

Summary
Hyperactivation of Ras-ERK1/2 signaling is critical to the development of many human malignancies but little is known regarding the specific contribution of ERK1 or ERK2 to oncogenic processes. We demonstrate that ERK2 but not ERK1 signaling is necessary for Ras-induced epithelial-to-mesenchymal transformation (EMT). Further, ERK2 but not ERK1 overexpression is sufficient to induce EMT. Many ERK1/2 interacting proteins contain amino acid motifs, e.g. DEF or D-motifs, which regulate docking with ERK1/2. Remarkably, ERK2 signaling to DEF motif-containing targets is required to induce EMT and correlates with increased migration, invasion and survival. Importantly, the late response gene product Fra1 is necessary for Ras- and ERK2-induced EMT through up-regulation of ZEB1/2 proteins. Thus, an apparent critical role for ERK2 DEF motif signaling during tumorigenesis is the regulation of Fra1, and the subsequent induction of ZEB1/2, suggesting a potential therapeutic target for Ras-regulated tumorigenesis.
Introduction
The Ras-ERK1/2 (Extracellular Signal-Regulated Kinases 1/2)-MAP kinase pathway plays a critical role in numerous cellular processes, including proliferation, differentiation, survival and motility. These processes are regulated spatially and temporally, and by protein-protein interactions to generate specific cell behavior (Murphy and Blenis, 2006; Murphy et al., 2002). Deregulation of the Ras-ERK pathway has been implicated in greater than 30% of human cancers (Hoshino et al., 1999) and is typically manifested in gain-of-function mutations in components located upstream of ERK1/2, such as ErbB, Ras and Raf (Downward, 2003).
The small GTPase Ras is activated by multiple mechanisms and regulates the ERK, PI3-K, and Ral signaling pathways. The ERK-MAPK cascade is initiated when the Raf family of Ser/Thr kinases binds to Ras-GTP. Activated Raf then phosphorylates and activates MEK1/2 (MAPK/ERK-activating Kinases 1/2), which in turn phosphorylate and activate ERK1/2-MAP kinases. At the level of ERK1/2, this linear cascade splits into separate paths as a result of at least two distinct docking motifs. The DEF (Docking site for ERK, F-X-F) motif and D-domain (Docking domain) are found within ERK1/2 interactors and provide spatial localization information and direct signaling to specific substrates. ERK1/2 interactors containing only DEF motifs, D-domains or a combination of the two motifs have been described thus allowing for intricate regulation of downstream biological processes. For example, the DEF motif is necessary for the interaction of activated ERK with several IEG (immediate early gene) products (Murphy et al., 2004; Murphy et al., 2002). Recent studies using peptide library screening in vitro have shown that the optimal DEF motif sequence is F/W-X-F/Y/W with proline at the +4 position weakly favored (Sheridan et al., 2008). The D-domain motif is needed for activation of RSK (90 kDa ribosomal S6 kinase) and may also play a role in the inactivation of ERK by MKPs (MAPK phosphatases) (Roux and Blenis, 2004; Anjum and Blenis, 2008). ERK2 interacts with DEF- and D-motif substrates through two distinct molecular surfaces. The DBP (DEF motif Binding Pocket) on ERK2 was recently described by Ahn and coworkers (Lee et al., 2004) while the CD (Common Docking) domain, found within all MAPKs, regulates interaction with D-domain motifs (Tanoue et al., 2000).
The existence of two closely related ERK isoforms, ERK1 and ERK2, which demonstrate 83% amino acid identity and are co-expressed in a majority of tissues and cell lines examined, was described nearly 20 years ago (Boulton et al., 1991). It has been suggested that the isoforms possess overlapping, if not redundant, function (Cobb and Goldsmith, 2000). Indeed, ERK1 and ERK2 are generally co-activated in response to multiple stimuli, but at least two reports have described preferential activation of a single ERK isoform (Papkoff et al., 1994; Sarbassov et al., 1997). A few studies have proposed distinct roles for ERK1 or ERK2 in the regulation of differentiation, gene expression or proliferation under certain contexts (Li and Johnson, 2006; Vantaggiato et al., 2006). Recent evidence demonstrates that some regions of the mouse brain specifically express ERK1 or ERK2 mRNA suggesting distinct roles for the individual isoforms (Di Benedetto et al., 2007). Data from Erk1 or Erk2 knockout mice have also suggested that the isoforms may possess distinct biological roles. Whereas Erk1-/- mice are viable but demonstrate impaired thymocyte development (Pages et al., 1999), ablation of Erk2 results in embryonic lethality due to improper placental development (Hatano et al., 2003; Saba-El-Leil et al., 2003; Yao et al., 2003). Interestingly, when the placental defect was rescued by tetraploid-aggregation, Erk2-/- mice grew as well as wild-type littermates (Hatano et al., 2003) suggesting that expression of ERK1 could compensate for the absence of Erk2 with respect to some biological processes. More recent evidence from Lenormand, Pouysségur and colleagues further suggests that expression of ERK1 can compensate for the absence of Erk2 (Lefloch et al., 2008). Thus, although relatively few in number, these reports suggest that ERK1 and ERK2 may direct specific biological functions under certain contexts, but likely possess redundant functions with regards to most processes.
Recently we demonstrated that we could specifically impair ERK2 signaling to DEF motif or D-domain containing interactors via single point mutations in the ERK2-DBP or the ERK2-CD domain, respectively, without affecting overall kinase activity (Dimitri et al., 2005). We now utilize these docking deficient mutants to examine how ERK regulates EMT, EMT-associated phenotypes and cell survival in MCF-10A cells, a non-transformed human mammary epithelial cell line (Soule et al., 1990). We demonstrate that ectopic expression of ERK2 results in a dramatic change in MCF-10A cell morphology, reduces E-cadherin expression and increases mesenchymal markers similar to cells expressing oncogenic RasV12, which is known to induce epithelial-to-mesenchymal transformation (EMT) in several cell types (Thiery, 2003). Surprisingly, cells expressing exogenous ERK1 resemble control cells in morphology and EMT markers. In addition, specific knockdown of endogenous ERK2 levels inhibits RasV12-induced EMT whereas knockdown of ERK1 expression has no effect. Importantly, these results suggest that ERK2 specifically regulates EMT in MCF-10A cells. The use of partially defective ERK2 signaling mutants (Dimitri et al., 2005) revealed further that ERK2-DEF motif interactions are necessary for ERK2-induced EMT. Significantly, expression and activation of the late response gene Fra1 is ERK2-DEF motif-specific and is required for Ras- and ERK2-induced EMT. Finally, we show that ZEB1/2 expression, known to play an important role in EMT, is a critical downstream effector of the Ras-ERK2-Fra1 pathway.
Results
ERK2 specifically regulates EMT in MCF-10A cells
We infected MCF-10A cells (Soule et al., 1990) with retrovirus encoding wild-type T7-tagged-ERK1, HA-tagged-ERK2 or RasG12V. Following infection, HA-ERK2 and RasV12 over-expressing cells demonstrated an elongated fibroblast-like morphology with decreased cell-cell contacts; conversely, control cells appeared cuboidal and cobblestone-like (Fig. 1A). Interestingly, cells stably expressing similar to higher levels of T7-ERK1 (Fig. S1A) resembled control cells (Fig. 1A), suggesting that ERK2 specifically regulated the morphological alterations observed. The distinct morphology evident in the ERK2 and RasV12 overexpressing cells was reminiscent of cells having undergone EMT.
Figure 1.
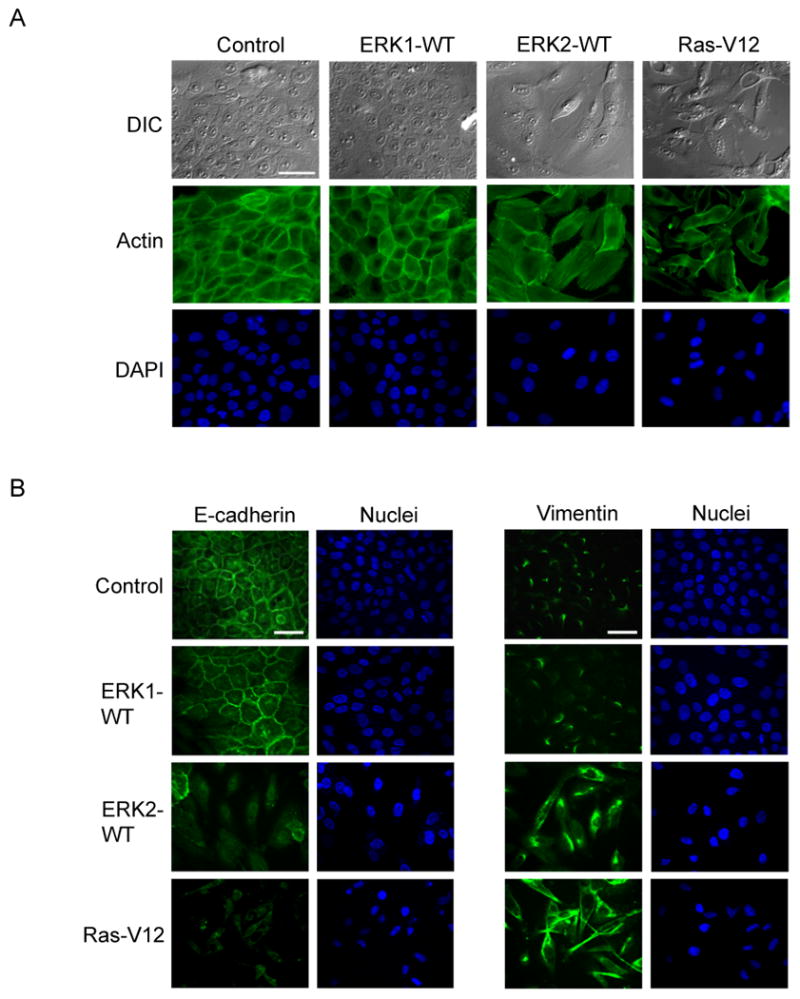
ERK2 specifically regulates EMT in MCF-10A cells
A) Cells were infected with retrovirus encoding for T7-ERK1, HA-ERK2 or RasV12 alleles. Actin was stained as described in materials and methods. Bar scale, 50 μm.
B) Stable cells were fixed and processed for immunofluorescence staining. Bar scale, 50 μm.
To investigate this possibility, we examined the expression of epithelial and mesenchymal markers associated with EMT (Kang and Massague, 2004). Cells overexpressing ERK2 or RasV12 displayed reduced expression of E-cadherin whereas ERK1 overexpressing cells resembled control cells (Fig. 1B, Fig. S1A). Importantly, we also observed increased expression of the mesenchymal marker vimentin in ERK2 and RasV12 overexpressing cells but not in ERK1 expressing cells (Fig. 1B, Fig. S1A). ERK2 and Ras also increased levels of N-cadherin and fibronectin, additional mesenchymal markers, whereas ERK1 did not (Fig S1A). These data suggest that overexpression of ERK2, like RasV12, induces morphological and cell biological alterations in MCF-10A cells that are consistent with EMT. We also asked whether ERK2 induces EMT in other epithelial cells. Expression of ERK2, but not ERK1, was sufficient to induce an EMT phenotype in normal murine mammary gland (NMuMG) and ureteric bud (UB) cells (Fig. S1B, S1C). ERK2 also induced morphological changes in mouse cortical tubule (MCT) epithelial cells although the phenotype was less dramatic (Fig. S1D). Some epithelial cell lines that were tested did not exhibit an EMT phenotype with Ras or ERK2. The reason for the cell type-specific responses to Ras and ERK2 is unclear.
As Ras can activate multiple signaling pathways, we sought to determine to what extent activation of the ERK-MAPK pathway was required for RasV12-mediated EMT using pharmacological and RNAi-based approaches. RasV12-infected cells maintained in the presence of U0126 demonstrated a similar morphology and levels of E-cadherin and vimentin to control cells, whereas treatment of cells with LY294002 demonstrated no effect (Fig. S2A, S2B). Thus, our data suggests, 1) that the ERK-MAPK and not the PI3K/mTOR pathway is necessary for RasV12 induced EMT, and 2) overexpression of ERK2 is sufficient to induce EMT.
To confirm that ERK2 specifically regulated EMT, we stably knocked down endogenous ERK1 or ERK2 expression using lentiviral shRNA constructs and then expressed RasV12. Consistent with our overexpression data, partial knockdown of endogenous ERK2 impaired the induction of EMT following RasV12 expression (Fig. 2, S2C). Knockdown of ERK1 had no apparent effect on RasV12-induced EMT (Fig. 2, S2C). Together, both the overexpression and knockdown studies demonstrate a distinct role for ERK2 in regulating the induction of EMT downstream of RasV12.
Figure 2.
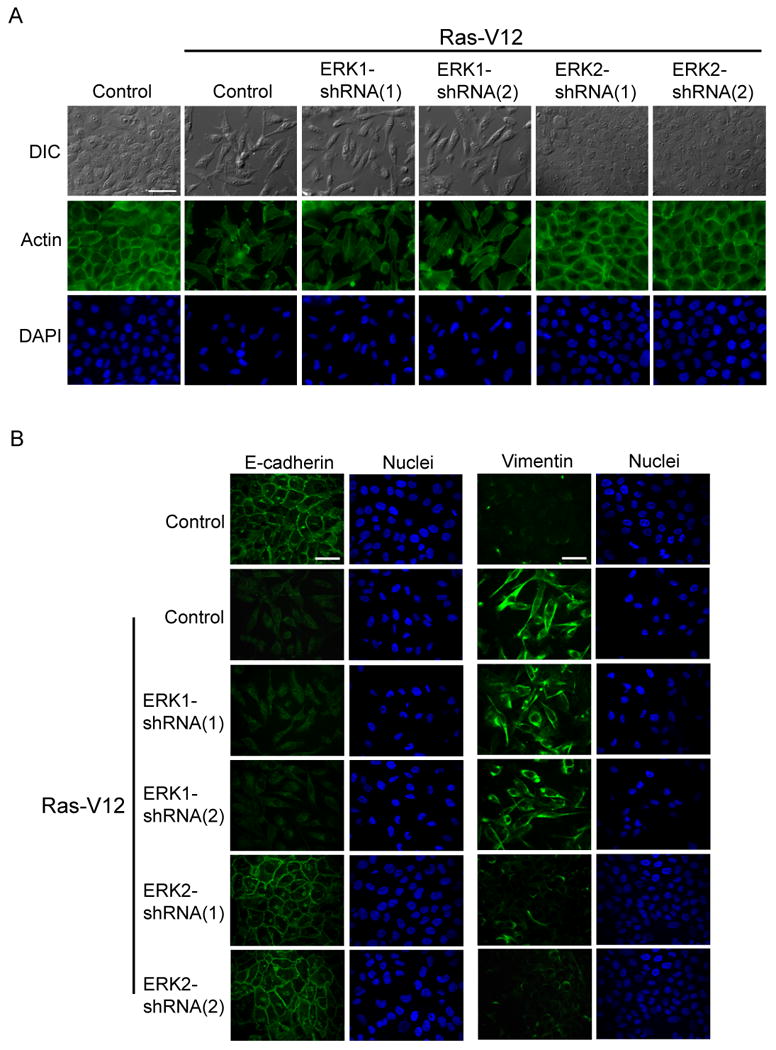
RasV12-induced EMT requires ERK2
A) MCF-10A cells were infected with ERK1 or ERK2 shRNA constructs. Cells were then infected with RasV12 and stained for actin cytoskeleton. Bar scale, 50 μm.
B) After generating cells as described in Fig. 2A, cells were fixed and processed for immunofluorescence staining. Bar scale, 50 μm.
Mutations in ERK2 docking motifs specifically impair ERK2 downstream signaling
Next, we asked if ERK2 signaling via either D-domain or DEF motif interactions was necessary for ERK2-induced EMT. We generated MCF-10A cells stably expressing ERK1 wild-type, ERK2 wild-type, or various ERK2 point mutant alleles and then assayed the ability of these cells to signal to ERK effectors. To examine ERK signaling via a D-domain interaction, we assessed phosphorylation of human RSK1 at Thr573, which is necessary for RSK activation and occurs in a D-domain dependent manner (Roux and Blenis, 2004; Anjum and Blenis, 2008). As shown previously (Dimitri et al., 2005), cells stably expressing the ERK2-WT allele or a DBP (DEF motif binding pocket) mutant ERK2 allele, ERK2-Y261A, demonstrated increased phosphorylation of Flag-HA-RSK1 at Thr573 (data not shown). Conversely, cells expressing a CD domain mutant allele, ERK2-D319N, were greatly impaired in RSK1 phosphorylation compared to ERK2-WT (data not shown).
We next examined the ability of ERK to signal to DEF motif containing substrates using a Gal4-Elk-1 luciferase reporter (Sugimoto et al., 1998; Dimitri et al., 2005). Consistent with the requirement of a functional DEF motif interaction for Elk-1 transactivation (Fantz et al., 2001), cells expressing the ERK2-Y261A mutant were impaired in their ability to induce Elk-1 transactivation compared to ERK2-WT (data not shown). Conversely, D-domain interactions were not found to be necessary for Elk-1 transactivation as expression of ERK2-D319N did not impair Elk-1 transactivation (data not shown). ERK2-D319N consistently induced Elk-1 transactivation more than wild-type, perhaps due to decreased cytoplasmic retention and/or decreased phosphatase inactivation as suggested previously (Tanoue et al., 2000). These results are consistent with previous biochemical studies in HeLa cells demonstrating that single point mutations can impair ERK2 signaling through one effector pathway without inhibiting overall kinase activity (Dimitri et al., 2005). Thus, these alleles provide valuable tools to assess whether biological processes regulated by ERK2 require DEF motif or D-domain signaling. Interestingly, cells overexpressing an ERK1-WT allele demonstrated similar levels of phosphorylation of Flag-HA-RSK at Thr573 and Elk-1 transactivation as ERK2-WT expressing cells (data not shown). Importantly, these results suggest that the differences in the induction of EMT observed between ERK1 and ERK2 are not due to signaling differences at the level of RSK1 or Elk-1.
ERK2-DEF motif signaling is necessary for EMT
Since ERK2 specifically regulates EMT (Fig. 1, 2, S1, S2), we asked if ERK2 utilized targets possessing one or both interaction motifs to modulate this process. Strikingly, cells stably expressing the CD domain mutant ERK2 allele, ERK2-D319N, displayed more pronounced morphological alterations and were no longer restrained by contact inhibition, similar to characteristics observed in RasV12 expressing cells (Fig. 3A). Similar results were also seen in MCT cells where ERK2-D319N expression induced a more pronounced EMT-like morphology than expression of ERK2-WT (Fig. S3A). Conversely, cells expressing the ERK2-DBP mutant, ERK2-Y261A, did not exhibit morphological changes in MCF-10A cells (Fig. 3A), MCT cells (Fig. S3A) or NMuMG cells (data not shown). ERK1 DBP and CD-domain mutants, ERK1-Y281A and –D339N respectively, were also generated and characterized. The ERK1 mutants demonstrated similar signaling defects as previously described for the corresponding ERK2 mutant alleles (data not shown). Interestingly, expression of either ERK1-WT or the ERK1 mutant alleles did not induce phenotypic changes resembling EMT in MCT cells and MCF-10A cells (Fig. S3A, S3B, respectively).
Figure 3.
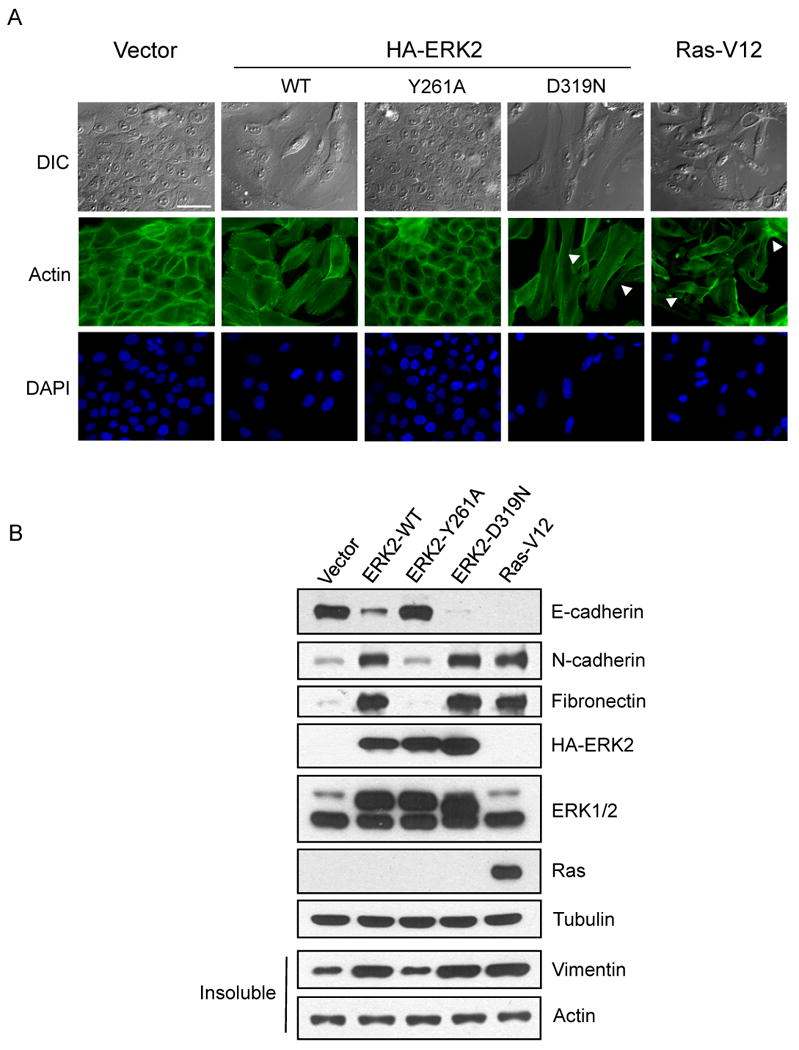
ERK2-DEF motif interactions are necessary for EMT
A) Stable MCF-10A cells were fixed and processed for actin cytoskeleton and nuclear staining. Loss of contact inhibition and cell-cell overlap can be seen in some cells (arrowheads). Bar scale, 50 μm.
B) Stable cells were lysed and immunoblot analysis of epithelial and mesenchymal makers was performed.
To further investigate the link between ERK2-DEF motif interactions and EMT, we examined expression of the epithelial marker, E-cadherin, and mesenchymal markers, N-cadherin, fibronectin, and vimentin. Following stable expression of the specified ERK2 alleles, cells were either examined by immunoblotting (Fig. 3B) or immunofluorescence staining (Fig. S3C, S3D). Expression of ERK2-WT, ERK2-D319N or RasV12 in MCF-10A cells demonstrated decreased E-cadherin protein levels (Fig. 3B, S3C) and increased expression of vimentin, N-cadherin and fibronectin (Fig. 3B, S3D). We also found that ERK2-D319N and RasV12 increased FSP1 expression, a mesenchymal marker expressed during tubular EMT (Pozzi et al., 2006; Okada et al., 1997) in MCT cells (Fig. S3A). Cells expressing ERK2-Y261A, ERK1-WT or the ERK1 mutants resembled vector control cells with regards to expression of EMT markers (Fig. 3B, S3C, S3D). Thus, ERK2 induces EMT and ERK2-DEF motif signaling is necessary for this process as shown by both morphological and cell biological changes.
Phenotypes associated with EMT or metastasis correlate with increased ERK2-DEF motif signaling
In addition to morphological and cell biological changes, induction of EMT in vitro has been correlated with increased migration and invasion. Indeed, ERK2-WT expressing cells demonstrated increased migration in response to EGF compared to control cells (Fig. 4A). Cells expressing ERK2-D319N also displayed increased migration in response to EGF, similar to RasV12 expressing cells (Fig. 4A). Importantly, migration of ERK2-Y261A expressing cells was similar to vector control and ERK1 expressing cells (Fig. 4A). These results were consistent with knockdown data demonstrating that ERK2 expression was necessary for full migration of RasV12 stable cells, whereas ERK1 expression was dispensable (Fig. 4B). Building upon a previous report suggesting a role for ERK1/2 in MCF-10A cell migration (Irie et al., 2005), we demonstrate that ERK2 specifically regulates EGF-induced migration, and importantly, that ERK2-DEF motif signaling plays a significant role in this cellular process.
Figure 4.
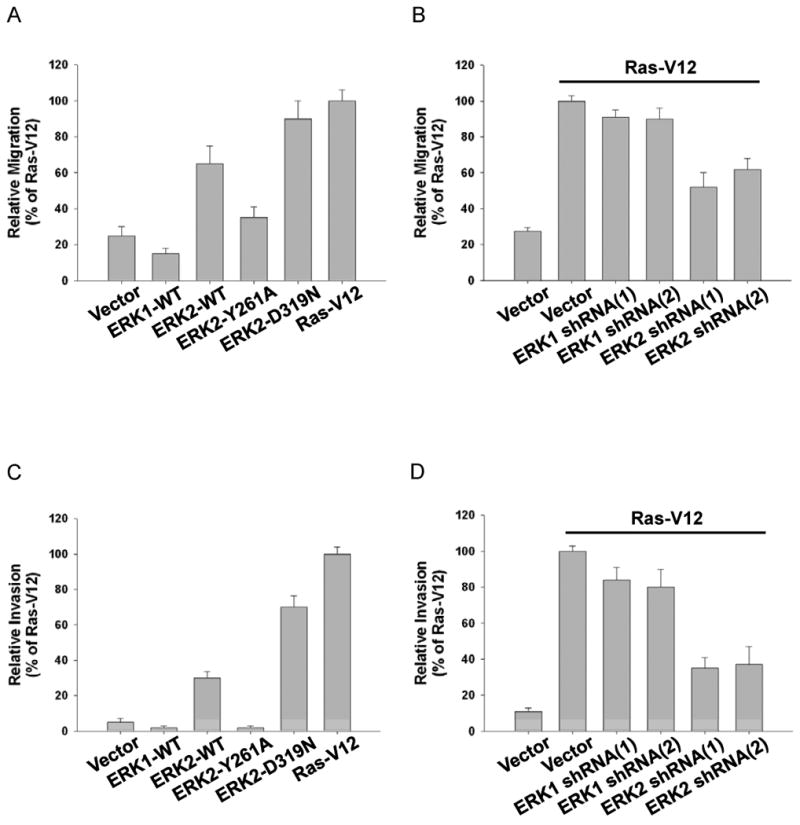
Increased migration and invasion correlate with ERK2-DEF motif signaling and an EMT phenotype
Migration (A, B) and invasion (C, D) assays were performed as described in the materials and methods. Cells were assessed for migratory and invasive potential after generation of stable overexpression cell lines (A, C) or knockdown of endogenous ERK1 or ERK2 and expression of RasV12 (B, D). Data are the means+/- SEM of four separate experiments.
We also examined the invasive properties of MCF-10A cells. Expression of ERK2-WT or ERK2-D319N increased the number of invasive cells compared to control cells (Fig. 4C). RasV12 expressing cells were even more invasive suggesting that other Ras effector pathways also contribute to invasion (Fig. 4C). Conversely, cells expressing ERK2-Y261A or ERK1 failed to invade effectively and resembled control cells (Fig. 4C). These results were consistent with ERK1 and ERK2 knockdown experiments demonstrating that specifically ERK2 was necessary for full invasion of RasV12 stable cells (Fig. 4D).
Resistance to cell death is essential for migratory cells to survive in sub-optimal environments during development and metastasis. We examined the ability of the stable cells to survive in suspension via an anoikis assay. ERK2-WT or ERK2-D319N expressing cells demonstrated increased survival in suspension compared to control cells and similar levels of survival compared to RasV12 cells (Fig. S4A). Conversely, expression of ERK2-Y261A provided a weak survival signal compared to ERK2-WT and ERK2-D319N cells (Fig. S4A). ERK1 partially enhanced survival under these conditions, suggesting that ERK1 and ERK2 may activate unique survival signals and/or have different activities for one or more shared substrates. These results indicate ERK2-DEF motif signaling was necessary for the increased survival of ERK2-WT cells in an anoikis assay and that ERK2-D-domain signaling contributes but was not sufficient to fully prevent anoikis.
It has been reported that ERK1/2 may regulate anoikis through inhibition of the proapoptotic protein BimEL (Ley et al., 2005; Reginato et al., 2003). Consistent with the anoikis data, elevated levels of BimEL were detected in vector control cells and ERK2-Y261A stable cells when cultured in suspension (Fig. S4B). However, BimEL expression levels were reduced in cells expressing ERK2-WT, ERK2-D319N or RasV12 (Fig. S4B). Whereas ERK1 overexpression weakly enhanced cell survival, BimEL levels were similar to control cells suggesting that ERK1 may provide Bim-independent survival signals (Fig. S4A, S4B). Thus, ERK2-DEF motif signaling is necessary for the degradation of the pro-apoptotic protein BimEL, thereby promoting cell survival under conditions where cells are detached from a substratum. We next examined cellular anoikis in ERK1 and ERK2 knockdown cells expressing RasV12. Interestingly, RasV12 expression in cells with partial knockdown of ERK2 resulted in a slight increase in anoikis compared to RasV12 expression alone (Fig. S4C). Accordingly, BimEL expression was not detectable in RasV12 cells whereas BimEL was detected upon ERK2 knockdown (Fig. S4D). Although more complete knockdown of ERK1 was achieved, ERK1 knockdown did not significantly alter the anti-anoikis effect of RasV12 expression. These results suggest that ERK2 contributes to the anti-anoikis effect of Ras, but other factors may be required for Ras to exert its full anti-anoikis function.
Growth in 3D culture can also be used to monitor cell survival and anchorage-independent growth of MCF-10A cells. In Matrigel™, MCF-10A cells undergo a morphogenetic program leading to the development of spherical acini-like structures whereas cells undergoing the process of cellular transformation can form more complex foci-like structures (Debnath and Brugge, 2005). Using this system, we examined the effect of ERK1 and ERK2 expression in MCF-10A cells. Cells expressing ERK2-WT and ERK2-D319N formed complex structures that were considerably larger than control acini (Fig. S4E). ERK1-WT overexpression resulted in small foci intermediate in size between ERK2-WT expressing cells and vector control cells. These results suggest that ERK2 is an important contributor to cell survival and growth in MCF-10A cells grown in 3D. Unlike ERK2-WT expressing cells, RasV12 cells formed large colonies exhibiting widespread protrusive extensions and disruption of acinar morphogenesis (Fig. S4E, S4F) that correlates with enhanced cell proliferation and invasion through matrigel (Debnath and Brugge, 2005). This result is consistent with our observation that RasV12 expressing cells are more invasive than ERK2 expressing cells in the invasion assays (Fig. 4C). Importantly, down-regulation of ERK2, but not ERK1, dramatically inhibited this invasive RasV12 phenotype (Fig. S4F). However, knockdown of ERK2 only partially inhibited RasV12-mediated survival and proliferation, in comparison to vector control cells, as larger non-invasive foci were still observed (Fig. S4F). These results indicate that knockdown of ERK2 prevents Ras-induced invasion and partially inhibits cell survival/proliferation in the 3D culture system.
Recently, it was reported that EMT leads to the generation of breast cancer cells with stem cell-like characteristics including the ability to self-renew and initiate tumors (Mani et al., 2008). Cultured normal human mammary epithelial cells contained a population of CD44high/CD24low cells that displayed a mesenchymal morphology and expressed characteristic mesenchymal markers. Consistent with this, Shipitsin et al showed that CD44+ cells expressing known stem cell markers are predicted to be more invasive, angiogenic, and higher risk of distant metastasis (Shipitsin et al., 2007). Therefore, we examined the consequences of ERK and Ras signaling on the expression of cell surface CD44 and CD24. We found that ERK2-WT, ERK2-D319N, and RasV12 expression increased cell surface CD44 (Fig. S4G); ERK1 and ERK2-Y261A did not increase CD44 cell surface levels. We also found that knockdown of ERK2, but not ERK1, inhibited increased CD44 expression by RasV12 (Fig. S4H). Examination of CD24 revealed that RasV12 expression significantly reduced CD24 cell surface levels; ERK2-D319N expression also demonstrated a decrease in detected CD24 (Fig. S4I). Interestingly, whereas expression of ERK2-WT did not greatly alter cell surface levels of CD24, knockdown of ERK2 in RasV12 expressing cells inhibited the RasV12-mediated decrease in CD24 levels (Fig. S4J). Taken together, the knockdown data indicate that the ERK2 is required for full acquisition of stem cell-like characteristics, with ERK2-DEF signaling differentially contributing to CD44 and CD24 expression (Fig. S4G-S4J).
Previous reports have demonstrated that concurrent activation of the Ras and TGF-β pathways cooperate to induce EMT in several cell types (Janda et al., 2002). Raf activation and an inducible FosER oncoprotein have been shown to induce TGF-β autocrine production (Eger et al., 2004), suggesting that ERK activation and downstream signaling may play a role in the regulation of TGF-β1. We therefore examined whether ERK overexpression resulted in increased TGF-β1 production. While TGF-β1 levels were not appreciably different immediately after retroviral infection (1-3 days), ERK2-D319N expression resulted in a dramatic increase in TGF-β1 levels 8-10 days post-infection; expression of ERK2-WT or ERK1-WT modestly increased TGF-β1 levels at the same time point (Fig. S4K). Interestingly, knockdown of TGF-β1 only mildly impaired ERK2-induced morphology and EMT marker changes (data not shown). Thus, although ERK2-DEF motif signaling plays a role in the regulation of TGF-β1 autocrine production, additional mechanisms must exist that explain why specifically ERK2 induces EMT in MCF-10A cells.
Fra1 is necessary for ERK2-induced EMT and is differentially regulated by ERK1 and ERK2
In addition to TGF-β1, we attempted to knock down the expression of Snail, Egr-1, c-Fos, and Fra1 by shRNA lentiviral expression to determine if these gene products, which have been implicated in regulating EMT or related oncogenic processes, might contribute to the induction of EMT downstream of ERK2. Knockdown of Snail or c-Fos did not prevent the reduction in E-cadherin levels following ERK2-D319N expression whereas knockdown of Egr-1, like TGF-β, mildly impaired the loss in E-cadherin expression (data not shown). This suggests that these gene products are not major contributors, that knockdown was not sufficient, and/or that there are redundant or overlapping signaling mechanisms contributing to ERK2-induced EMT in MCF-10A cells. Fra1, however, was clearly required for ERK2-induced EMT. Although we only achieved partial knockdown of Fra1 at the protein level, this was sufficient to impair ERK2-D319N-induced and Ras-induced EMT (Fig. 5A, 5B, and data not shown). In addition to changes in EMT markers and morphology, we asked if Fra1 expression was necessary for ERK2-D319N- or RasV12-mediated cell migration, invasion and survival in an anoikis assay. Fra1 knockdown suppressed the effect of ERK2-D319N and RasV12 on these biological processes (Fig. S5A-S5F). Similar knockdown efficiency of Fra2 did not prevent EMT (data not shown). These results suggest that Fra1 is a critical and necessary downstream target of ERK2 for the induction of EMT.
Figure 5.
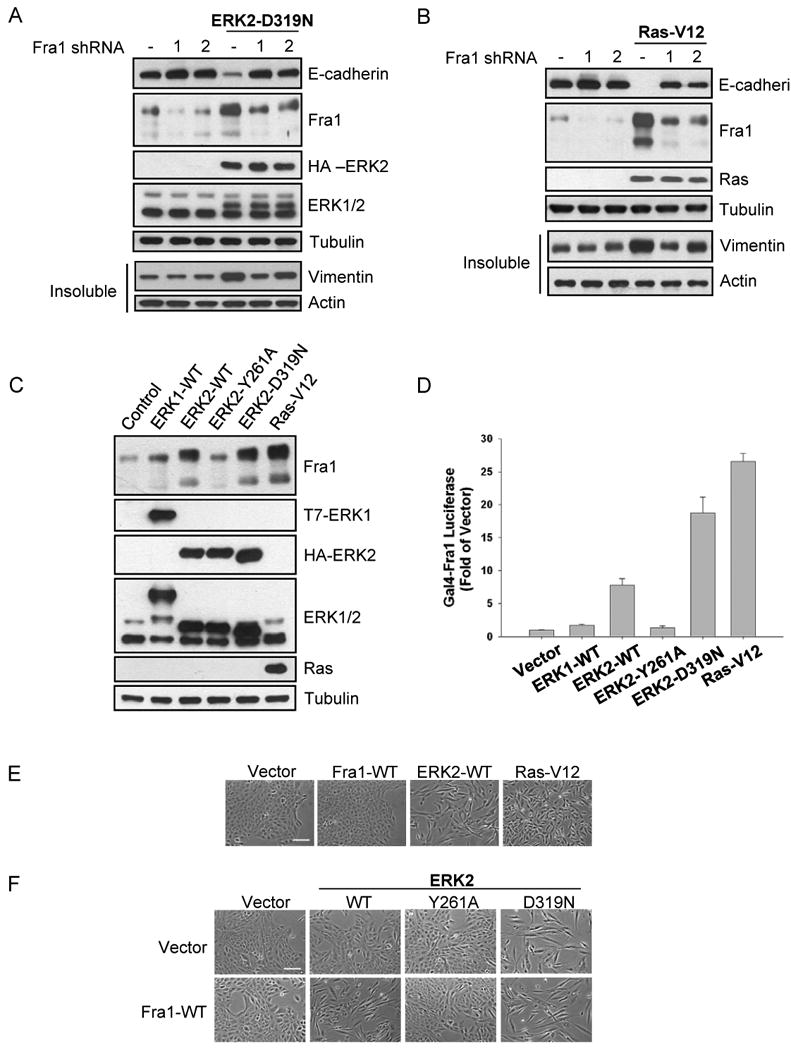
Fra1 expression is necessary for ERK2-induced EMT and is differentially regulated by ERK1 and ERK2
A-B) Following infection of MCF-10A cells with two separate lentiviral shRNA constructs, stable cells were infected with HA-ERK2-D319N (A) or RasV12 (B) encoding retrovirus. Stable cells were lysed for immunoblot analysis.
C) Immunoblot analysis was performed with cell lysates from stable ERK1, ERK2, or RasV12 overexpressing cells.
D) Fra1 activity was measured as described in the materials and methods. Data shown are the means +/- SEM of three separate assays preformed in duplicate.
E) Stable cells expressing Fra1, ERK2, or RasV12 for 7-10 days after selection were fixed for phase-contrast microscopy. Bar scale, 100 μm.
F) Fra1-WT expressing cells were infected with retrovirus encoding various HA-ERK2 alleles for 4 days and fixed for phase-contrast microscopy. Bar scale, 100 μm.
As a possible explanation for the disparity in the induction of EMT by ERK1 and ERK2, we asked whether ERK1 and ERK2 might differentially regulate Fra1. Cells stably expressing ERK2-WT or ERK2-D319N demonstrated increased levels of Fra1 compared to control cells, consistent with results observed in RasV12 expressing cells, which displayed an even greater increase in Fra1 levels (Fig. 5C). Expression of ERK2-Y261A did not appreciably increase Fra1 expression indicating that ERK2-DEF motif interactions are necessary for proper regulation of Fra1. ERK1-WT expressing cells demonstrated reduced expression of Fra1 compared to ERK2-WT cells (Fig. 5C), suggesting that ERK1 and ERK2 differentially regulate Fra1 expression. Consistent with these results, cells expressing RasV12 and ERK2 shRNA demonstrated reduced Fra1 expression whereas expression of GFP shRNA or ERK1 shRNA had no effect on the RasV12-mediated increase in Fra1 levels (Fig. S5G).
We also assessed the ability of ERK1 and ERK2 to activate Fra1 in a Gal4-Fra1 luciferase reporter assay (Young et al., 2002). ERK2-WT and ERK2-D319N expressing cells were able to activate Gal4-Fra1, whereas ERK1-WT and ERK2-Y261A expression did not result in increased luciferase expression (Fig. 5D). Additionally, RasV12 expression displayed a greater activation of Fra1 than ERK2-WT, consistent with the levels of protein expression observed (Fig. 5C). Together, these results strongly suggest that ERK1 and ERK2 differentially regulate the expression and activation of Fra1, that Fra1 induction occurs downstream of ERK-DEF motif signaling and that Fra1 is a critical regulator of EMT downstream of Ras and ERK2.
The observation that ERK2 specifically increases Fra1 protein level and activity (Fig. 5C, 5D, respectively) prompted us to further investigate the molecular basis of Fra1 regulation and its role in EMT. First, we expressed wild-type Fra1 in MCF-10A cells and examined morphological changes. Interestingly, expression of Fra1 alone for 7-10 days after selection was not sufficient to induce changes in morphology (Fig. 5E) or expression of EMT markers (data not shown). Thus we hypothesized that activation of Fra1 by ERK2 was also required, which would be consistent with the Gal4-Fra1 luciferase reporter data (Fig. 5D). To examine this possibility, we expressed Fra1 and ERK2 together, and assessed morphological changes at early time points when ERK2-WT alone does not induce the mesenchymal phenotype. Co-expression of Fra1 and ERK2-WT accelerated the appearance of the EMT phenotype compared to ERK2-WT alone (Fig. 5F, S5H). Second, our previous work had shown that Fra1 hyperphosphorylation is dependent on ERK signaling and an intact DEF motif, and that Fra1 stability also requires ERK activity (Murphy et al., 2004). Furthermore, we have shown that activation of a Fra1 reporter construct requires ERK2-DEF signaling (Fig. 5D). To provide biochemical evidence that ERK2 is capable of directly phosphorylating Fra1 in a DEF motif-dependent fashion, we examined in vitro phosphorylation of Fra1-WT and Fra1-F235A by ERK in vitro. As shown, the phosphorylation of Fra1 by ERK in vitro was dependent on an intact DEF motif (Fig. S5I) and ERK2 with a mutated DEF binding pocket was inefficient at phosphorylating Fra1-WT in vitro (Fig. S5J). Thus, ERK2-DEF motif signaling is required to increase Fra1 levels (Fig. 5C) and activity (Fig. 5D) in cells and our biochemical data support the conclusion that the DEF motif is critical for efficient phosphorylation to occur (Fig. S5I, S5J). Combined, these results further support a mechanism by which ERK2-DEF motif signaling is required for the phosphorylation and activation of Fra1, and induction of EMT.
ERK2/Fra1 induce EMT via regulation of ZEB protein expression
To further investigate the molecular basis for Fra1-regulated EMT, we asked if Snail, Slug, Twist, and ZEB, transcription factors known to regulate EMT in various cell systems, acted downstream of Fra1. First, we assessed whether these molecules are regulated by ERKs. Interestingly, ZEB1 and ZEB2 mRNA levels were clearly increased by ERK2 but not ERK1, while mRNA levels of Snail, Slug and Twist were not affected (Fig 6A). In agreement with the mRNA data, ERK2 increased ZEB1 protein levels while ERK1 did not (Fig. 6B). RasV12 also increased ZEB1 protein expression level through an ERK2-dependent mechanism (Fig. S6A). Additionally, the increase in ZEB1 and ZEB2 occurred in an ERK2-DEF-motif dependent manner (Fig. 6B). These results are consistent with the ERK2-mediated induction of EMT and suggest that ZEB1 and ZEB2 may be downstream effectors of ERK2-DEF motif signaling. Having shown that ZEB1 and ZEB2 are induced downstream of ERK2-DEF motif signaling, we next asked whether ZEB proteins are necessary for ERK2-induced EMT. Similar to knockdown of Fra1, knockdown of ZEB1 or ZEB2 dramatically impaired the induction of EMT by ERK2-D319N (Fig. 6C, 6D) and RasV12 (Fig. S6B, S6C).
Figure 6.
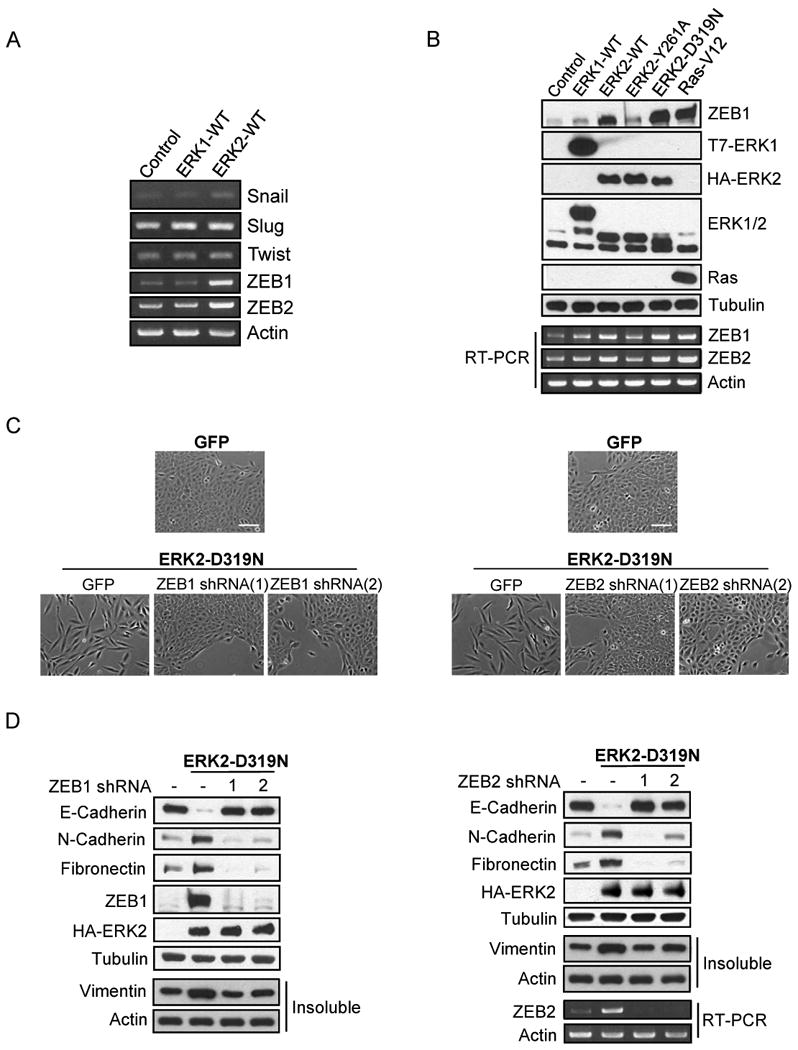
ZEB1/2 expression is differentially regulated by ERK1 and ERK2 and is necessary for ERK2-induced EMT
A) Snail, Slug, Twist, ZEB1/2, and Actin mRNA levels in stable ERK1-WT and ERK2-WT cells were assessed by RT-PCR.
B) Stable ERK1, ERK2, or RasV12 cells were lysed and immunoblot analysis was performed. For the detection of ZEB1/2 mRNA, RT-PCR was performed.
C-D) Stable ZEB1 or ZEB2 knockdown cells were infected with HA-ERK2-D319N encoding retrovirus. Cell morphology and protein expression were assessed by phase-contrast images (C) and immunoblot analysis (D). Bar scale, 100 μm (C).
Having demonstrated that ERK2 regulates Fra1, we examined whether Fra1 is necessary for ERK2 regulation of ZEB. Expression of ERK2-D319N resulted in increased Fra1, ZEB1 and ZEB2, however these increases were impaired in cells with Fra1 knockdown (Fig. 7A). RasV12 also increased ZEB1 protein expression through a Fra1-dependent mechanism (Fig. 7B). Finally, we extended this analysis to cell migration and invasion. Knockdown of ZEB1 or ZEB2 impaired cell migration and invasion in ERK2-D319N (Fig. 7C, 7D respectively) and RasV12 (Fig. S7A, S7B respectively) expressing cells. Together, these data suggest that ZEB1 and ZEB2 are downstream of ERK2/Fra1 signaling and are necessary for ERK2-DEF motif-mediated induction of the EMT phenotype.
Figure 7.
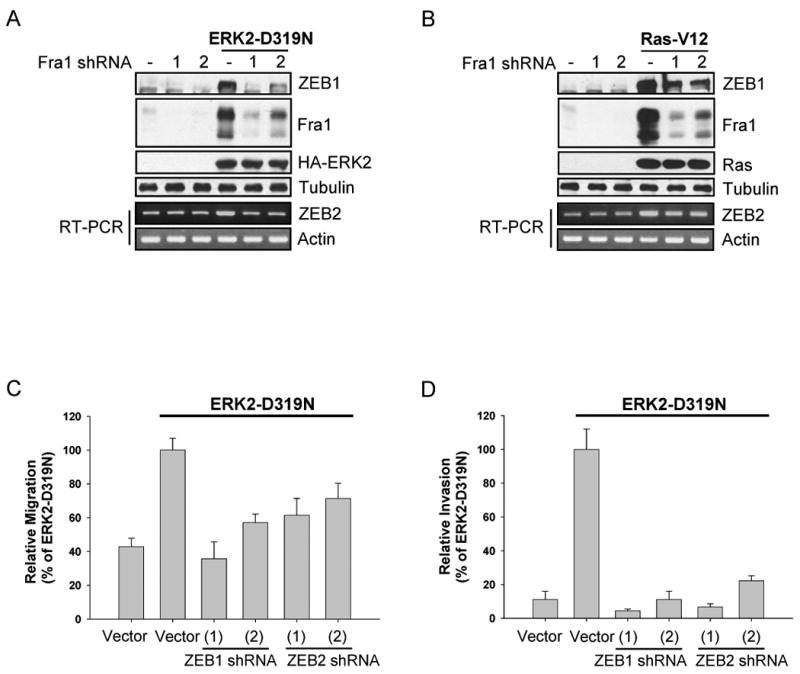
ZEB1/2 expression is regulated by ERK2/Fra1 and is necessary for ERK2-induced migration and invasion
A-B) Stable Fra1 knockdown cells were infected with HA-ERK2-D319N (A) or RasV12 (B) encoding retrovirus. Proteins and RNA were extracted from these cells and the expression of ZEB1 protein and ZEB2 mRNA were assessed by immunoblot and RT-PCR analysis, respectively.
C-D) Migration (C) and invasion (D) assays were performed as described in the materials and methods. Data are the means +/- SEM of three separate experiments performed in duplicate.
Discussion
Previous studies have established a critical role for receptor tyrosine kinase signaling and the Ras pathway in EMT (Thiery, 2003). Expanding on previous reports that ERK activation is necessary for EMT downstream of Ras (Janda et al., 2002), we found that ERK2, but not ERK1, is a critical regulator of EMT downstream of RasV12 in MCF-10A cells. This ERK1/2 specificity is also observed in other epithelial cells such as NMuMG, UB, and MCT cells. Isoform specific roles for Akt1 and Akt2 in the regulation of EMT and growth-factor induced migration have also been suggested (Irie et al., 2005), demonstrating the intricacy of cellular signaling networks. We then examined whether ERK2-induced EMT occurred through interactions via specific docking motifs found in downstream interactors. In the current study, we demonstrate that interaction with DEF motif containing substrates is necessary for ERK2-mediated induction of EMT, characterized by morphological changes, decreased expression of E-cadherin and increased expression of mesenchymal markers. In addition, the ERK2 requirement for Ras-induced changes in cell surface expression of CD44/CD24 are consistent with ERK2-mediated acquisition of a stem cell-like phenotype associated with EMT. Critically, we establish that Fra1 is required for ERK2-induced EMT and provide evidence that ERK2 and ERK1 differentially regulate Fra1 expression and activation. Finally, we identify ZEB1/2 as critical mediators of Fra1-induced EMT.
Although previous research has implicated some downstream targets of ERK1/2 in the regulation of EMT-like processes, our results are the first suggesting that interactions between ERK2 and DEF motif containing proteins are necessary for ERK2-induced EMT. As many immediate-early and late-response gene products contain DEF motifs (Murphy et al., 2004; Murphy et al., 2002), it is possible that ERK2 is regulating EMT and other biological processes assayed herein through the coordinated regulation of several of these gene products. Whereas downregulation of several immediate early or late response genes previously implicated in the regulation of EMT did not prevent ERK2-induced EMT, downregulation of Fra1 dramatically impaired the ability of ERK2-D319N or RasV12 to induce EMT. Under conditions of Ras transformation, Fra1 has been shown to be among the most highly upregulated transcriptional targets (Zuber et al., 2000) and accumulation of Fra1 requires ERK-dependent stabilizing phosphorylation events (Casalino et al., 2003). Overexpression of Fra1 in vitro results in cellular alterations consistent with a transformed phenotype, including morphological changes and anchorage-independent growth (Kakumoto et al., 2006).
We now show that accumulation and activation of Fra1 downstream of RasV12 occurs in an ERK2-specific manner and requires an intact ERK2-DEF motif binding pocket, consistent with the requirement of an intact Fra1 DEF motif for its full phosphorylation following growth factor stimulation (Murphy et al., 2004). Thus, Fra1 is a critical regulator of EMT downstream of ERK2 and our results suggest that ERK2 predominantly regulates Fra1 protein levels and its phosphorylation. Whether this occurs via regulation of Fra1 transcription, mRNA stability, protein stabilization and/or Fra1 activation will require further investigation.
Several transcription factors are known to contribute to EMT and tumor progression by regulating cell-cell adhesion, cell motility and invasiveness. Among them, Snail is regulated by transcription, post-translational modifications and/or nuclear transport. However, much less is known about the regulation of the other factors. In this study, we show that Fra1 is necessary for ERK2-mediated upregulation of ZEB mRNA and protein expression. Based on the potential clinical importance of these transcription factors, it will be interesting to determine the molecular basis of how signaling regulates these factors in different contexts.
While ERK2 expression altered the levels of several EMT markers and increased cell survival, cell migration and invasion, the effect of RasV12 expression was more potent with regards to cell invasion, upregulation of Fra1, and induction of stem cell-like properties. Additionally, in 3D cultures, ERK2 expressing cells formed disorganized colonies but did not exhibit the invasive characteristics of RasV12 expressing cells. Knockdown of ERK2 also did not fully impair the anti-anoikis effect of RasV12 expression. Incomplete knockdown of ERK2 and/or survival signaling by ERK1, however, may also account for the partial survival. While our results are consistent with the requirement of ERK2 for RasV12-mediated EMT, activation of additional pathways appears necessary to produce the full Ras phenotype. Interestingly, expression of ERK1 did not resemble expression of ERK2 with regards to the assays conducted in this study, suggesting that ERK1 and ERK2 are not functionally redundant molecules. The contributions of ERK1, RSK, Akt, and other Ras-modulated pathways to proliferation, survival and invasion will be the subject of future studies.
The Ras-ERK pathway has been implicated in greater than 30% of human tumors (Hoshino et al., 1999). Although oncogenic activation of the Ras-ERK pathway can occur via a number of distinct mechanisms, little is known regarding the functional consequences of signaling by specific ERK isoforms, by ERKs from specific subcellular locations, by ERKs at different times during the cell cycle or by the ERKs utilizing specific interaction motifs. We demonstrate that ERK2-DEF motif signaling via Fra1 and ZEB1/2 is critical for the induction of EMT in human breast epithelial cells. Furthermore, cells that underwent EMT also displayed increased migratory and invasive properties and an increased resistance to anoikis, characteristics that are necessary for metastasis by aggressive cancers. Our results suggest that ERK2-DEF motif signaling is a critical regulatory step in ERK-mediated tumorigenesis. Additional studies are needed to further examine the mechanisms downstream of ERK2-DEF motif signaling that regulate EMT and the potential implications on tumorigenesis and metastasis in animal models.
Experimental Procedures
Plasmids
pBabe-T7-ERK1 was constructed by subcloning T7-tagged murine ERK1 from pcDNA3-T7-ERK1 into the pBabe retroviral vector. pBabe-HA-ERK2 constructs were constructed by subcloning HA-rat ERK2 constructs (Dimitri et al., 2005) into pBabe. Human Fra1 plasmid was purchased from Open Biosystems and subcloned into pLNCX2; a Flag-tag was introduced at the amino terminus. Specific point mutations were introduced using the Quickchange kit (Stratagene) and sequences were verified.
Supplementary Material
Acknowledgments
We thank Drs. Kun-Liang Guan, Nancy Colburn, William Hahn, Raghu Kalluri, Roy Zent for generously providing reagents. We thank Drs. Joan Brugge, Stephanie Walker, Leon Murphy and members of the Blenis lab for helpful discussions, especially Michelle Mendoza, Yuki Abe, Greg Hoffman and Andrew Choo for critical comments on the manuscript. We also thank Jennifer Waters, Lara Petrak, Cassandra Rogers, and the Nikon Imaging Center at Harvard Medical School for microscopy support. This work was supported by National Institutes of Health grant RO1CA46595 (J.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- Casalino L, De Cesare D, Verde P. Accumulation of Fra-1 in ras-transformed cells depends on both transcriptional autoregulation and MEK-dependent posttranslational stabilization. Molecular and cellular biology. 2003;23:4401–4415. doi: 10.1128/MCB.23.12.4401-4415.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. Dimerization in MAP-kinase signaling. Trends Biochem Sci. 2000;25:7–9. doi: 10.1016/s0968-0004(99)01508-x. [DOI] [PubMed] [Google Scholar]
- Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- Di Benedetto B, Hitz C, Holter SM, Kuhn R, Vogt Weisenhorn DM, Wurst W. Differential mRNA distribution of components of the ERK/MAPK signalling cascade in the adult mouse brain. J Comp Neurol. 2007;500:542–556. doi: 10.1002/cne.21186. [DOI] [PubMed] [Google Scholar]
- Dimitri CA, Dowdle W, MacKeigan JP, Blenis J, Murphy LO. Spatially separate docking sites on ERK2 regulate distinct signaling events in vivo. Curr Biol. 2005;15:1319–1324. doi: 10.1016/j.cub.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Eger A, Stockinger A, Park J, Langkopf E, Mikula M, Gotzmann J, Mikulits W, Beug H, Foisner R. beta-Catenin and TGFbeta signalling cooperate to maintain a mesenchymal phenotype after FosER-induced epithelial to mesenchymal transition. Oncogene. 2004;23:2672–2680. doi: 10.1038/sj.onc.1207416. [DOI] [PubMed] [Google Scholar]
- Fantz DA, Jacobs D, Glossip D, Kornfeld K. Docking sites on substrate proteins direct extracellular signal- regulated kinase to phosphorylate specific residues. The Journal of biological chemistry. 2001;276:27256–27265. doi: 10.1074/jbc.M102512200. [DOI] [PubMed] [Google Scholar]
- Hatano N, Mori Y, Oh-hora M, Kosugi A, Fujikawa T, Nakai N, Niwa H, Miyazaki J, Hamaoka T, Ogata M. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells. 2003;8:847–856. doi: 10.1046/j.1365-2443.2003.00680.x. [DOI] [PubMed] [Google Scholar]
- Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene. 1999;18:813–822. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grunert S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002;156:299–313. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakumoto K, Sasai K, Sukezane T, Oneyama C, Ishimaru S, Shibutani K, Mizushima H, Mekada E, Hanafusa H, Akagi T. FRA1 is a determinant for the difference in RAS-induced transformation between human and rat fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5490–5495. doi: 10.1073/pnas.0601222103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- Lefloch R, Pouyssegur J, Lenormand P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol. 2008;28:511–527. doi: 10.1128/MCB.00800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R, Ewings KE, Hadfield K, Cook SJ. Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell death and differentiation. 2005;12:1008–1014. doi: 10.1038/sj.cdd.4401688. [DOI] [PubMed] [Google Scholar]
- Li J, Johnson SE. ERK2 is required for efficient terminal differentiation of skeletal myoblasts. Biochemical and biophysical research communications. 2006;345:1425–1433. doi: 10.1016/j.bbrc.2006.05.051. [DOI] [PubMed] [Google Scholar]
- Mailleux AA, Overholtzer M, Brugge JS. Lumen formation during mammary epithelial morphogenesis: insights from in vitro and in vivo models. Cell Cycle. 2008;7:57–62. doi: 10.4161/cc.7.1.5150. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Murphy LO, MacKeigan JP, Blenis J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Molecular and cellular biology. 2004;24:144–153. doi: 10.1128/MCB.24.1.144-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nature cell biology. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- Okada H, Danoff TM, Kalluri R, Neilson EG. Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol. 1997;273:F563–574. doi: 10.1152/ajprenal.1997.273.4.F563. [DOI] [PubMed] [Google Scholar]
- Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- Papkoff J, Chen RH, Blenis J, Forsman J. p42 mitogen-activated protein kinase and p90 ribosomal S6 kinase are selectively phosphorylated and activated during thrombin-induced platelet activation and aggregation. Molecular and cellular biology. 1994;14:463–472. doi: 10.1128/mcb.14.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi A, Coffa S, Bulus N, Zhu W, Chen D, Chen X, Mernaugh G, Su Y, Cai S, Singh A, et al. H-Ras, R-Ras, and TC21 differentially regulate ureteric bud cell branching morphogenesis. Mol Biol Cell. 2006;17:2046–2056. doi: 10.1091/mbc.E05-08-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nature cell biology. 2003;5:733–740. doi: 10.1038/ncb1026. [DOI] [PubMed] [Google Scholar]
- Roux PP, Richards SA, Blenis J. Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates extracellular signal-regulated kinase docking and RSK activity. Molecular and cellular biology. 2003;23:4796–4804. doi: 10.1128/MCB.23.14.4796-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba-El-Leil MK, Vella FD, Vernay B, Voisin L, Chen L, Labrecque N, Ang SL, Meloche S. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 2003;4:964–968. doi: 10.1038/sj.embor.embor939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Jones LG, Peterson CA. Extracellular signal-regulated kinase-1 and -2 respond differently to mitogenic and differentiative signaling pathways in myoblasts. Mol Endocrinol. 1997;11:2038–2047. doi: 10.1210/mend.11.13.0036. [DOI] [PubMed] [Google Scholar]
- Sheridan DL, Kong Y, Parker SA, Dalby KN, Turk BE. Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. The Journal of biological chemistry. 2008;283:19511–19520. doi: 10.1074/jbc.M801074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer research. 1990;50:6075–6086. [PubMed] [Google Scholar]
- Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- Sugimoto T, Stewart S, Han M, Guan KL. The kinase suppressor of Ras (KSR) modulates growth factor and Ras signaling by uncoupling Elk-1 phosphorylation from MAP kinase activation. The EMBO journal. 1998;17:1717–1727. doi: 10.1093/emboj/17.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nature cell biology. 2000;2:110–116. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. Journal of biology. 2006;5:14. doi: 10.1186/jbiol38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Li W, Wu J, Germann UA, Su MS, Kuida K, Boucher DM. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12759–12764. doi: 10.1073/pnas.2134254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MR, Nair R, Bucheimer N, Tulsian P, Brown N, Chapp C, Hsu TC, Colburn NH. Transactivation of Fra-1 and consequent activation of AP-1 occur extracellular signal-regulated kinase dependently. Molecular and cellular biology. 2002;22:587–598. doi: 10.1128/MCB.22.2.587-598.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MR, Colburn NH. Fra-1 a target for cancer prevention or intervention. Gene. 2006;379:1–11. doi: 10.1016/j.gene.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Zuber J, Tchernitsa OI, Hinzmann B, Schmitz AC, Grips M, Hellriegel M, Sers C, Rosenthal A, Schafer R. A genome-wide survey of RAS transformation targets. Nature genetics. 2000;24:144–152. doi: 10.1038/72799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.