

Abstract
Shape is a fundamentally important molecular feature that often determines the fate of a compound in terms of molecular interactions with preferred and non-preferred biological targets. Complementarity of binding in small molecule-protein, peptide-receptor, antigen-antibody and protein-protein interactions is key to life and survival, but also to targeting molecules with bioactivity. We review the application of shape in various biological systems such as substrate recognition, ligand specificity / selectivity and antibody recognition in the context of computational methods such as docking, quantitative structure activity relationships, classification models and similarity search algorithms. These in silico pharmacology methods have recently demonstrated the importance and applicability of determining molecular shape in drug discovery, virtual screening and predictive toxicology. The results from recently published studies show that shape and shape-based descriptors are at least as useful as other traditional molecular descriptors.
Keywords: Antibody, Depth, Descriptors, Dopamine receptors, Molecular shape, Nuclear hormone receptor, Pharmacophore
Introduction
Form follows function - that has been misunderstood. Form and function should be one, joined in a spiritual union.
Frank Lloyd Wright, 1908
It has been long recognized that determining molecular shape (Box 1) and changes in this property is essential in order to understand, molecules involved in chemical reactions 1. Enzymes can differentiate between functional groups in a molecule through shape recognition; also, natural products produced through biosynthetic pathways involve shape recognition for selective oxidation 2. More recently, chiral recognition was shown at the single-molecule level to involve mutually induced conformational adjustments 3. At lower concentrations of magnesium ions, ribozymes from Escherichia coli and Bacillus subtilis recognize cloverleaf shape RNAs rather than hairpin shape RNAs, indicating shape recognition 4. Chemical shape interaction has a key role in the senses 5: smell (via hundreds of olfactory receptors), sight (via receptors responsible for the perception of color), and taste (via receptors responsible for the perception of bitter, sour, sweet, salt and umami), and all of these receptors are G-protein coupled receptors. Various studies have reinforced the idea that molecular shape plays a major role in biological activity (6 and references therein). Addition of other features to molecular shape would be expected to increase specificity such as complementary electrostatic or steric interactions.
Box 1. Shape and depth.
The shape (OE. sceap Eng. created thing) of an object located in some space refers to the part of space occupied by the object as determined by its external boundary — abstracting from other aspects the object may have such as its color, content, or the object’s position and orientation in space, and its size. Shape can also be more generally defined as "the appearance of something, especially its outline". However, the definition of shape in molecular pharmacology does encompass structural features like depth, size and surface. As can be visualized by a simple Tupperware toy in which small shapes fit into complementary holes (Box Figure A), the importance of shape comes into play when a protein packs against another protein (Box Figure B) or when a small molecule is desired to fit into a binding site on a protein surface (Box Figure C), where size and shape complementarity may be essential in addition to favorable electrostatic and steric interactions, to fulfill the “lock and key” or “induced fit” hypotheses 63.
Analogous to the term shape is “depth” which can be defined as the distance of an atom from the nearest surface water molecule 64 that is usually applicable when describing shape based features in proteins such as grooves in DNA or binding pockets buried in membranes 65. However, the term depth is more abstract and provides only a limited quantitative measure of shape.
Box 1. Examples to illustrate the importance of shape.

Figure A. simplified example to demonstrate how a protein represented by the Tupperware toy, could have multiple potential binding sites of different shape and size, into which specific shaped molecules may fit. B. Analogous to the Tupperware toy is an example of favorable protein-protein interactions that makes the homotetrameric structure of turkey beta adrenergic receptor. C. An example of a protein with multiple binding sites: surface representation of dihydroorotate dehydrogenase of plasmodium falciparum with bound inhibitor A26 colored red, flavin mononucleotide colored yellow, orotic acid colored green and N8E colored blue.
Irrespective of the type of definition used, the essence of shape is therefore very useful in describing molecule(s) by themselves or the nature of interactions between molecules. Hence, the study of shape in molecular pharmacology has gained importance due to its applicability in drug design process in silico techniques (Table 1) widely employed to decrease the costs of drug discovery and development. These computational methods can enable rapid comparisons between small molecules, or small molecules with protein receptor sites, mainly based on their shape and other properties such as electrostatics. This review explores the various definitions of shape generally used when describing a molecule or interaction between molecules and provides examples of biological systems where the concept of shape plays a major role. The applicability of shape in in silico techniques is also detailed along with future developments for in silico pharmacology. Several published studies illustrate that shape-based methods and descriptors in various classification and other modeling schemes are as useful as traditional molecular properties, like 2D descriptors 7-9.
Table 1.
Computational methods incorporating shape
| Method | Examples | Uses | Pros and Cons |
|---|---|---|---|
| Docking using explicit shape | DOCK 26, GOLD 27, GLIDE 12 and AutoDock 28 | Small molecule – protein interactions and protein-protein interactions | Value in database screening is linked with scoring algorithm used. Docking may be poorer at finding actives than ligand based methods 46 |
| Docking using implicit shape | soft docking method 29, PUZZLE 30 | Small molecule – protein interactions and protein-protein interactions | As above but the shape complementarity may be a useful filter. |
| Molecular shape descriptors | Topological torsions, Wiener indices, atom pair representations for small fragments 61, Shape Signatures 53, Zernicke descriptors 67, local intersection volume 68, path-space ratio 69, ROCS 37, 43-47, TAE48, 49, PEST 52, Shape Signatures54 and others 70. | Use with Molecular similarity indices, QSAR, QSPR, machine learning methods for classification | Descriptor use may depend widely on data modeled, algorithm used and combination of descriptors evaluated. Some descriptors may require longer to calculate than others. Applicability domain of models may also be a limitation depending on the training set and the coverage of chemical space. |
| Pharmacophores | DISCO, GASP, Catalyst, Apex 3D, GALAHAD, Phase 24, 25 | Database searching, QSAR | The method is applicable when there is no target 3D structure available. Pharmacophore searching is faster than docking but likely not as fast as descriptor based methods for filtering compound libraries. Potential to find structural matches that may not look similar in 2D, but possess the key features for 3D mapping. |
| Pharmacophore fingerprints | ChemX/ ChemDiverse, PharmPrint, OSPREYS, 3D Keys and Tuplets 24, 25 | Use with Molecular similarity indices, QSAR, QSPR, machine learning methods for classification | Fingerprints are useful for very rapid comparisons of molecules and similarity analyses. |
| Pseudoreceptor models | QUASAR, RAPTOR, and many others see previous review36. | Exploration of ligand receptor interactions | As with pharmacophores this method has utility when there is no target 3D structure available. Potential for finding novel binding modes. Alignment dependent. |
| Molecular Field Analysis | CoMFA and XED 24, 25 | QSAR, QSPR | Requirement for molecular alignment is a limitation and may also restrict use of such methods for database searching. |
Visualizing molecular shape in biological systems
There are many examples of applications of computational methods using shape (Table 2). Simple molecular shape analysis by determining the van der Waals volume of active and inactive compounds can be insightful for enzymes, for example, in elucidating substrate recognition by Pseudomonas fluorescens N3 dioxygenase 10 and in visualizing differences in inhibitors for human cytochrome P450 (CYP) 51 11. Shape descriptors (Box 2) have been found to be important in some recent computational models. For example, a model for protein-protein interaction inhibitors found the shape descriptor SHP2 at the top of the decision tree 12. Sammon and Kohonen mapping human ether-ago-go potassium ion channel models contained Wiener and Balaban index descriptors, suggesting that molecular shape or topological characteristics were important for binding to this ion channel 13.
Table 2.
Examples showing the importance of shape
| Role of shape | Examples |
|---|---|
| Substrate recognition | Pseudomonas fluorescens N3 dioxygenase 10, human cytochrome CYP51 11 |
| Classification models | Protein-protein interaction inhibitor decision tree 12. Sammon and Kohonen mapping hERG models 13, PEST descriptors to distinguish musk and non-musk compounds52, models of the 5-HT2B receptor, the hERG ion channel, blood brain barrier penetration31, 32 and PXR 9 |
| QSAR models | ADME/Tox related datasets using TAE descriptors 50 |
| Virtual screening | ROCS used to discriminate between cruzain and cathepsin L inhibitors 43, Comparisons of different methods 37, 44-47, Shape signatures used to find novel antiestrogens 80 and enrich screening for serotonin ligands 54 |
| Co-evolution of molecule–protein interactions | NHRs (pregnane X receptor, farnesoid X receptor and liver X receptor) 15, 16, 18 |
| Ligand-receptor interactions | Dopamine D4 receptor 19 |
| Immunoassay cross reactivity | Using 2D similarity searching and 3D pharmacophore searching for barbitures, benzodiazepines, and tricylic antidepressants |
| Bioisosterism | Angiotensin-II analogs 57 |
| Masking molecular shape | Compartmentalization of reagents 71 |
| Inducing protein disorder | Rifampicin binding to human PXR 73 |
| Molecule encryption | Extending methods using structural fragments 74 and similarity based approaches 75 |
Box 2. Shape as a molecular descriptor.
Many different molecular shape descriptors have been proposed so far in the literature for small molecules and polymers. A review of all the different topological indices and their application to drug discovery is discussed in 66 and is beyond the scope of this current review. Fragment or substructure based indices (also called Free-Wilson-analysis) are the 2D descriptors commonly used to describe molecular shape 61. Field based descriptors and others such as Shape Signatures 53, Zernicke descriptors 67, local intersection volume 68 and path-space ratio 69 use 3D information of the molecule and are generally more efficient and computationally intensive forms describing the molecule. Field methods in general have been broadly classified as quantum mechanics (QM) based descriptors (PEST and TAE) 70 and non-QM methods such as Comparative Molecular Field Method (CoMFA) 42. Shape descriptors represent only an essence of the molecular shape by reducing the three dimensions to a set of numbers. Hence, these descriptors cannot be qualitatively used to ascertain ligand atoms responsible for hydrogen bonding to protein donor atoms.
Ligand-protein interactions
The co-evolution of molecule protein interactions with regard to shape has been explored in the study of nuclear hormone receptors (NHRs) that recognize bile salts 14, 15. Bile salts are the main end-metabolites of cholesterol in vertebrate animals. Evolutionarily early vertebrates such as jawless fish (lampreys and hagfish) use planar 5α (‘allo’) bile salts 15. In contrast, many other vertebrates, including humans and most mammals, use 5β bile salts that have a ‘bend’ at the junction of the A and B steroid rings. Cross-species comparisons of the selectivity of the farnesoid X receptor (FXR; ‘bile acid receptor’) for structurally diverse bile salts showed that FXR has changed selectivity for bile salts from preference for 5α (flat) bile salts (‘ancestral’ pattern in sea lamprey and zebrafish) to a preference for 5β bile salts in humans and mice (‘recent’ pattern). Computational homology models predicted that this selectivity change was mediated by altering the shape and size of the ligand binding pocket 15. Using similar computational approaches, vertebrate liver X receptors (LXR, ‘oxysterol receptor’) have also been found to diverge from invertebrate LXR in their ligand specificity 16. Analyses of cross-species differences in receptor binding sites can also be inferred using a ligand-based approach such as a pharmacophore. For example, pregnane X receptors (PXRs) (broad specificity nuclear hormone receptor (NHR) involved in regulation of liver metabolism 17), show significant cross-species differences in ligand specificity, with a broadening of ligand specificity from teleost fish to mammals and birds14, 18. These NHRs represent robust model systems to explore the co-evolution of receptors and ligands in terms of shape and size.
In a recent study on identifying highly selective dopamine D4 receptor agonists and antagonists, shape and charge complementarity between the ligand and the receptor microdomain was found to play a major role in the functioning of the 1,4-disubstituted aromatic piperidines and piperazine inhibitors (Figure 1) 19. Thus, the presence of structurally compatible regions between the receptor and its ligand is required for the functioning of these compounds.
Figure 1. Models to show favorable and unfavorable ligand-protein interactions.

A. structural model of the wild type Dopamine D4 receptor with bound ligand L750,667 (stick model and colored atom type with C-green, O-red, N-blue, H-white, I-purple) 19. The residues F2.61 and V3.28 in the second and third transmembrane domain (seen as orange sticks) aid in favorable interactions with the ligand. B. structural model of the wild type Dopamine D2 receptor with bound ligand L750,667. The residues V2.61 and F3.28 residues in TM2 and TM3 poses steric hindrance to the ligand.
Antibody-antigen interactions
Immunoassays based on antibodies are widely used in clinical medicine 20. Common applications include drug of abuse (DOA) screening, endocrinology testing, and therapeutic drug monitoring (TDM). Immunoassays are also employed as sensors for the detection of chemical warfare agents such as nerve gases and environmental pollutants 21. Immunoassays may have either narrow specificity for a single target compound (e.g., a drug, vitamin, toxin, or hormone) or broader specificity for a group of structurally related target compounds (e.g., benzodiazepines, opiates). Antibodies use molecular shape to recognize the antigen and this may present problems with similar shaped but distinct molecules. Immunoassays are limited by the occurrence of false positives (or ‘cross-reactive’ compounds), defined as a positive result in the absence of the target compound(s). False positives are a particular limitation of DOA screening assays. For example, many drugs with structural similarity to amphetamine and methamphetamine such as pseudoephedrine and bupropion can cross-react with amphetamine screening tests. There have been some studies of the three-dimensional structure of antibodies bound to drugs that are the target of DOA screening tests or TDM assays, providing insight into the specificity of antibody-drug interactions. For example, in X-ray crystallographic structures of antibodies complexed with cocaine 22 (additionally supported by molecular modeling studies 23), the antibody interacts with all portions of the target molecule.
There have been few efforts to use computational methods to predict and identify cross-reactive compounds for clinically used immunoassays. In initial studies, we have looked at molecular shape in terms of 2D similarity of test compounds to that of the antigen, using the MDL public keys fingerprint descriptors. A database of frequently used Food and Drug Administration (FDA)-approved drugs derived from the Clinician’s Pocket Drug Reference supplemented with drugs of abuse and drug metabolites (n = 813) important in clinical toxicology was used for searching. We found that ‘within-class’ true positives for three urine toxicology screening assays (barbitures, benzodiazepines, and tricylic antidepressants) tend to have high similarity to the target compound (Figure 2A). Particularly for benzodiazepines, however, some within-class compounds (e.g., clobazam and clonazepam) have lower similarity to the target compound diazepam, reflecting the diversity of structural modifications on the basic benzodiazepine core structure. Similarity methods provide a means of quantitatively rationalizing why compounds like clobazam and clonazepam generate ‘false negative’ results in an assay where diazepam was used to generate the assay antibodies.
Figure 2. Using 2D-Structural similarity to understand immunoassay false positives.
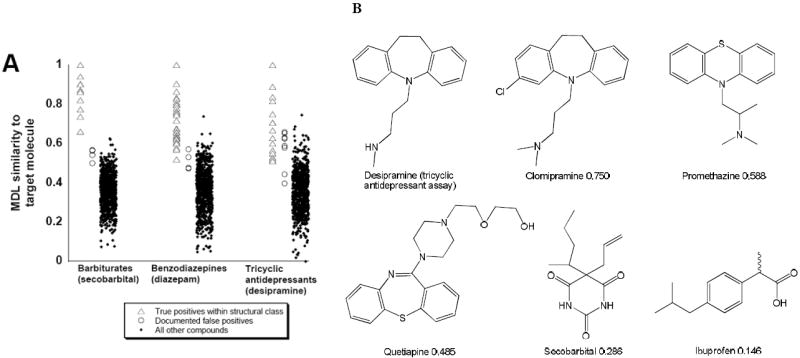
A. 2D similarity of compounds in the database (FDA approved drugs and some metabolites) to the target compounds of several DOA immunoassays. B. Similarity of five compounds to the target compound desipramine (target of some tricyclic antidepressant screening assays) calculated by MDL public keys using the Tanimoto similarity measure. Of the five compounds, clomipramine is the most similar (Tanimoto similarity = 0.750) while ibuprofen is the least similar (Tanimoto similarity = 0.146).
In addition, other computational approaches could be used such as 3D methods that require the development of a pharmacophore or pharmacophoric pattern representing the arrangement of the chemical features and distances between them important for binding 24, 25 (e.g. alignment of desipramine and amitrityline). The advantage of the 3D similarity approach is that it is able to find structural matches that may not look similar in 2D, but possess the key features for 3D mapping. For example, a pharmacophore for amitriptyline and desipramine that was used to search the same database of over 800 drugs and metabolites retrieved over 150 hits including many non-tricyclic compounds (Figure 3). The number of hits can be filtered further to 79 by addition of a van der Waals surface around one of the molecules accounting for shape of the molecule (Figure 3B, 3C). The approaches described above represent unique ways to search for potential cross reacting compounds based on shape that corresponds to some degree with the antibody-antigen interaction.
Figure 3. Using 3D pharmacophores to understand immunoassay false positives.

A. 3D-pharmacophore of amitriptyline and desipramine (yellow) generated with Catalyst Discovery Studio 2.0 (Accelrys, San Diego, CA) to illustrate how these could represent the shape of the antigen. B. Mapping of protriptyline (Fit value 3.964 and shape similarity 0.786) the best fitting molecule to the pharmacophore features and inside the van der Waals shape (shaded volume) after performing the pharmacophore-shape search. C. Mapping of clomipramine (Fit value 3.926 and shape similarity 0.761) to the pharmacophore features and inside the van der Waals shape. The cyan blobs represent hydrophobic features and the red blob represents a positive ionizable feature.
Shape representations in in silico methods
Given the enormous domain of shape-based features, it is challenging to characterize shape in algorithms for drug discovery. Several docking and scoring algorithms (Table 1) typically use shape either in implicit form or explicitly. In an explicit representation, the shape of the ligand is used to position it in the binding site of the target (usually a protein) with multiple conformer evaluations. This feature can be used to guide the rest of the docking process. Examples of the many available docking algorithms that use an explicit shape method are DOCK 26, GOLD 27, GLIDE 12 and AutoDock 28. Although some of these methods may also use shape implicitly through their scoring functions (the reader is referred to the individual references for further detail on each method). In an implicit representation, the shape of a molecule is used as a first level of filter or in a “screening” mode to identify if the molecule fits into the target or not. The soft docking method 29 and PUZZLE 30 (which use the shape complementarity between the surface of two interacting molecules as a filter) and many other programs use various representations for shape implicitly.
Another way to describe shape is to represent it in the form of molecular descriptors. Many different molecular shape descriptors have been proposed so far in the literature for small molecules and polymers (Box-2) It had been suggested over twenty years ago that a descriptor should obey all of the following properties to be useful, namely it should: a) describe local shape b) be invariant to coordinate transformations and c) enable determination of shape complementarity between 2 or more compounds 31. One group 32 added an additional requirement that the shape descriptor must provide a means of positioning the shape that it describes in a few canonical orientations. These shape descriptors can be used together with a distance metric such as the Tanimoto coefficient and/or quantitative structure activity relationship (QSAR) and quantitative structure property relationship (QSPR) algorithms (Table 2).
Similar to these descriptor notations is another collection of shape representations called the “shape catalog”. For a set of active compounds, a database or catalog of fingerprints can be created by considering all possible shapes that arise from the conformations of these compounds 33. The main advantage of this catalog, apart from its application as a repository of molecular shapes, is that it can be customized by the user for the desired level of similarity cutoff between the compounds 34, 35.
A minimal description of molecular shape may also be rendered as a pharmacophore or pharmacophore fingerprints 24, 25, whereby the angle and distance between key molecular features imparts some degree of information on size and shape required for favorable molecular interactions. Another approach that can incorporate shape details (of both ligand and indirectly the protein binding site) is pseudoreceptor modelling, which represents a protein binding site using one or more molecules or conformations, and has been recently discussed in detail 36. Pseudoreceptor models can be used for searching databases for molecules with complementarity to the pseudoreceptor model.
Applications of shape descriptors to molecular pharmacology
Shape descriptors have been applied to many pharmacologically relevant problems as briefly summarized below. Shape descriptors are highly useful for classification purposes 37 and can be modified to act as weights to scoring functions 9. Further, shape descriptors have been also shown to be applicable to clustering molecules into groups that share similar overall shapes and hence to find analogs of lead compounds in a drug discovery application.
Shape similarity searches can be performed at two levels of complexity namely: global shape analysis and local shape analysis 38. If a complete structure of one molecule is matched with another, it is called a global shape analysis method. Examples of algorithms that use global shape analysis are the distance geometry method 39 and the molecular shape analysis (MSA) method that uses steric and van der Waals volumes as a shape descriptor 40. If a sub-structure of a complete molecule matches with another molecule then such an analysis is called local shape analysis. Many algorithms have incorporated the local shape analysis to identify compounds that not only share a part of their core structure with the query molecule but also other scaffolds, that may have shape similarity and not necessarily structural similarity 38. Be it the local or global similarity, the issue narrows down to finding “neighborhood” molecules using many clues such as a receptor structure 41 or QSAR based classification results 42. 3D similarity or classification studies can be performed using the following global alignment and alignment free methods.
Alignment-based methods
One approach to classifying compounds based on shape is to overlay them (alignment-based) using a set of guide points. An example is the shape-based and ligand centric approach called rapid overlay of chemical structures (ROCS, Openeye Scientific Software, Sante Fe) which performs overlays of conformers of a molecule of interest 37, 43-47. The overlays are performed quickly as the molecules are described as atom-centered Gaussian functions and conformers are compared using the Tanimoto coefficient. Adding chemical feature information is also possible and has been found useful for improving virtual screening results. ROCS has generally been found to perform as well as docking and other virtual screening methods (or better in the case of most targets) 37, 38, 40 although with some exceptions 46. ROCS has also been used to discriminate between cruzain and cathepsin L inhibitors, as well as replicating the X-ray conformation of a known cruzain inhibitor 43.
Alignment-free methods
Quantum chemical derived molecular descriptors are normally considered computationally costly; however, fragment-based approaches including transferable atom equivalent (TAE) descriptors surmount this limitation 48, 49 and are alignment free. The TAE descriptors have been used with machine learning methods to model 26 absorption, distribution, metabolism, excretion and toxicity related datasets demonstrating good internal model validation statistics in most cases 50. These descriptors have also been used to describe ligands and binding sites of proteins in the Protein Database 51. Using a k nearest neighbor (kNN) pattern recognition approach and variable selection, the active site structure could identify its complementary ligand after screening 1% of the database in over 90% of cases when a representative family protein was present in the training set. The alignment free TAE descriptors can also be used to generate property-encoded surface translation (PEST) descriptors using a ray tracing approach reflected on the inside of the electron density isosurface. This provides 2D histograms of distances versus surface property, and each bin of the histogram becomes a molecular descriptor. These descriptors have been used with an olfactory database to train a genetic algorithm to distinguish between musk and non-musk compounds52. A very similar approach to PEST is the alignment free Shape Signatures method which results in compact histograms that encode for molecular shape, shape and polarity or other properties to produce signatures53. This method has been used for database similarity searching in a series of drug discovery applications 54. The approach can also be used to build enriched or biased databases of small molecules by customizing the screening database to a particular drug target such as G-protein coupled receptors 49. For every molecule, the heights of the corresponding normalized molecular shape, polarity and shape signature bins comprise sets of molecular descriptors which have been used for machine learning models of the 5-HT2B receptor, the human ether-a-go-go potassium ion channel 7, and blood-brain barrier penetration 8. These models were also evaluated against and in combination with additional descriptors in the MOE suite of software (Chemical Computing Group, Montreal, Canada). It was found that Shape Signature descriptors describing molecular shape and charge slightly outperformed shape descriptors alone. These descriptors have been used recently to build classification models for PXR , as 9 well as a hybrid docking and molecular descriptor-based approach, coupling the GoldScore with other shape-based scoring functions.
Bioisosterism
The concept of bioisosterism (i.e. molecules with similar shapes may share similar biological activities) has been applied to drug discovery 55, 56. In a study identifying angiotensin-II analogs, four query molecules were used to search for bioisosteres in a database of ~1000 compounds. Based on the search results, 425 compounds were synthesized and tested for angiotensin II inhibition. Of these 425 compounds, only 63 compounds that were identified by shape similarity search as being most similar to any of the four query structures were found to be active 57. A good correlation between the shape scores and the inhibitory activity was found among all 425 compounds. However, algorithms that implement this concept have often been criticized for using the shape descriptor derived from a single representative conformer to search databases of large numbers of compounds. To overcome this limitation, many groups have now resorted to using at least 3-10 unique conformers and also to perform enrichment studies on the virtual libraries of compounds as an initial filter. Although successful stories like the one above validate bioisosterism, this hypothesis does not necessarily hold true with other types of descriptor or even some targets 58, 59.
These studies and others (Table 2) using different computational methods illustrate the broad applicability of shape-based descriptors which are of value to the pharmaceutical and environmental sciences fields. Shape can adequately describe the ligand-protein interaction indirectly and be used for screening databases to obtain good enrichments with active compounds. Most of these algorithms also have the advantage of enabling fast comparisons 6 and are broadly applicable.
Concluding remarks
Shape is a fundamentally important molecular feature important for describing ligands interacting with receptors, ion channels, enzymes and transporters and an array of other proteins and complex biological processes. The shape of the protein or a (pseudo)receptor binding site can also be used to find molecules with complementarity or by using the crystal structure conformation of the ligands in the PDB as a shape-based search query. This might point to potential off-targets or alternate targets that may represent repurposing opportunities for known drugs. Shape-based approaches have many potential areas for development in the future as applied to in silico pharmacology 60 (Box 3).
Box 3. Future uses of molecular shape.
The following represent some potential future uses of molecular shape:
Masking molecular shape - developing prodrugs that have improved physicochemical processes such as improved absorption or BBB permeability, or cloaking small molecules in nucleic acids and proteins 71.
Mimicking molecular shape - using computational methods to find new bioisosteres 55, 56.
Molecular phylogeny - while protein binding sites tend to have geomorphic or other shape descriptions 72, small molecules could be described as rods, cyclic, cylindrical, spherical, curved, T-shaped, Y-shaped, crosses, asymmetrical etc, which can be used for simple classifications.
Molecular shape chimeras - molecules could be developed that can change the shape of the protein by inducing order to disorder changes such as that seen when rifampicin binds to human PXR 73, for example.
Molecular shape confidentiality - Molecular shape descriptors could be used to share molecules between groups while the encryption key is confidential, building on the approaches used with structural fragments 74 and similarity based approaches 75.
Development of faster methods - required for mining shapes of multiple molecules simultaneously and potential creation of multiconformer shape databases consisting of tens to hundreds of millions of compounds 6, 76, 77. To date the databases searched have been much smaller and there has been little discussion of time required to create them or limitations of these algorithms 6. Many of the studies currently benchmark against ROCS in terms of speed of molecular shape comparisons 6. This may also require the creation of freely available standardized test databases for future shape-based evaluations.
Shape fragment based discovery - the above computational descriptors of shape and corresponding algorithms could be used to develop methods for shape based drug design analogous to fragment based drug discovery 78. This may also enable analysis of marketed drugs based on molecular shapes rather than as fragments 79 (e.g. answer the question of whether some molecular shapes more drug-like)
In the same way that an architect like Frank Lloyd Wright had an excellent appreciation of shape, thus it appears that his concept of form and function is generally applicable to shape and its role in molecular pharmacology.
Acknowledgments
SE kindly acknowledges Penelope Ekins for lending her Tupperware toy and Accelrys for providing Discovery Studio. MDK is supported by K08-GM074238 from the National Institutes of Health . We gratefully acknowledge our collaborators from many of the studies described.
Glossary
- CoMFA
Comparative molecular field analysis 42 is a widely used 3D-QSAR method that is alignment dependent. This method is useful for suggesting steric and electrostatic interactions between molecule and protein but is not used widely for virtual screening
- Decision tree
is a predictive data mining method with a tree-like structure consisting of leaves which represent classifications and branches representing molecular features or descriptors that lead to the classifications 13
- Docking Methods
such as DOCK 26, GOLD 27, GLIDE 12 and AutoDock 28, soft docking method 29 and PUZZLE 30 etc are generally computational methods to place a (small) molecule into a binding site (normally a protein) in a manner such that optimal molecule-protein interactions result
- k nearest neighbor
A method for classifying molecular bioactivity / properties based on similarity of molecular descriptors for a test molecule to those of the most similar molecules (nearest neighbors) in a training set
- MDL public keys
Molecular Design Limited (MDL now Symyx) created a key based fingerprint which uses a pre-defined set of definitions (relating to structural features). A fingerprint is created based on pattern matching of the structure to this set of 166 keys
- Molecular descriptors
are the end result of an experimental or mathematical procedure transforming chemical information into a useful number 61
- Pharmacophore
the essential molecular features that contribute to bioactivity
- QSAR and QSPR
Quantitative structure activity relationship and quantitative structure property relationship are algorithmic methods to relate molecular descriptors with bioactivity (e.g. Ki values) or biological / physicochemical property (e.g logP) data, respectively. These methods will provide a quantitative prediction once descriptors for a new molecule are input
- ROCS
rapid overlay of chemical structures, enables search for similarity of molecular 3D shape. A recent study has suggested this method is slower than the Ultrafast shape recognition method 6
- Sammon and Kohonen Mapping
are dimensionality reduction techniques frequently used with cheminformatics data. Sammon non-linear maps are unbiased in reproducing the topology and structure of the data space. Kohonen maps are a type of neural networks known as self-organizing networks consisting of artificial neurons connected by a distance dependent function. These neurons self-organize until their pairwise neighborhoods represent the correct topology of the original data set 13
- Scaffold hopping
use computational methods to find novel compounds with similar bioactivity or physicochemical property to a known reference ligand but with different structural chemotype/s
- Shape SignaturesTAE, PEST descriptors
all use a ray-tracing algorithm to explore the volume enclosed by the surface of a molecule, using the output to construct compact histograms 48, 49, 53
- Tanimoto coefficient
a measure of molecular similarity frequently used for comparing molecular fingerprint descriptors 62
References
- 1.Mezey PG. Shape in Chemistry. VCH Publishers Inc; 1993. [Google Scholar]
- 2.Hili R, Yudin AK. Making carbon-nitrogen bonds in biological and chemical synthesis. Nat Chem Biol. 2006;2:284–287. doi: 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- 3.Lingenfelder M, et al. Tracking the chiral recognition of adsorbed dipeptides at the single-molecule level. Angew Chem Int Ed Engl. 2007;46:4492–4495. doi: 10.1002/anie.200700194. [DOI] [PubMed] [Google Scholar]
- 4.Ando T, et al. Bacterial ribonuclease P reaction is affected by substrate shape and magnesium ion concentration. Nucleic Acids Res Suppl. 2003:293–294. doi: 10.1093/nass/3.1.293. [DOI] [PubMed] [Google Scholar]
- 5.Atkins PW. Molecules. Scientific American Library; 1987. [Google Scholar]
- 6.Ballester PJ, Richards WG. Ultrafast shape recognition to search compound databases for similar molecular shapes. J Comput Chem. 2007;28:1711–1723. doi: 10.1002/jcc.20681. [DOI] [PubMed] [Google Scholar]
- 7.Chekmarev DS, et al. Shape signatures: new descriptors for predicting cardiotoxicity in silico. Chem Res Toxicol. 2008;21:1304–1314. doi: 10.1021/tx800063r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kortagere S, et al. New Predictive Models for Blood-Brain Barrier Permeability of Drug-like Molecules. Pharm Res. 2008;25:1836–1845. doi: 10.1007/s11095-008-9584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kortagere S, et al. Hybrid scoring and shape based classification approaches for human pregnane X receptor. submitted. 2008 doi: 10.1007/s11095-008-9809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Gennaro P, et al. Specificity of substrate recognition by Pseudomonas fluorescens N3 dioxygenase. The role of the oxidation potential and molecular geometry. J Biol Chem. 1997;272:30254–30260. doi: 10.1074/jbc.272.48.30254. [DOI] [PubMed] [Google Scholar]
- 11.Ekins S, et al. Three-Dimensional Quantitative Structure-Activity Relationship Analysis of Human CYP51 Inhibitors. Drug Metab Dispos. 2007;35:493–500. doi: 10.1124/dmd.106.013888. [DOI] [PubMed] [Google Scholar]
- 12.Neugebauer A, et al. Prediction of protein-protein interaction inhibitors by chemoinformatics and machine learning methods. J Med Chem. 2007;50:4665–4668. doi: 10.1021/jm070533j. [DOI] [PubMed] [Google Scholar]
- 13.Ekins S, et al. Insights for human Ether-a-Go-Go-Related Gene Potassium Channel inhibition using recursive partitioning, Kohonen and Sammon mapping Techniques. J Med Chem. 2006;49:5059–5071. doi: 10.1021/jm060076r. [DOI] [PubMed] [Google Scholar]
- 14.Krasowski MD, et al. Evolution of the pregnane X receptor: adaptation to cross-species differences in biliary bile salts. Mol Endocrinol. 2005;19:1720–1739. doi: 10.1210/me.2004-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reschly EJ, et al. Evolution of the bile salt nuclear receptor FXR in vertebrates. J Lipid Res. 2008;49:1577–1587. doi: 10.1194/jlr.M800138-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reschly EJ, et al. Ligand specificity and evolution of liver X receptors. J Steroid Biochem Mol Biol. 2008;110:83–94. doi: 10.1016/j.jsbmb.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Synold TW, et al. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Medicine. 2001;7:584–590. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 18.Ekins S, et al. Evolution of pharmacologic specificity in the Pregane X Receptor. BMC Evol Biol. 2008;8:103. doi: 10.1186/1471-2148-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kortagere S, et al. Certain 1,4-disubstituted aromatic piperidines and piperazines with extreme selectivity for the dopamine D4 receptor interact with a common receptor microdomain. Mol Pharmacol. 2004;66:1491–1499. doi: 10.1124/mol.104.001321. [DOI] [PubMed] [Google Scholar]
- 20.Kricka LJ. Principles of immunochemical techniques. In: Burtis CA, et al., editors. Tietz textbook of clinical chemistry and molecular diagnostics. 4. Elsevier: Saunders; 2006. [Google Scholar]
- 21.Arduini F, et al. Fast, sensitive and cost-effective detection of nerve agents in the gas phase using a portable instrument and an electrochemical biosensor. Anal Bioanal Chem. 2007;388:1049–1057. doi: 10.1007/s00216-007-1330-z. [DOI] [PubMed] [Google Scholar]
- 22.Larsen NA, et al. Crystal structure of a cocaine-binding antibody. J Mol Biol. 2001;311:9–15. doi: 10.1006/jmbi.2001.4839. [DOI] [PubMed] [Google Scholar]
- 23.Paula S, et al. Three-dimensional structure-activity relationship modeling of cocaine binding to two monoclonal antibodies by comparative molecular field analysis. J Mol Biol. 2003;325:515–530. doi: 10.1016/s0022-2836(02)01235-4. [DOI] [PubMed] [Google Scholar]
- 24.Langer T, Hoffman RD. Pharmacophores and pharmacophore searches. Wiley-VCH; 2006. [Google Scholar]
- 25.Guner OF, editor. Pharmacophore, perception, development, and use in drug design. University International Line; 2000. [Google Scholar]
- 26.Ewing TJ, et al. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 27.Jones G, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 28.Goodsell DS, Olson AJ. Automated docking of substrates to proteins by simulated annealing. Proteins. 1990;8:195–202. doi: 10.1002/prot.340080302. [DOI] [PubMed] [Google Scholar]
- 29.Jiang F, Kim SH. “Soft docking”: matching of molecular surface cubes. J Mol Biol. 1991;219:79–102. doi: 10.1016/0022-2836(91)90859-5. [DOI] [PubMed] [Google Scholar]
- 30.Helmer-Citterich M, Tramontano A. PUZZLE: a new method for automated protein docking based on surface shape complementarity. J Mol Biol. 1994;235:1021–1031. doi: 10.1006/jmbi.1994.1054. [DOI] [PubMed] [Google Scholar]
- 31.Connolly ML. Shape complementarity at the hemoglobin alpha 1 beta 1 subunit interface. Biopolymers. 1986;25:1229–1247. doi: 10.1002/bip.360250705. [DOI] [PubMed] [Google Scholar]
- 32.Goldman BB, Wipke WT. QSD quadratic shape descriptors. 2. Molecular docking using quadratic shape descriptors (QSDock) Proteins. 2000;38:79–94. [PubMed] [Google Scholar]
- 33.Lanctot JK, et al. Using ensembles to classify compounds for drug discovery. J Chem Inf Comput Sci. 2003;43:2163–2169. doi: 10.1021/ci034129e. [DOI] [PubMed] [Google Scholar]
- 34.Putta S, et al. A novel shape-feature based approach to virtual library screening. J Chem Inf Comput Sci. 2002;42:1230–1240. doi: 10.1021/ci0255026. [DOI] [PubMed] [Google Scholar]
- 35.Srinivasen J, et al. Evaluation of a novel shape-based computational filter for lead evolution: application to thrombin inhibitors. J Med Chem. 2002;45:2494–2500. doi: 10.1021/jm010494q. [DOI] [PubMed] [Google Scholar]
- 36.Tanrikulu Y, Schneider G. Pseudoreceptor models in drug design: bridging ligand- and receptor-based virtual screening. Nat Rev Drug Discov. 2008;7:667–677. doi: 10.1038/nrd2615. [DOI] [PubMed] [Google Scholar]
- 37.Ebalunode JO, et al. Novel approach to structure-based pharmacophore search using computational geometry and shape matching techniques. J Chem Inf Model. 2008;48:889–901. doi: 10.1021/ci700368p. [DOI] [PubMed] [Google Scholar]
- 38.Robinson DD, et al. Partial molecular alignment via local structure analysis. J Chem Inf Comput Sci. 2000;40:503–512. doi: 10.1021/ci990272p. [DOI] [PubMed] [Google Scholar]
- 39.Crippen GM. Distance geometry approach to rationalizing binding data. J Med Chem. 1979;22:988–997. doi: 10.1021/jm00194a020. [DOI] [PubMed] [Google Scholar]
- 40.Hopfinger AJ. A general QSAR for dihydrofolate reductase inhibition by 2,4-diaminotriazines based upon molecular shape analysis. Arch Biochem Biophys. 1981;206:153–163. doi: 10.1016/0003-9861(81)90076-x. [DOI] [PubMed] [Google Scholar]
- 41.Babine RE, Bender SL. Molecular Recognition of Proteinminus signLigand Complexes: Applications to Drug Design. Chem Rev. 1997;97:1359–1472. doi: 10.1021/cr960370z. [DOI] [PubMed] [Google Scholar]
- 42.Cramer RD, et al. Comparative Molecular Field Analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 43.Freitas RF, et al. 2D QSAR and similarity studies on cruzain inhibitors aimed at improving selectivity over cathepsin L. Bioorg Med Chem. 2008;16:838–853. doi: 10.1016/j.bmc.2007.10.048. [DOI] [PubMed] [Google Scholar]
- 44.Hawkins PC, et al. Comparison of shape-matching and docking as virtual screening tools. J Med Chem. 2007;50:74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 45.Kirchmair J, et al. Fast and efficient in silico 3D screening: toward maximum computational efficiency of pharmacophore-based and shape-based approaches. J Chem Inf Model. 2007;47:2182–2196. doi: 10.1021/ci700024q. [DOI] [PubMed] [Google Scholar]
- 46.McGaughey GB, et al. Comparison of topological, shape, and docking methods in virtual screening. J Chem Inf Model. 2007;47:1504–1519. doi: 10.1021/ci700052x. [DOI] [PubMed] [Google Scholar]
- 47.Moffat K, et al. A comparison of field-based similarity searching methods: CatShape, FBSS, and ROCS. J Chem Inf Model. 2008;48:719–729. doi: 10.1021/ci700130j. [DOI] [PubMed] [Google Scholar]
- 48.Breneman C, et al. New developments in PEST shape/property hybrid descriptors. J Comp-Aided Mol Des. 2003;17:231–240. doi: 10.1023/a:1025334310107. [DOI] [PubMed] [Google Scholar]
- 49.Whitehead C, et al. Transferable atom equivalent multicentered multipole expansion method. J Comput Chem. 2003;24:512–529. doi: 10.1002/jcc.10240. [DOI] [PubMed] [Google Scholar]
- 50.Ekins S, et al. Novel applications of Kernel-partial least squares to modeling a comprehensive array of properties for drug discovery. In: Ekins S, editor. Computational Toxicology: Risk assessment for pharmaceutical and environmental chemicals. Wiley-Interscience; 2007. pp. 403–432. [Google Scholar]
- 51.Oloff S, et al. Chemometric analysis of ligand receptor complementarity: identifying Complementary Ligands Based on Receptor Information (CoLiBRI) J Chem Inf Model. 2006;46:844–851. doi: 10.1021/ci050065r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lavine BK, et al. Electronic van der Waals surface property descriptors and genetic algorithms for developing structure-activity correlations in olfactory databases. J Chem Inf Comput Sci. 2003;43:1890–1905. doi: 10.1021/ci030016j. [DOI] [PubMed] [Google Scholar]
- 53.Zauhar RJ, et al. Shape signatures: a new approach to computer-aided ligand- and receptor-based drug design. J Med Chem. 2003;46:5674–5690. doi: 10.1021/jm030242k. [DOI] [PubMed] [Google Scholar]
- 54.Nagarajan K, et al. Enrichment of ligands for the serotonin receptor using the Shape Signatures approach. J Chem Inf Model. 2005;45:49–57. doi: 10.1021/ci049746x. [DOI] [PubMed] [Google Scholar]
- 55.Jennings A, Tennant M. Selection of molecules based on shape and electrostatic similarity: proof of concept of “electroforms”. J Chem Inf Model. 2007;47:1829–1838. doi: 10.1021/ci600549q. [DOI] [PubMed] [Google Scholar]
- 56.Olesen PH. The use of bioisosteric groups in lead optimization. Curr Opin Drug Discov Devel. 2001;4:471–478. [PubMed] [Google Scholar]
- 57.Cramer RD, et al. Prospective identification of biologically active structures by topomer shape similarity searching. J Med Chem. 1999;42:3919–3933. doi: 10.1021/jm990159q. [DOI] [PubMed] [Google Scholar]
- 58.Cramer RD, et al. Bioisosterism as a molecular diversity descriptor: steric fields of single “topomeric” conformers. J Med Chem. 1996;39:3060–3069. doi: 10.1021/jm960291f. [DOI] [PubMed] [Google Scholar]
- 59.Patterson DE, et al. Neighborhood behavior: a useful concept for validation of “molecular diversity” descriptors. J Med Chem. 1996;39:3049–3059. doi: 10.1021/jm960290n. [DOI] [PubMed] [Google Scholar]
- 60.Ekins S, et al. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol. 2007;152:9–20. doi: 10.1038/sj.bjp.0707305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Todeschini R, Consonni V. Handbook of molecular descriptors. Wiley-VCH; 2000. [Google Scholar]
- 62.Willett P. Similarity-based approaches to virtual screening. Biochem Soc Trans. 2003;31:603–606. doi: 10.1042/bst0310603. [DOI] [PubMed] [Google Scholar]
- 63.Lill MA. Multi-dimensional QSAR in drug discovery. Drug Discov Today. 2007;12:1013–1017. doi: 10.1016/j.drudis.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 64.Varrazzo D, et al. Three-dimensional computation of atom depth in complex molecular structures. Bioinformatics. 2005;21:2856–2860. doi: 10.1093/bioinformatics/bti444. [DOI] [PubMed] [Google Scholar]
- 65.Coleman RG, Sharp KA. Travel depth, a new shape descriptor for macromolecules: application to ligand binding. J Mol Biol. 2006;362:441–458. doi: 10.1016/j.jmb.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 66.Ciubotariu D, et al. Molecular van der Waals space and topological indices from the distance matrix. Molecules. 2004;9:1053–1078. doi: 10.3390/91201053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mak L, et al. An extension of spherical harmonics to region-based rotationally invariant descriptors for molecular shape description and comparison. J Mol Graph Model. 2008;26:1035–1045. doi: 10.1016/j.jmgm.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 68.Verli H, et al. Local intersection volume: a new 3D descriptor applied to develop a 3D-QSAR pharmacophore model for benzodiazepine receptor ligands. Eur J Med Chem. 2002;37:219–229. doi: 10.1016/s0223-5234(02)01334-x. [DOI] [PubMed] [Google Scholar]
- 69.Edvinsson T, et al. Path-space ratio as a molecular shape descriptor of polymer conformation. J Chem Inf Comput Sci. 2003;43:126–133. doi: 10.1021/ci020269x. [DOI] [PubMed] [Google Scholar]
- 70.Urbano-Cuadrado M, et al. New quantum mechanics-based three-dimensional molecular descriptors for use in QSSR approaches: application to asymmetric catalysis. J Chem Inf Model. 2007;47:2228–2234. doi: 10.1021/ci700181v. [DOI] [PubMed] [Google Scholar]
- 71.Hof F, Rebek J., Jr Molecules within molecules: recognition through self-assembly. Proc Natl Acad Sci U S A. 2002;99:4775–4777. doi: 10.1073/pnas.042663699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee M, et al. Shapes of antibody binding sites: qualitative and quantitative analyses based on a geomorphic classification scheme. J Org Chem. 2006;71:5082–5092. doi: 10.1021/jo052659z. [DOI] [PubMed] [Google Scholar]
- 73.Chrencik JE, et al. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol Endocrinol. 2005;19:1125–1134. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 74.Trepalin S, Osadchiy N. The centroidal algorithm in molecular similarity and diversity calculations on confidential datasets. J Comput Aided Mol Des. 2005;19:715–729. doi: 10.1007/s10822-005-9023-1. [DOI] [PubMed] [Google Scholar]
- 75.Kaiser D, et al. Similarity-based descriptors (SIBAR)--a tool for safe exchange of chemical information? J Comput Aided Mol Des. 2005;19:687–692. doi: 10.1007/s10822-005-9000-8. [DOI] [PubMed] [Google Scholar]
- 76.Fontaine F, et al. Fast 3D shape screening of large chemical databases through alignment-recycling. Chem Cent J. 2007;1:12. doi: 10.1186/1752-153X-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mansfield ML, et al. A new class of molecular shape descriptors. 1. Theory and properties. J Chem Inf Comput Sci. 2002;42:259–273. doi: 10.1021/ci000100o. [DOI] [PubMed] [Google Scholar]
- 78.Siegal G, et al. Integration of fragment screening and library design. Drug Discov Today. 2007;12:1032–1039. doi: 10.1016/j.drudis.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 79.Siegel MG, Vieth M. Drugs in other drugs: a new look at drugs as fragments. Drug Discov Today. 2007;12:71–79. doi: 10.1016/j.drudis.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 80.Wang CY, et al. Identification of previously unrecognized antiestrogenic chemicals using a novel virtual screening approach. Chem Res Toxicol. 2006;19:1595–1601. doi: 10.1021/tx060218k. [DOI] [PMC free article] [PubMed] [Google Scholar]