1) whereas leaf gene 34 had strong effect (β34 = 10).
Table 2 suggests that L1-SVM generated sparse models that gave the most accurate predictions under both the scenarios. However, on average, it missed about 2.5 out of 4 informative genes and identified only around 0.5 out of 3 disease genes. In contrast, NG-SVM yielded models that contained every gene. Even though DGC-SVMs led to bigger test errors than L1-SVM, they detected most of the informative and disease genes. In particular, DGC-SVM-PT detected all the informative and disease genes. The different weights led to similar results. Figure 4 displays the pathways most frequently identified by each method ( when a weight was involved) except the standard SVM in each scenario. It is obvious that L1-SVM most frequently captured the gene that exerted the largest influence on the response (genes 32 and 34 in scenarios 1 and 2 respectively). NG-SVM included every gene in each run, that is, it did not distinguish informative genes from the noise genes. After examining the output closely, we find that the DGC-SVM-PT selected all the informative genes as well as all the three disease genes in each run even though the three disease genes affected the response weakly. Although this method generated larger models and made less accurate predictions compared with L1-SVM, it exactly realized our attempt to identify all the disease genes along a pathway, including those with only weak effects.
Table 2.
Simulation results (averaged over 100 runs) for a complicated network with p = 13 genes. There were 4 informative genes in each scenario.
| Scenario | Method | Test Error (SE) | # False Negative (SE) | Model Size (SE) | # Disease Genes (SE) |
|---|---|---|---|---|---|
| 1 | STD | 0.131 (0.002) | 0.00 (0.00) | 13.00 (0.00) | 3.00 (0.00) |
| L1 | 0.089 (0.003) | 2.59 (0.07) | 3.04 (0.25) | 0.41 (0.07) | |
| NG (w = 1) | 0.214 (0.005) | 0.00 (0.00) | 13.00 (0.00) | 3.00 (0.00) | |
|
|
0.210 (0.004) | 0.00 (0.00) | 13.00 (0.00) | 3.00 (0.00) | |
| NG (w = d) | 0.216 (0.005) | 0.00 (0.00) | 13.00 (0.00) | 3.00 (0.00) | |
| PW (w = 1) | 0.109 (0.003) | 1.10 (0.14) | 9.58 (0.36) | 1.90 (0.14) | |
|
|
0.115 (0.003) | 0.86 (0.12) | 9.70 (0.34) | 2.14 (0.12) | |
| PW (w = d) | 0.117 (0.003) | 0.46 (0.09) | 10.65 (0.25) | 2.54 (0.09) | |
| PT (w = 1) | 0.146 (0.004) | 0.00 (0.00) | 10.79 (0.20) | 3.00 (0.00) | |
|
|
0.145 (0.004) | 0.00 (0.00) | 10.86 (0.18) | 3.00 (0.00) | |
| PT (w = d) | 0.145 (0.004) | 0.00 (0.00) | 10.62 (0.18) | 3.00 (0.00) | |
| 2 | STD | 0.137 (0.003) | 0.00 (0.00) | 13.00 (0.00) | 3.00 (0.00) |
| L1 | 0.095 (0.003) | 2.46 (0.08) | 3.75 (0.29) | 0.54 (0.08) | |
| NG (w = 1) | 0.217 (0.005) | 0.00 (0.00) | 13.00 (0.00) | 3.00 (0.00) | |
|
|
0.213 (0.004) | 0.00 (0.00) | 13.00 (0.00) | 3.00 (0.00) | |
| NG (w = d) | 0.215 (0.004) | 0.00 (0.00) | 13.00 (0.00) | 3.00 (0.00) | |
| PW (w = 1) | 0.101 (0.004) | 2.12 (0.13) | 5.52 (0.44) | 0.88 (0.13) | |
|
|
0.099 (0.003) | 1.56 (0.14) | 6.79 (0.44) | 1.44 (0.14) | |
| PW (w = d) | 0.107 (0.004) | 1.16 (0.13) | 8.03 (0.40) | 1.84 (0.13) | |
| PT (w = 1) | 0.151 (0.004) | 0.00 (0.00) | 9.62 (0.25) | 3.00 (0.00) | |
|
|
0.150 (0.004) | 0.00 (0.00) | 9.97 (0.23) | 3.00 (0.00) | |
| PT (w = d) | 0.152 (0.005) | 0.00 (0.00) | 9.98 (0.22) | 3.00 (0.00) | |
Figure 4.

Most frequently recovered pathways. Rows from top to bottom: L1-SVM, NG-SVM, DGC-SVM-PW, and DGC-SVM-PT. Columns from left to right: scenario 1 (S1) {1, 2, 3, 32}, and scenario 2 (S2) {1, 2, 3, 34}. Frequencies of the recovered pathways are included in parentheses.
4 Applications to microarray data
To evaluate their performance in the real world, we applied the proposed DGC-SVMs to two microarray data sets related to breast cancer metastasis (Wang et al 2005; Chuang et al 2007) and Parkinson’s disease (Scherzer et al 2007) (Gene Expression Omnibus: GSE6613; ttp://www.lncbi.nlm.ni.gov/geo/).
4.1 Breast cancer metastasis
The breast cancer metastasis (BC) data set contains expression levels of 8,141 genes from 286 patients, 106 of whom developed metastasis within a 5-year follow-up after surgery. The data set includes three tumor suppressor genes, TP53, BRCA1, and BRCA2, which are known for preventing uncontrolled cell proliferation, and for playing a critical role in repairing the chromosomal damage. The malfunction of these genes leads to an increased risk of breast cancer. The protein-protein interaction (PPI) network previously used by Chuang et al (2007) was adopted as our prior biological information, which was obtained by assembling a pooled data set comprising 57,235 interactions among 11,203 proteins and curation of the literature. We considered a subnetwork consisting of the direct neighbors of the three tumor suppressor genes, denoted as BC-1nb-net. Eexpression levels of a total of 294 genes that belong to BC-1nb-net were observed. In our analysis, due to its prominent role, TP53 was considered as the center gene of the network.
In each run, we randomly split the data into training, tuning, and testing set with 95, 95 and 96 observations respectively. The expression level of each gene was normalized to have mean 0 and standard deviation 1 across samples. Given any value from a prespecified set of wide-ranging values for the tuning parameter λ, we obtained the classifier f̂λ on the training set and applied f̂λ on tuning data. The value of λ that yielded the minimal classification error on the tuning data was chosen as λ̂. Then we applied the classifier f̂λ̂ on the test set to evaluate its performance. We compared the performance of DGC-SVM with that of STD-SVM, L1-SVM, and NG-SVM in terms of classification error, selection of mutant genes (genes with available mutation frequencies), and model sparsity, averaged over 50 runs. Since genes with large mutation frequencies are more likely to malfunction, disturbing the aggregate activity of the network, a method that can detect more such mutant genes may be considered to perform better. The mutation frequencies of 227 genes are available (Chuang et al 2007). Among the 294 genes in BC-1nb-net were 40 genes with mutation frequencies (named as cancer genes), 7 of which (ABL1, JAK2, p53, PTEN, p14ARF, PTCH, and RB) with a mutation frequency larger than 0.10 (named as cancer genes with large mutation frequency).
Table 3 indicates that all the methods had similar predictive accuracies even though DGC-SVM-PT with w = 1 was slightly better. However, an improvement in detecting clinically relevant genes was significant by incorporating the prior network information. Compared with L1-SVM, NG-SVM with w = d detected almost twice as many frequently mutant cancer genes and more than 1.5 times cancer genes with models less than 1.5 times larger. DGC-SVM-PW with w = d generated a more sparse model with 3 genes less than L1-SVM while detected the same number of cancer genes and almost twice as many frequently mutant cancer genes as L1-SVM. The advantage of DGC-SVM-PT with w = d in gene selection was evident. It captured almost 3.5 times as many frequently mutant cancer genes and almost twice cancer genes by a model only 1.2 times larger than L1-SVM. The proposed DGC-SVM prevailed the other methods in identifying clinically important genes.
Table 3.
Breast cancer metastasis data with 294 genes in BC-1nb-net including 40 cancer genes, and 7 cancer genes with mutation frequencies larger than 0.10: classification error, number of selected cancer genes (CA), number of selected cancer genes with mutation frequencies larger than 0.10 (CA-LMF), number of selected genes, and their standard errors (SE in parentheses) obtained by averaging over 50 runs.
| Method | Error (SE) | # CA-LMF | # CA | # Genes | |
|---|---|---|---|---|---|
| STD | 0.396 (0.006) | 7.00 (0.00) | 40.00 (0.00) | 294.00 (0.00) | |
| L1 | 0.384 (0.007) | 0.46 (0.10) | 3.92 (0.46) | 26.48 (3.01) | |
| NG (w = 1) | 0.385 (0.008) | 0.58 (0.10) | 3.82 (0.45) | 29.38 (2.91) | |
|
|
0.388 (0.006) | 0.64 (0.11) | 4.90 (0.60) | 31.82 (2.95) | |
| NG (w = d) | 0.404 (0.006) | 0.86 (0.11) | 6.42 (0.58) | 36.88 (2.45) | |
| PW (w = 1) | 0.380 (0.007) | 0.78 (0.15) | 4.86 (0.55) | 42.32 (4.27) | |
|
|
0.382 (0.006) | 0.62 (0.12) | 4.48 (0.62) | 30.50 (3.64) | |
| PW (w = d) | 0.396 (0.006) | 0.86 (0.12) | 3.96 (0.49) | 23.80 (2.89) | |
| PT (w = 1) | 0.373 (0.006) | 1.76 (0.16) | 14.90 (1.09) | 88.18 (6.31) | |
|
|
0.395 (0.006) | 1.84 (0.18) | 12.52 (0.61) | 63.88 (2.12) | |
| PT (w = d) | 0.396 (0.005) | 1.58 (0.16) | 7.20 (0.60) | 31.76 (2.64) |
4.2 Parkinson’s Disease
The Parkinson’s disease (PD) data set includes disease status and expression levels of 22,283 genes from 105 patients, 50 cases and 55 controls (Scherzer et al 2007). The gene network information was obtained from two sources: (1) the network in Li and Li (2008), which combines 33 KEGG regulatory pathways and contains 1,523 genes and 6,865 edges; (2) the Parkinson’s disease KEGG pathway (PD-KEGG, ttp://www.genome.ad.jp/kegg/patway/hsa/hsa05020.html), which uncovers the interactions of 27 PD disease genes as of November 2008. A total of 12 out of the 27 PD disease genes are contained in the network of Li and Li: UBB, UBE1, CASP9, CASP3, APAF1, CYCS, PARK2, GPR37, SEPT5, SNCAIP, SNCA, and TH. We focused our analysis on a subnetwork expanded from the 12 disease genes with the network of Li and Li, denoted as PD-net (Figure 5), which consists of four components: (1) the 6th-order-neighbor-subnetwork of UBB (A direct neighbor of UBB is defined as a 1st-order-neighbor; a direct neighbor of a 1st-order-neighbor of UBB as a 2nd-order-neighbor; and so on.); (2) the 3rd-order-neighbor-subnetwork of CASP9; (3) the isolated four-gene-subnetwork including PARK2, GPR37, SEPT5, and SNCAIP; and (4) the isolated two-gene-subnetwork including SNCA and TH. A total of 181 genes belong to PD-net. Note that PARK2/GPR37/SEPT5/SNCAIP and SNCA/TH form two islands respectively. The DAG of PD-net (Figure 6) was obtained by merging the DAG of UBB and that of CASP9 at the common node SMAD7. Note the two islands in Figure 6.
Figure 5.

PD-net: subnetworks grown from the 12 PD disease genes (UBB, UBE1, CASP9, CASP3, APAF1, CYCS, PARK2, GPR37, SEPT5, SNCAIP, SNCA, and TH); 181 genes in total. PD genes are in red.
Figure 6.

DAG transformed from PD-net: 12 PD disease genes (UBB, UBE1, CASP9, CASP3, APAF1, CYCS, PARK2, GPR37, SEPT5, SNCAIP, SNCA, and TH); 181 genes in total. PD genes are in red.
In each run, the data set was randomly split into training, tuning, and test sets with 40, 20, and 45 observations respectively. The expression level of each gene was normalized to have mean 0 and standard deviation 1 across samples. The performance of each method was evaluated on the test set by the classification error, the selection of PD genes, and model sparsity averaged over 50 runs. Again five methods were compared: STD-SVM, L1-SVM, NG-SVM, DGC-SVM-PW (w = 1, , or w = d), and DGC-SVM-PT (w = 1, , or w = d). To obtain the final model for each method, we used a new tuning dataset by combining, in each run, the previous tuning and test data, leading to a sample size as large as 65. Here λ̂ was identified on the new tuning set from a wide range of prespecified values. We averaged the classification errors corresponding to each tuning parameter value over 50 runs. The value λ̂ leading to the minimal average error was used to fit the final model to all the data. Note that the classification error from the final model was likely to be biased due to reuse of the data for training/tuning and test; the purpose of fitting the final model was to yield a set of selected genes.
As Table 4 indicates, the methods that incorporate the prior gene network information improved gene selection while maintaining a predictive accuracy comparable to that of the STD-SVM and L1-SVM. L1-SVM generated models with an average of 16.86 genes including 1.50 PD genes. NG-SVM with w = 1 detected more than twice as many PD genes with models having 1.28 less genes than L1-SVM. The improvement in gene selection of DGC-SVM-PW with w = 1 was more significant. It produced models with 3.76 less genes while detected more than three times as many PD genes as L1-SVM. Both the methods had a predictive accuracy at a comparable level to that of L1-SVM. DGC-SVM-PT with w = 1 improved both prediction accuracy and gene selection. It reduced the classification error by 17% and also selected three times as many PD genes with a model size not so much larger than that of L1-SVM. Overall, DGC-SVM-PT with w = 1 outperformed the other methods.
Table 4.
Parkinson’s disease: classification error, number of selected disease genes, number of selected genes, and their standard errors (SE in parentheses) obtained by averaging over 50 runs.
| Method | Error (SE) | # Disease Genes | # Genes | |
|---|---|---|---|---|
| STD | 0.448 (0.011) | 12.00 (0.00) | 181.00 (0.00) | |
| L1 | 0.490 (0.008) | 1.50 (0.19) | 16.86 (1.58) | |
| Final | - | 5 | 70 | |
| NG (w = 1) | 0.500 (0.011) | 3.32 (0.39) | 15.58 (1.72) | |
| Final | - | 8 | 83 | |
|
|
0.501 (0.009) | 0.66 (0.19) | 12.00 (1.34) | |
| Final | - | 12 | 102 | |
| NG (w = d) | 0.493 (0.008) | 0.00 (0.00) | 14.10 (1.44) | |
| Final | - | 5 | 133 | |
| PW (w = 1) | 0.481 (0.010) | 4.80 (0.43) | 13.10 (1.52) | |
| Final | - | 9 | 97 | |
|
|
0.493 (0.009) | 3.26 (0.41) | 13.22 (1.70) | |
| Final | - | 9 | 103 | |
| PW (w = d) | 0.493 (0.007) | 1.52 (0.28) | 14.70 (1.88) | |
| Final | - | 7 | 97 | |
| PT (w = 1) | 0.407 (0.011) | 4.56 (0.41) | 42.34 (4.64) | |
| Final | - | 6 | 108 | |
|
|
0.445 (0.013) | 2.40 (0.18) | 45.04 (3.80) | |
| Final | - | 6 | 113 | |
| PT (w = d) | 0.456 (0.011) | 2.42 (0.15) | 55.56 (4.08) | |
| Final | - | 5 | 117 | |
The selected genes of the final model obtained from each method (L1-SVM, NG-SVM with w = 1, DGC-SVM-PW with w = 1, and DGC-SVM-PT with w = 1) are displayed in the networks in Figure 7, Figure 8, Figure 9 and Figure 10. L1-SVM generated the sparsest final model and missed the most PD genes. NG-SVM selected more leaves, some of which were from the two islands, while neglecting around a half of the nodes. According to DGC-SVM-PT, nodes located at higher level are more likely to have nonzero estimates. The majority of nodes were detected and some of the genes from the two islands were included by this method. DGC-SVM-PW identified the most PD genes among the four final models, though it neglected most part of the subnetwork derived from UBB.
Figure 7.

L1-SVM final model: selected PD genes (UBB, UBE1, CASP9, CASP3, and SNCA) in red; missed PD genes (APAF1, CYCS, PARK2, GPR37, SEPT5, SNCAIP, and TH) in green; other selected genes in yellow; unselected genes in white; 181 genes in total.
Figure 8.

NG-SVM with w = 1 final model: selected PD genes (UBB, UBE1, CASP9, APAF1, CYCS, SEPT5, SNCA, and TH) in red; missed PD genes (CASP3, PARK2, GPR37, and SNCAIP) in green; other selected genes in yellow; unselected genes in white; 181 genes in total.
Figure 9.

DGC-SVM-PW with w = 1 final model: selected PD genes (CASP9, CASP3, SNCA, CYCS, APAF1, TH, PARK2, SEPT5, and SNCAIP) in red; missed PD genes (UBB, UBE1, and GPR37,) in green; other selected genes in yellow; unselected genes in white; 181 genes in total.
Figure 10.

DGC-SVM-PT with w = 1 final model: selected PD genes (UBB, UBE1, PARK2, CASP3, SNCA, and CASP9) in red; missed PD genes (APAF1, CYCS, GPR37, SEPT5, SNCAIP, and TH) in green; other selected genes in yellow; unselected genes in white; 181 genes in total.
5 Discussion and conclusion
The availability of various repositories of gene networks and the accumulating knowledge on genes central to diseases make it possible to use these two sources of prior biological information to help build microarray-based classifiers. Employing such a network-based perspective not only sheds insight on deciphering the complexity within the network modules but also offers the possibility to detect genes that play a critical role in mediating multiple biological processes but have weak direct effects on the outcome by themselves. Such genes are often ignored by gene-expression-alone analysis, as pointed out by Chuang et al (2007).
In this paper, we were motivated to devise a penalty that encourages gene selection along pathways or within subnetworks. The identification of any gene triggers the search for disease genes along gene pathways or within subnetworks all the way toward genes central to the disease. The penalty boosts investigation of relationships within collectives of genes that contain both the selected genes and the central genes. By converting the undirected gene network into DAG, we imposed an upper-lower hierarchy on the gene network depending on each gene’s distance to the assumed network center, typically gene(s) central to the disease. The DAG facilitated the definition of gene groups according to one of two different ways of grouping, one based on grouping genes along linear pathways, and a second on grouping genes with all their downstream genes. The penalty term was constructed from the L∞-norm being applied to each group. Combined with the hinge loss, we obtained our proposed method: DGC-SVM-PW and DGC-SVM-PT. The former attempts to realize selection along linear pathways in the network. The latter ensures the selection of an upper-level gene if any of its downstream genes is selected, thus realizing hierarchical selection. Due to this property, genes that have more downstream genes and are closer to the center are very likely to be detected. In addition, selection of any gene guarantees the inclusion of at least one central gene. According to the simulation studies, DGC-SVM detected more disease genes even if they potentially affected the outcome only weakly. Two real data applications showed that the new method prevailed STD-SVM and L1-SVM in capturing clinically relevant genes while making either more accurate or comparable predictions on the outcome. We conclude that DGC-SVM has the potential to be an effective classification tool for microarray data.
Even though its strength in improving gene selection has been established, DGC-SVM has weaknesses in four aspects. First, specification of the network center is somewhat arbitrary. Specifying different central gene(s) results in different group structures and thus different penalties. Second, how to define DAG when more than one center gene exist is worth debating. We suggested an approach that is admittedly somewhat arbitrary. Third, the computational cost of DGC-SVM-PW is expensive for a large network since a large number of groups are derived. Fourth, use of the weights may improve gene selection but also reduce predictive accuracy or vice versa; it is not guaranteed to better both at the same time. A further investigation may focus on developing a simpler and more effective way to incorporate a prior gene network.
Acknowledgments
YZ and WP were partially supported by NIH grants HL65462 and GM081535; XS supported by NIH grant GM081535 and NSF grants IIS-0328802 and DMS-0604394. The Minnesota Supercomputing Institute (MSI) provided computing resource. We thank Dr Hongzhe Li and Dr Trey Ideker for providing the KEGG network and PPI network data respectively. We appreciate the helpful comments from the reviewers.
References
- Alfarano C, Andrade CE, Anthony K, Bahroos N, Bajec M, Bantoft K, Betel D, et al. The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res Database Issue. 2005;33:D418CD424. doi: 10.1093/nar/gki051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson M, Carlsson L, Guillot G, Jernas M, Langston MA, Rudemo M, Andersson B. A network-based analysis of allergen-challenged CD4+ T cells from patients with allergic rhinitis. Genes and Immunity. 2006;7:514–521. doi: 10.1038/sj.gene.6364322. [DOI] [PubMed] [Google Scholar]
- Bondell HD, Reich BJ. Simultaneous regression shrinkage, variable selection, and supervised clustering of predictors with OSCAR. Biometrics. 2008;64:115–123. doi: 10.1111/j.1541-0420.2007.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Børresen-Dale AL. TP53 and breast cancer. Human Mutation. 2003;21:292–300. doi: 10.1002/humu.10174. [DOI] [PubMed] [Google Scholar]
- Brown MPS, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M, Haussler D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvano SE, Xiao WZ, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF. A network-based analysis of systematic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- Cortes C, Vapnik V. Support-vector networks. Machine Learning. 1995;20:273–297. [Google Scholar]
- Chuang HY, Lee EJ, Liu YT, Lee DH, Ideker T. Network-based classification of breast cancer metastasis. Molecular Systems Biology. 2007;3 doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey T, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16:906–914. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res Database Issue. 2004;32:D277CD280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MW, Liberzon A, Kong SW, Lai WR, Park PJ, Kohane IS, Kasif S. Network-based analysis of affected biological processes in type 2 diabetes models. PLoS Genetics. 2007;3:958–972. doi: 10.1371/journal.pgen.0030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Li H. Network-constrained regularization and variable selection for analysis of genomic data. Bioinformatics. 2008;24:1175–1182. doi: 10.1093/bioinformatics/btn081. [DOI] [PubMed] [Google Scholar]
- Pan W, Xie B, Shen X. To appear Biometrics. Division of Biostatistics, University of Minnesota; 2009. Incorporating predictor network in penalized regression with application to microarray data. Available at http:http://www.biostat.umn.edu./rrs.php as Research Report 2009-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri S, Navarro JD, Kristiansen TZ, Amanchy R, Surendranath V, Muthusamy B, Gandhi TK, et al. Human Protein Reference Database as a discovery resource for proteomics. Nucleic Acids Res Database Issue. 2004;32:D497CD501. doi: 10.1093/nar/gkh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzer CR, Eklund AC, Morse LJ, Liao Z, Locascio JJ, Fefer D, Schwarzschild MA, Schlossmacher MG, Hauser MA, Vance JM, Sudarsky LR, Standaert DG, Growdon JH, Jensen RV, Gullans SR. Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc Natl Acad Sci U S A. 2007;104:955–960. doi: 10.1073/pnas.0610204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussi T, Béroud C. Significance of TP53 mutations in human cancer: a critical analysis of mutations at CpG dinucleotides. Human Mutation. 2003;21:192–200. doi: 10.1002/humu.10189. [DOI] [PubMed] [Google Scholar]
- Tibshirani R. Regression Shrinkage and Selection via the Lasso. Journal of the Royal Statistical Society, B. 1996;58:267–288. [Google Scholar]
- Vapnik V. The Nature of Statistical Learning Theory. Springer-Verlag; 1995. [Google Scholar]
- Wang L, Shen X. On L1-Norm multiclass support vector machines: Methodology and theory. Journal of the American Statistical Association. 2007;102:583–594. [Google Scholar]
- Wang L, Zhu J, Zou H. The doubly regularized support vector machine. Statistica Sinica. 2006;16:589–615. [Google Scholar]
- Wang Y, Klijin JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Wu S, Shen X, Geyer CJ. Adaptive regularization through entire solution surface. 2008 doi: 10.1093/biomet/asp038. Manuscript. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Rocha G, Yu B. The composite absolute penalties family for grouped and hierarchical variable selection. The Annals of Statistics, to appear [Google Scholar]
- Zhu J, Rosset S, Hastie T, Tibshirani R. 1-norm support vector machines. Neural Information Processing Systems 2003 [Google Scholar]
- Zhu Y, Shen X, Pan W. Network-based support vector machine for classification of microarray samples. BMC Bioinformatics. 2009;10(Suppl 1):S21. doi: 10.1186/1471-2105-10-S1-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Hastie T. Regularization and variable selection via the elastic net. Journal of Royal Statistical Society: Series B. 2005;67:301–320. [Google Scholar]
- Zou H, Yuan Ming. The F∞-norm Support Vector Machine. Statistica Sinica. 2008;18:379–398. [Google Scholar]