

Abstract
Nogo is a myelin-derived protein that limits axonal regeneration after CNS injury. A short hydrophilic Nogo-66 loop between two hydrophobic domains of Nogo binds to a Nogo-66 receptor (NgR) to inhibit axonal outgrowth. Inhibition of axon outgrowth and cell spreading by a second Nogo domain, termed Amino-Nogo-A, is thought to be mediated by a distinct receptor complex. Here, we define a novel Nogo-A-specific domain in Amino-Nogo that binds to NgR with nanomolar affinity. This second domain of 24 amino acids does not alter cell spreading or axonal outgrowth. Fusion of the two NgR-binding Nogo-A domains creates a ligand with substantially enhanced affinity for NgR and converts a NgR antagonist peptide to an agonist. Thus, NgR activation by Nogo-A involves multiple sites of interaction between Nogo-A and NgR.
Keywords: myelin, axon, regeneration, spinal cord injury, outgrowth inhibitor, cell spreading
Introduction
Inhibition of axon outgrowth by CNS myelin contributes to CNS regeneration failure after injury. Three CNS myelin proteins are capable of inhibiting axonal growth in vitro: Nogo, MAG, and OMgp (McGee and Strittmatter, 2003). Nogo exists in three isoforms, sharing a C-terminal segment that contains two hydrophobic domains (Chen et al., 2000; GrandPre et al., 2000; Prinjha et al., 2000; McGee and Strittmatter, 2003). The three isoforms have distinct hydrophilic N-terminal segments, and Nogo-A is the primary form produced by oligodendrocytes in CNS myelin (Chen et al., 2000; GrandPre et al., 2000; Huber et al., 2002; X. Wang et al., 2002). Nogo-A has been shown to possess two inhibitory domains. The inhibitory Nogo-66 domain in the carboxyl region is flanked by the two hydrophobic segments and is detectable on the surface of oligodendrocytes (GrandPre et al., 2000; Fournier et al., 2001; Oertle et al., 2003b). The N-terminal segment of Nogo-A (Amino-Nogo-A) independently inhibits axonal outgrowth and spreading of non-neuronal cells (Chen et al., 2000; Fournier et al., 2001); a central Δ20 region appears most critical for this activity (Oertle et al., 2003b). The Amino-Nogo domain, like the Nogo-66 domain, has been detected on the surface of oligodendrocytes, and two conformations for Nogo-A have been proposed (Chen et al., 2000; GrandPre et al., 2000; Oertle et al., 2003b). In one, the N and C termini are cytosolic, and the Nogo-66 loop is extracellular with two transmembrane segments. In an alternate topology, the first hydrophobic segment loops into and out of the plasma membrane without forming a transmembrane segment, so that the Amino-Nogo and Nogo-66 are located on the same side of the lipid bilayer.
Antibody or peptide perturbation of the Nogo pathway leads to an enhanced axonal growth, plasticity, and functional recovery after spinal injury or stroke (Schnell and Schwab, 1990; Bregman et al., 1995; GrandPre et al., 2002; Li and Strittmatter, 2003; Wiessner et al., 2003; Lee et al., 2004). Genetic studies of Nogo function have provided conflicting data on the essential role for Nogo in axonal regeneration (Kim et al., 2003; Simonen et al., 2003; Zheng et al., 2003). A receptor for the Nogo-66 domain was identified by expression cloning [Nogo-66 receptor (NgR)] (Fournier et al., 2001). Remarkably, MAG and OMgp bind to the leucine-rich repeat (LRR) domain of the same NgR protein to inhibit axonal growth in vitro (Domeniconi et al., 2002; Liu et al., 2002; K. C. Wang et al., 2002). In vivo, genetic deletion of NgR or the addition of an NgR decoy receptor allows certain axonal fibers to sprout and enhances functional recovery after spinal cord transection (Fischer et al., 2004; Kim et al., 2004; Lee et al., 2004; Li et al., 2004).
Here, we show that the Amino-Nogo domain of Nogo-A harbors a region (Nogo-A-24) that interacts with NgR. The combination of Nogo-66 with this Amino-Nogo domain creates a substantially higher-affinity NgR ligand. Thus, axon inhibition and NgR activation by Nogo-A in vivo are likely to require simultaneous binding of Nogo-66 and Nogo-A-24.
Materials and Methods
Recombinant proteins and peptides. To generate alkaline phosphatase (AP) fusion proteins, human Amino-Nogo fragments were amplified and ligated to the pcAP6 vector digested with restriction enzymes EcoRI and XhoI as described previously (Fournier et al., 2001). The EcoRI/BamHI-digested Y4C fragment and BamHI/XhoI-digested Nogo-66 fragment were ligated to EcoRI- and XhoI-digested pcAP6 to generate the AP-Y4C66 plasmid. Plasmids were then transfected into human embryonic kidney 293T (HEK293) cells, and conditioned media were collected after 7 d. To generate glutathione S-transferase (GST) fusion proteins, Amino-Nogo fragments were cloned in pGEX2T (Amersham Biosciences, Piscataway, NJ). Native and soluble GST fusion proteins were expressed and purified as described previously (GrandPre et al., 2000). Biotin-labeled Nogo-A-24 (B24; biotin-IFSAELSKTSVVDLLYWRDIKKTG) and biotin-labeled Nogo-A-24-32 (B24-32; biotin-IFSAELSKTSVVDLLYWRDIKKTGGRIYKGV-IQAIQKSDEGHPFRAYLESEVAISEE) were synthesized and purified by the W. M. Keck facility at Yale University.
COS-7 binding assay, fibroblast spreading assay, and chick DRG outgrowth assay. COS-7 binding assays were done as described previously (Fournier et al., 2001). Conditioned media with AP ligands were incubated with untransfected or mouse NgR-transfected COS-7 cells overnight at 4°C before fixation and heat inactivation of endogenous AP. Bound AP to COS-7 cells was measured using NIH Image software. Fibroblast spreading and chick DRG outgrowth assay were done as described previously (Fournier et al., 2001) with some modifications. Fifty microliters of purified GST fusion protein or peptides diluted in PBS were pipetted into polylysine-precoated 96-well plates (Biocoat plates; Becton Dickinson, Mountain View, CA) and dried overnight at room temperature. For fibroblast spreading assays, subconfluent COS-7 cells were cultured on this surface for 1 h in serum-containing medium before fixation and staining with rhodamine-phalloidin. For outgrowth assays, dissociated embryonic day 13 (E13) chick DRG neurons were plated for 6 h before fixation. Neurons were stained with anti-Neurofilament (catalog #N4142; Sigma, St. Louis, MO) and anti-human neuronal protein HuC/HuD (A-21271; Molecular Probes, Eugene, OR) antibodies. Cell area, the number of attached cells, and neurite length were measured using the Imagexpress Automated Cellular Imaging and Analysis System (Molecular Devices, Union City, CA).
ELISA. NgR(344)ecto-Fc was provided by Dr. Daniel Lee (BiogenIdec, Boston, MA). Twenty nanograms of NgR(344)ecto-Fc in 50 μl of PBS were allowed to bind overnight to each well of 96-well Maxisorb plates (Nunc, Naperville, IL) at 4°C. The plates were then blocked with 10 mg/ml BSA in PBS for 1.5 h at room temperature. Conditioned media with AP fusion ligands were diluted in HBSS plus 0.1% BSA and 20 mm HEPES, pH 7.3 (HBH) and incubated with immobilized NgR for 2 h at room temperature. Unbound AP ligand was washed away with HBH. Activities of bound AP ligands were then measured by absorbance in the presence of p-nitrophenyl phosphate substrate.
Results
Amino-Nogo fragments bind to NgR
While developing the means to identify the mechanism of Amino-Nogo-A action, we examined several AP fusion proteins containing various Nogo-A segments derived from regions between the N terminus and the first hydrophobic segment. None of these fragments binds with high affinity to nontransfected COS-7 cells. While examining presumed control conditions, we unexpectedly observed that the carboxyl half of the Amino-Nogo-A (fragment Y) exhibits high-affinity binding to COS-7 cells expressing NgR (Fig. 1). The amino half of Amino-Nogo-A (fragment X) and the Δ20 region do not bind to NgR (Fig. 1). Binding of Amino-Nogo-A fragment Y is saturable with a Kd indistinguishable from that for AP-Nogo-66 association with NgR (Table 1) (Fournier et al., 2001). To better define the region responsible for Amino-Nogo-A interaction with NgR, a range of truncation mutants of Amino-Nogo-A were examined as AP fusion proteins. Subdivision of the Y fragment into overlapping 150 aa segments reveals that the NgR interaction site is localized to the most C-terminal segment (Y4 and Y4C) (Fig. 2A). In fact, the NgR-interacting segment of Amino-Nogo-A is fully accounted for in the extreme carboxyl 24 aa (amino acids 995-1018, Nogo-A-24) (Table 1; Fig. 2A,B). The Ile residue located at amino acid 995 is necessary for high-affinity binding, and 19 aa from residue 995 to residue 1013 seem to be sufficient for binding to NgR (Table 1).
Figure 1.

Binding of Amino-Nogo fragment Y to NgR. A, Schematic drawing of Nogo-A and Amino-Nogo (AmNg) fragments X, Y, and Δ20. TM, Transmembrane. B, Binding of AP-fused Amino-Nogo fragment Y (AP-Y) but not fragments X (AP-X) or Δ20 to COS-7 cells expressing NgR. Conditioned media from HEK293T cells containing the AP fusion protein of indicated concentrations were applied to untransfected (None) or COS-7 cells expressing NgR, and bound AP was stained.
Table 1.
Binding affinity of Amino-Nogo fragments to NgR
|
Amino acid number |
Amino acid sequence |
NgR Kd (nm) |
|---|---|---|
| 181-864 (fragment X) | No binding at 150 nm | |
| 622-1018 (fragment Y) | 6.66 ± 1.49 | |
| 877-1018 (fragment Y4) | 9.01 ± 6.36 | |
| 950-1018 (fragment Y4C) | 3.51 ± 3.36 | |
| 971-1018 | 2.69 ± 1.32 | |
| 995-1018 (Nogo-A-24) | IFSAELSKTSVVDLLYWRDIKKTG | 2.43 ± 0.51 |
| 995-1015 | IFSAELSKTSVVDLLYWRDIK | 4.55 ± 3.66 |
| 995-1014 | IFSAELSKTSVVDLLYWRDI | 3.19 ± 0.12 |
| 995-1013 | IFSAELSKTSVVDLLYWRD | 2.48 ± 0.72 |
| 996-1018 | FSAELSKTSVVDLLYWRDIKKTG | 26.59 ± 6.86 |
| 1000-1018 | LSKTSVVDLLYWRDIKKTG | No binding at 25 nm |
| 1005-1018 | VVDLLYWRDIKKTG | No binding at 25 nm |
| 950-1004 | .........IFSAELSKTS | No binding at 400 nm |
| Amino of Nogo-C | MDGQKKNWKDKVVDLLYWRDIKKTG | No binding at 400 nm |
| Amino of Nogo-B |
|
No binding at 50 nm
|
Binding Kd values for AP-fused Amino-Nogo fragments were measured by applying conditioned media containing the AP fusion protein to NgR-expressing COS-7 cells. Bound AP was stained and measured.
Figure 2.

Nogo-A-24 is the NgR-binding domain in Amino-Nogo-A. A, Different fragments of Amino-Nogo as indicated were fused to AP, and their binding to NgR was determined in COS-7 cell binding assays. B, Top, AP-Nogo-A-24 binding to NgR-expressing COS-7 cells measured as a function of AP-Nogo-A-24 concentration. Bottom, Replotted data from top panel. Binding Kd was determined from four independent measurements. C, Binding of AP-fused Amino-Nogo fragments to dissociated E13 chick DRG neurons (arrows). Conditioned media from HEK293T cells containing the AP fusion protein as indicated were applied to dissociated E13 chick DRG neurons, and bound AP was stained. AP-X, AP-fused fragment X of Amino-Nogo-A.
The 24 aa NgR-binding residues of Amino-Nogo-A are encoded by nucleotides that span the splice site (amino acids 1004/1005) between the Nogo-A-specific exon of the nogo gene and the 5′ common exon of the gene (Fig. 1A) (Chen et al., 2000; GrandPre et al., 2000; Oertle et al., 2003a). AP fusion proteins comprising amino acids from the Nogo-A-specific region alone do not bind to NgR (amino acids 950-1004). Amino-Nogo residues of Nogo-B or Nogo-C also fail to associate with NgR-expressing cells at 50 nm (Table 1). Thus, this second high-affinity NgR-interacting domain is Nogo-A-isoform specific and is immediately N-terminal to the hydrophobic segment that separates it from Nogo-66.
If these Amino-Nogo-A fragments are to play a role in regulating neurite outgrowth, then they would be expected to bind to neuronal processes. Previously, we have shown that AP-Nogo-66 binds to NgR on DRG processes (Fournier et al., 2001). As expected from COS-7 NgR binding experiments, the C-terminal 24 aa of Amino-Nogo can also mediate AP fusion protein binding to DRG axons, but a shorter fragment (amino acids 999-1018) of Nogo-A fails to interact with DRG neurons (Fig. 2C). The N-terminal X fragment of Amino-Nogo also binds to DRG axons, presumably through NgR-independent mechanisms.
Carboxyl region of Amino-Nogo-A binds to the LRR domain of NgR
Binding of Nogo-66, MAG, or OMgp to NgR requires the LRR domain of NgR (Fournier et al., 2001; K. C. Wang et al., 2002; Barton et al., 2003). As for Nogo-66, MAG, and OMgp, deletion of any two LRRs eliminates binding to NgR for the Amino-Nogo-Y and -Y4 fragments (Fig. 3A). Similarly, the cysteine-rich LRR-N-terminal and LRR-C-terminal capping domains are essential for Amino-Nogo-Y4 binding. In contrast, deletion of the unique signaling domain of NgR extending from the LRR region to the glycosylphosphatidyl inositol anchorage site (C-terminal domain) does not alter Amino-Nogo-Y4 binding. NgR is part of the gene family that includes NgR2 and NgR3. When expressed on the surface of COS-7 cells, these related proteins do not bind AP-Nogo-66, AP-MAG, or AP-OMgp (Barton et al., 2003). Similarly, NgR2 and NgR3 are not binding partners for Amino-Nogo-A (Fig. 3B). By these measures, the NgR requirements for Nogo-66 and Amino-Nogo-Y4C binding are indistinguishable.
Figure 3.
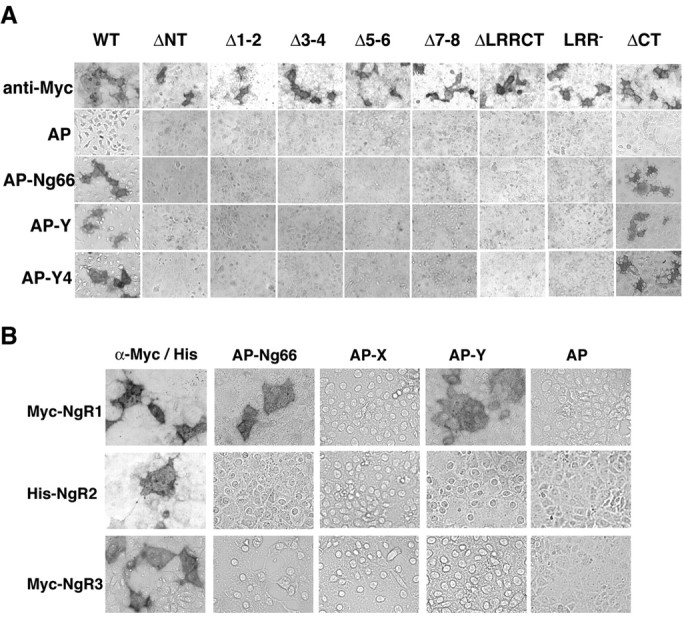
Amino-Nogo-A does not bind NgR LRR deletion mutants, NgR2, or NgR3. A, Binding of AP or AP-fused Nogo fragments to COS-7 cells expressing NgR mutants (Fournier et al., 2002) as indicated. Surface expression of NgR mutants was detected using anti-Myc antibodies. AP-Y and AP-Y4, AP fusion protein of the Y or Y4 fragment of Amino-Nogo-A, respectively. B, Amino-Nogo-A does not bind to NgR2 or NgR3. Conditioned media from transfected HEK293T cells containing a 20 nm concentration of the indicated AP fusion protein were applied to COS-7 cells expressing mouse NgR1, human NgR2, or mouse NgR3, and bound AP was stained. Surface expression of NgRs was detected using anti-Myc or anti-His antibodies. AP-X and AP-Y, AP fusion protein of the X or Y fragment of Amino-Nogo-A, respectively.
Inhibition of cell spreading and axon outgrowth by Amino-Nogo is separable from NgR binding
It has been recognized that the Amino-Nogo-A protein inhibits non-neuronal cell spreading and axonal outgrowth when the protein is substrate bound (Chen et al., 2000; Fournier et al., 2001). Work by Oertle et al. (2003b) has suggested that a specific amino acid stretches near the N terminus and the middle of Amino-Nogo-A is responsible for this activity. The later domain has been termed Δ20. To determine whether the NgR-interacting amino acids of Amino-Nogo-A regulate cell spreading and axonal outgrowth, various fragments were expressed as GST fusion proteins and purified from Escherichia coli. Fragments containing portions of the Δ20 region significantly reduce COS-7 cell attachment and spreading (Fig. 4A-C). Fragments consisting of the C-terminal 75 aa (Y4C) or 150 aa (Y4) lack the Δ20 region but possess the entire 24 aa NgR-binding region (Fig. 2A). The Y4 and Y4C proteins do not alter COS-7 morphology when presented as a substrate (Fig. 4A-C). Thus, inhibition of fibroblast spreading is separable from NgR binding by Amino-Nogo-A.
Figure 4.
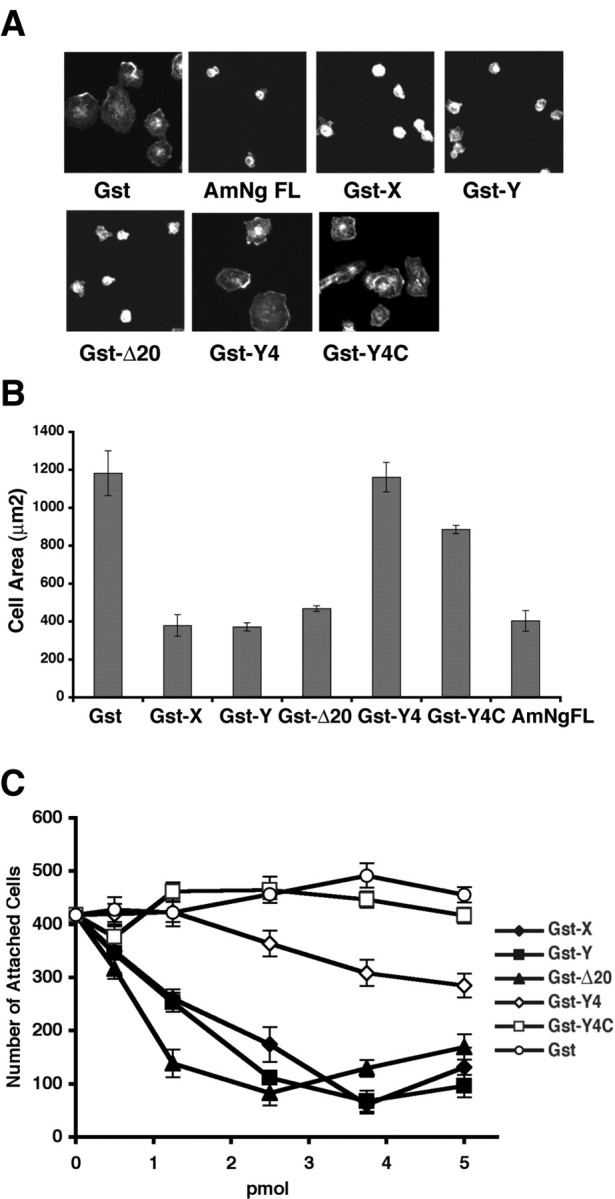
Effects of Amino-Nogo fragments on cell attachment and spreading. A, Different effects of Amino-Nogo fragments on cell spreading. COS-7 cells were allowed to attach and spread on slides with spots coated with 50 ng of dried GST fusion protein as indicated and stained for F-actin. B, COS-7 cell area for experiments as in A was measured and plotted. C, COS-7 cells were allowed to attach and spread on 96-well dishes coated with dried GST fusion proteins as indicated. The number of attached cells was counted and plotted as a function of the amount of various proteins dried per well in a 96-well dish. GST-X, GST-Y, GST-Δ20, GST-Y4, and GST-Y4C, Fusion protein of GST and X, Y, Δ20, Y4, or Y4C fragments (Figs. 1, 2) of Amino-Nogo-A, respectively; AmNg FL, full length of Amino-Nogo expressed and purified from HEK293T cells (Fournier et al., 2001).
The same GST-Amino-Nogo proteins were tested for their ability to reduce neurite outgrowth from chick E13 DRG neurons. As shown previously for the entire Amino-Nogo domain, those subfragments containing portions of the Δ20 region are inhibitory for neurite outgrowth (Fournier et al., 2001; Oertle et al., 2003b) (Fig. 5). Because these cultures are known to express NgR and respond to binding with Nogo-66, we tested whether the NgR-binding Y4 and Y4C fragments of Amino-Nogo would alter neurite outgrowth. Unexpectedly, substrates coated with the NgR-binding Y4 and Y4C fragments of Amino-Nogo-A are not inhibitory for axonal growth (Fig. 5). Thus, the NgR-binding domain of Amino-Nogo does not bind to or affect morphology of NgR-negative COS-7 cells and, when bound to NgR-positive neurons, does not alter axon growth.
Figure 5.

Effects of Amino-Nogo fragments on neurite outgrowth. A, Differential effects of Amino-Nogo fragments on neurite outgrowth. Dissociated neurons from E13 chick DRGs were plated on 96-well dishes coated with 1 pmol of protein per well and stained for neurofilament localization. B, Neurite length per neuron was measured and plotted as a percentage of GST control with increasing concentration of dried protein for the experiment described in A. GST-X, GST-Y, GST-Δ20, GST-Y4, and GST-Y4C, Fusion protein of GST and X, Y, Δ20, Y4, or Y4C fragments (Figs. 1, 2) of Amino-Nogo-A, respectively.
Juxtaposition of two NgR-binding domains from Nogo-A creates high-affinity agonist activity
For intact Nogo-A, Nogo-66 and Nogo-A-24 may be adjacent to one another at the plasma membrane surface, because they are separated in the primary structure only by a hydrophobic loop that extends into the lipid bilayer (Oertle et al., 2003b). It is possible that Nogo-66 and Nogo-A-24 could bind simultaneously to NgR to create a high-affinity ligand based on two-site binding. To create a soluble, tagged ligand resembling this conformation, we generated an AP fusion protein with the Y4C fragment of Amino-Nogo-A fused directly to Nogo-66. The affinity of this AP-Y4C-66 ligand for NgR is substantially greater than is that of AP-Y4C or AP-Nogo-66 (Fig. 6). The Kd for this binding is subnanomolar (Fig. 6). Thus, bivalent binding of two linked Nogo-A domains creates a significantly more potent NgR ligand.
Figure 6.

Fusion of Y4C with Nogo-66 creates a high-affinity ligand for NgR. A, AP-Y4C66, AP-Y4C, and AP-Nogo-66 binding to NgR-expressing COS-7 cells measured as a function of AP-Y4C66, AP-Y4C, or AP-Nogo-66 concentration. B, Replotted data from A. Binding Kd was determined from four independent measurements.
Next, we considered the effect of these two peptide domains on neurite outgrowth. Attempts to produce quantities of purified recombinant or synthetic Nogo 24-66 or Y4C-66 have been un-successful for technical reasons. Although a synthetic Nogo-66 peptide fragment inhibits neurite outgrowth by binding to NgR as an agonist, the N-terminal 40 aa of Nogo-66 (NEP1-40) bind to NgR as an antagonist (GrandPre et al., 2002). Similar NgR antagonistic results are obtained for peptides as short as 32 aa (NEP32) (data not shown). Fragments of Nogo-66 shorter than these 32 aa do not interact with NgR (GrandPre et al., 2002). We reasoned that fusing the 24 aa segment of Amino-Nogo-A to the NEP32 antagonist peptide might create a high-affinity antagonist with a potency similar to the binding of AP-Y4C-66 to NgR. To examine this hypothesis, a biotinylated peptide containing the Nogo-A-24 sequence fused at its C terminus to NEP32 (B24-32) was synthesized. The potency of this fusion peptide was first examined in an ELISA-type assay with immobilized NgR (Fig. 7). The AP fusion protein of Nogo-A-24 binds to immobilized NgR as AP-Nogo-66, but the AP control does not (Fig. 7A). The binding of AP-Nogo-66 to immobilized NgR can be displaced weakly by biotinylated Nogo-A-24 (B24) and NEP1-40 peptides with Ki values of ∼5 μm (Fig. 7B). The apparent Kd/Ki values are lower than in cell-based assays because of enhanced avidity at the cell surface compared with purified protein binding in solution, as observed previously (Li et al., 2004). Binding of AP-Nogo-A-24 to NgR can be displaced by B24 peptide with a Ki value ∼1 μm, and the binding cannot be displaced by NEP1-40 peptide, suggesting that Nogo-A-24 and Nogo-66 use at least partially separable residues in NgR for binding. The B24-32 peptide, with an apparent Ki value of 50 nm, is ∼20- to 100-fold more potent in displacing AP-Nogo-66 and AP-Nogo-A-24 binding to NgR than the B24 and NEP1-40 peptides (Fig. 7B,C). Thus, the B24-32 fusion peptide binds to NgR with much higher affinity than the two uncoupled NgR-binding peptides.
Figure 7.

B24-32 peptide displaces AP-Nogo-66 and AP-Nogo-A-24 binding to NgR with high affinity. A, AP-Nogo-66 and AP-Nogo-A-24 bind specifically to immobilized NgR. A concentration of 50 nm AP, 50 nm AP-Nogo-66, or 25 nm AP-Nogo-A-24 was allowed to bind to immobilized NgR(344)ecto-Fc, and the activity of bound AP was measured. B, C, B24-32 peptide displaces AP-Nogo-66 and AP-Nogo-A-24 binding to NgR. The experiment was done as in A in the presence of increasing concentrations of peptides.
To explore the potential antagonist activity of the high-affinity B24-32 peptide, we considered its effect on DRG neurite outgrowth. As predicted from studies with the GST-Y4C fusion protein (Fig. 5), the B24 peptide alone does not alter neurite outgrowth (Fig. 8). Unexpectedly, the B24-32 fusion peptide potently inhibits axon outgrowth from DRG neurons. This agonist activity requires covalent intermolecular linkage of the two domains, because an equimolar mixture of B24 and NEP32 peptides is not inhibitory (Fig. 8). As suggested by the purified NgR ELISA binding studies (Fig. 7), the Nogo-A-24 and Nogo-66 peptides have essentially distinct interaction sites with NgR, because the B24 peptide did not significantly alter the axon outgrowth inhibition by Nogo-66.
Figure 8.

B24-32 peptide inhibits neurite outgrowth. A, Dissociated neurons from E13 chick DRG were plated onto 96-well dishes coated with 500 pmol of dried peptides as indicated and stained for neurofilament localization. B, Neurite length per neuron was measured and plotted as a percentage of PBS control for experiments as in A.
Discussion
This study recognizes and characterizes a novel Nogo-A-specific NgR-binding domain. The Nogo-A-24 and Nogo-66 domains interact with NgR to create an isoform-selective high-affinity ligand. Nogo-A also has the capacity to inhibit cell spreading and axonal outgrowth through NgR-independent domains. The high potency of bivalent Nogo-A interactions, as well as the redundancy of myelin ligands and axonal receptors, are likely to contribute to the near total failure of adult axonal regeneration in vivo.
Second NgR-interacting domain in Nogo-A
Our initial functional analysis of Nogo-A activity had separated the Amino-Nogo-A domain from the Nogo-66 domain (Fournier et al., 2001). We had demonstrated that NgR is a receptor for Nogo-66 but that Amino-Nogo-A uses other mechanisms. Here, we have uncovered an additional activity not revealed in morphological assays. The C terminus of the Amino-Nogo-A domain interacts with NgR with high affinity. This NgR-binding domain of 24 aa (Nogo-A-24) does not alter cell spreading or axonal outgrowth, explaining why it was not detected in initial assays. Thus, for any cell to which Nogo-66 does not bind, Amino-Nogo-24 binding is irrelevant to neurite outgrowth. Because Nogo-66 binding and growth cone collapse are lost in NgR1-/- DRG neurons, Amino-Nogo-24 cannot act in the absence of NgR1 (Kim et al., 2004). The Amino-Nogo-24 domain is present only in Nogo-A, providing one basis for Nogo-A being a more potent inhibitor of axonal growth than Nogo-C (Chen et al., 2000; GrandPre et al., 2000). It has been reported that a fraction of Nogo-A in oligodendrocytes is situated in a conformation exposing both the N terminus and the Nogo-66 domain at the cell surface (Oertle et al., 2003b). The more N-terminal hydrophobic segment of Nogo-A is proposed to insert into the plasma membrane as a loop. This conformation is predicted to bring the Nogo-A-24 segment and the Nogo-66 domain at the cell surface into close proximity at the cell surface. The ability of both of these domains to interact with NgR is consistent with a physiological role for this conformation.
The N-terminal segment of Nogo-A enhances the potency of Nogo-66
Nogo-A-24 binding does not activate NgR to inhibit axonal outgrowth. However, fusion of this domain to Nogo-66 creates a bivalent ligand for NgR with substantially enhanced receptor affinity. This enhanced affinity may explain the finding that in vitro and in vivo assays indicate a greater role for Nogo-A than MAG in limiting axonal growth, despite the greater abundance of MAG protein in myelin preparations. Mice lacking MAG have been reported to lack CNS axonal regeneration (Bartsch et al., 1995), although peripheral regeneration may be enhanced in certain genetic backgrounds (Schafer et al., 1996).
Previous work had shown that the entire Nogo-66 domain was required for NgR activation and inhibition of axonal growth. Amino segments of Nogo-66 as short as 32 aa bind with high affinity to NgR but act as antagonists of Nogo-66, suggesting that the 33-66 region is required for receptor activation but not high-affinity binding (GrandPre et al., 2002). However, it is remarkable that fusion of the inactive Nogo-A-24 to the NEP1-32 antagonist creates a potent agonist peptide. Thus, the Nogo-66 (33-66) region is not essential for receptor activation. Instead, the results raise the possibility that bivalent interaction of ligands with NgR is critical for activation. Because NgR can bind to itself and is clustered in lipid rafts (Fournier et al., 2002; Liu et al., 2002), one hypothesis is that bivalent ligands may activate the receptor through modulation of its aggregation state in the plane of the bilayer. We examined this hypothesis by testing whether the B24-32 peptide alters the oligomerization status of NgR. The NgR aggregation state of purified or cellular NgR is not modulated by the B24-32 peptide or other NgR ligands (data not shown). Therefore, we favor the hypothesis that simultaneous binding of two Nogo-A domains causes a conformational change of NgR to an activated state. Single binding events appear ineffective in causing this shift in NgR conformation.
NgR-independent action of Amino-Nogo
We and others have documented previously that substrate-bound or aggregated Amino-Nogo-A inhibited fibroblast spreading and neurite outgrowth (Chen et al., 2000; Fournier et al., 2001; Oertle et al., 2003b). As suggested by these properties, we confirm that the Amino-Nogo-A domain responsible for these activities does not bind to NgR. The molecular basis for these actions remains unknown. At least a significant portion of this activity can be localized to a Δ20 segment near the middle of Amino-Nogo-A. The N terminus of Nogo has been recognized recently to have another NgR-independent action via an extreme N-terminal domain present in Nogo-B. This domain has a selective role in remodeling the vasculature after injury (Acevedo et al., 2004). Thus, Nogo appears to have multiple functional domains and receptors (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The Nogo-66 and Nogo-A-24 regions join to create a high-affinity ligand to inhibit axonal growth through NgR. The Δ20 region of Nogo-A does not bind to NgR but is nonpermissive as a substrate for multiple cell types, probably through the action of another receptor. The N-terminal segment of Nogo-B has no affinity for NgR but does regulate vascular endothelial and smooth muscle cell migration through an unidentified receptor.
Conclusions
The identification of a high-affinity bipartite interaction of Nogo-A with NgR has several implications. This interaction is Nogo-A isoform selective, consistent with the fact that Nogo-A is the predominant Nogo protein in myelin. The high affinity of Y4C-66 and B24-32 fusion peptides for NgR is consistent with Nogo-A being a prominent inhibitor in myelin under concentration-limited conditions.
Footnotes
This work was supported by grants to S.M.S. from the National Institutes of Health and BiogenIdec.
Correspondence should be addressed to Dr. Stephen M. Strittmatter, Department of Neurology, Yale University School of Medicine, P.O. Box 208018, New Haven, CT 06510. E-mail: stephen.strittmatter@yale.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255298-07$15.00/0
References
- Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, Tempst P, Strittmatter SM, Sessa WC (2004) A new role for Nogo as a regulator of vascular remodeling. Nat Med 10: 382-388. [DOI] [PubMed] [Google Scholar]
- Barton WA, Liu BP, Tzvetkova D, Jeffrey PD, Fournier AE, Sah D, Cate R, Strittmatter SM, Nikolov DB (2003) Structure and axon outgrowth inhibitor binding of the Nogo-66 receptor and related proteins. EMBO J 22: 3291-3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch U, Bandtlow CE, Schnell L, Bartsch S, Spillmann AA, Rubin BP, Hillenbrand R, Montag D, Schwab ME, Schachner M (1995) Lack of evidence that myelin-associated glycoprotein is a major inhibitor of axonal regeneration in the CNS. Neuron 15: 1375-1381. [DOI] [PubMed] [Google Scholar]
- Bregman BS, Kunkel-Bagden E, Schnell L, Dai HN, Gao D, Schwab ME (1995) Recovery from spinal cord injury mediated by antibodies to neurite growth inhibitors. Nature 378: 498-501. [DOI] [PubMed] [Google Scholar]
- Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME (2000) Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403: 434-439. [DOI] [PubMed] [Google Scholar]
- Domeniconi M, Cao Z, Spencer T, Sivasankaran R, Wang K, Nikulina E, Kimura N, Cai H, Deng K, Gao Y, He Z, Filbin M (2002) Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron 35: 283-290. [DOI] [PubMed] [Google Scholar]
- Fischer D, He Z, Benowitz LI (2004) Counteracting the Nogo receptor enhances optic nerve regeneration if retinal ganglion cells are in an active growth state. J Neurosci 24: 1646-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM (2001) Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 409: 341-346. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Gould GC, Liu BP, Strittmatter SM (2002) Truncated soluble Nogo receptor binds Nogo-66 and blocks inhibition of axon growth by myelin. J Neurosci 22: 8876-8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrandPre T, Nakamura F, Vartanian T, Strittmatter SM (2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403: 439-444. [DOI] [PubMed] [Google Scholar]
- GrandPre T, Li S, Strittmatter SM (2002) Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 417: 547-551. [DOI] [PubMed] [Google Scholar]
- Huber AB, Weinmann O, Brosamle C, Oertle T, Schwab ME (2002) Patterns of nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci 22: 3553-3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Li S, GrandPre T, Qiu D, Strittmatter SM (2003) Axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38: 187-199. [DOI] [PubMed] [Google Scholar]
- Kim JE, Liu BP, Park JH, Strittmatter SM (2004) Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron 44: 439-451. [DOI] [PubMed] [Google Scholar]
- Lee JK, Kim JE, Sivula M, Strittmatter SM (2004) Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J Neurosci 24: 6209-6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Strittmatter SM (2003) Delayed systemic Nogo-66 receptor antagonist promotes recovery from spinal cord injury. J Neurosci 23: 4219-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Liu BP, Budel S, Li M, Ji B, Walus L, Li W, Jirik A, Rabacchi S, Choi E, Worley D, Sah DW, Pepinsky B, Lee D, Relton J, Strittmatter SM (2004) Blockade of nogo-66, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein by soluble nogo-66 receptor promotes axonal sprouting and recovery after spinal injury. J Neurosci 24: 10511-10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BP, Fournier A, GrandPre T, Strittmatter SM (2002) Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 297: 1190-1193. [DOI] [PubMed] [Google Scholar]
- McGee AW, Strittmatter SM (2003) The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci 26: 193-198. [DOI] [PubMed] [Google Scholar]
- Oertle T, Huber C, van der Putten H, Schwab ME (2003a) Genomic structure and functional characterisation of the promoters of human and mouse nogo/rtn4. J Mol Biol 325: 299-323. [DOI] [PubMed] [Google Scholar]
- Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, Huber AB, Simonen M, Schnell L, Brosamle C, Kaupmann K, Vallon R, Schwab ME (2003b) Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J Neurosci 23: 5393-5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinjha R, Moore SE, Vinson M, Blake S, Morrow R, Christie G, Michalovich D, Simmons DL, Walsh FS (2000) Inhibitor of neurite outgrowth in humans. Nature 403: 383-384. [DOI] [PubMed] [Google Scholar]
- Schafer M, Fruttiger M, Montag D, Schachner M, Martini R (1996) Disruption of the gene for the myelin-associated glycoprotein improves axonal regrowth along myelin in C57BL/Wlds mice. Neuron 16: 1107-1113. [DOI] [PubMed] [Google Scholar]
- Schnell L, Schwab ME (1990) Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature 343: 269-272. [DOI] [PubMed] [Google Scholar]
- Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab ME (2003) Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron 38: 201-211. [DOI] [PubMed] [Google Scholar]
- Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z (2002) Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417: 941-944. [DOI] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM (2002) Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci 22: 5505-5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiessner C, Bareyre FM, Allegrini PR, Mir AK, Frentzel S, Zurini M, Schnell L, Oertle T, Schwab ME (2003) Anti-Nogo-A antibody infusion 24 hours after experimental stroke improved behavioral outcome and corticospinal plasticity in normotensive and spontaneously hypertensive rats. J Cereb Blood Flow Metab 23: 154-165. [DOI] [PubMed] [Google Scholar]
- Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier-Lavigne M (2003) Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron 38: 213-224. [DOI] [PubMed] [Google Scholar]