

Abstract
A robust surge of gonadotropin-releasing hormone (GnRH) release triggers the luteinizing hormone surge that induces ovulation. The GnRH surge is attributable to estradiol feedback, but the mechanisms are incompletely understood. Voltage-gated calcium channels (VGCCs) regulate hormone release and neuronal excitability, and may be part of the surge-generating mechanism. We examined VGCCs of GnRH neurons in brain slices from a model exhibiting daily luteinizing hormone surges. Mice were ovariectomized (OVX), and a subset was treated with estradiol implants (OVX+E). OVX+E mice exhibit negative feedback in the A.M. and positive feedback in the P.M. GnRH neurons express prominent high-voltage-activated (HVA) and small low-voltage-activated (LVA) macroscopic (whole-cell) Ca currents (ICa). LVA-mediated currents were not altered by estradiol or time of day. In contrast, in OVX+E mice, HVA-mediated currents varied with time of day; HVA currents in cells from OVX+E mice were lower than those in cells from OVX mice in the A.M. but were higher in the P.M. These changes were attributable to diurnal alternations in L- and N-type components. There were no diurnal changes in any aspect of HVA-mediated ICa in OVX mice. Acute in vitro treatment of cells from OVX and OVX+E mice with estradiol rapidly increased HVA currents primarily through L- and R-type VGCCs by activating estrogen receptor β and GPR30, respectively. These results suggest multiple mechanisms contribute to the overall feedback regulation of HVA-mediated ICa by estradiol. In combination with changes in synaptic inputs to GnRH neurons, these intrinsic changes in GnRH neurons may play critical roles in estradiol feedback.
Introduction
Gonadotropin-releasing hormone (GnRH) neurons form the final common pathway by which the CNS regulates fertility (Wildt et al., 1980). GnRH is released in a pulsatile pattern critical for secretion of gonadotropic hormones by the pituitary in both sexes, and in a surge mode of continuous release that triggers ovulation in females (Karsch et al., 1992). In females, these two modes of release are differentially regulated by estradiol negative and positive feedback, respectively. In rodents, estradiol interacts with a circadian signal so that the luteinizing hormone surge occurs at a specific time of day (Norman et al., 1973; Legan and Karsch, 1975; Christian et al., 2005). Although progress has been made in understanding synaptic mechanisms underlying surge generation (Miller et al., 2006; Dungan et al., 2007; Christian and Moenter, 2008b; Clarkson et al., 2008), intrinsic mechanisms are not well understood.
Calcium entry mediated by voltage-gated calcium channels (VGCCs) triggers a wide array of cellular responses ranging from muscle contraction and activation of calcium-dependent enzymes, to calcium-triggered exocytosis (Batra, 1987; Jarvis and Zamponi, 2007). High-voltage-activated (HVA) VGCCs require substantial depolarization to open, whereas low-voltage-activated (LVA) VGCCs open at more hyperpolarized potentials near the resting potential. The role of VGCCs in regulating excitability and secretion makes them candidates for altering GnRH neuron activity during different estradiol feedback states. Estrogens modulate calcium channel activity and expression in muscle and other neurons via direct and indirect mechanisms (Batra, 1987; Joëls and Karst, 1995; Johnson et al., 1997; Patterson et al., 1998; Kurata et al., 2001; Lee et al., 2002; Ullrich et al., 2007; Sarkar et al., 2008). There are few studies of VGCCs in GnRH neurons and fewer that consider changes in current through these channels with physiological state. Calcium currents in GnRH neurons have been studied in mouse embryonic olfactory placode cultures (Kusano et al., 1995), immortalized GnRH neurons (GT1-7 cells) (Bosma, 1993; Hiruma et al., 1997; Van Goor et al., 1999, 2000; Krsmanović et al., 2001; Watanabe et al., 2004), acutely dissociated GnRH neurons (Kato et al., 2003; Nunemaker et al., 2003b), and teleost and mouse brain slices (Haneda and Oka, 2004; Spergel, 2007). The current subtypes detected were dependent on both species and developmental stage. Although other studies suggest calcium channels are important for GnRH neuron function (Bourguignon et al., 1987; Krsmanović et al., 1992; Giri and Kaufman, 1994; Fukushima et al., 2003) and that estradiol might modulate their activity (Temple et al., 2004; Abe et al., 2008), the physiological regulation of calcium currents in these cells has not been studied beyond gross developmental changes.
Estradiol can act via multiple mechanisms (Nilsson et al., 2001; Edwards, 2005; Woolley, 2007). To better understand the mechanism of estradiol feedback on GnRH neurons, we tested how both in vivo estradiol, in an animal model exhibiting daily switches between estradiol negative and positive feedback (Christian et al., 2005), and acutely applied in vitro estradiol alter VGCCs. The data suggest estradiol and diurnal signals converge to modulate VGCCs on GnRH neurons.
Materials and Methods
Animals.
Adult female mice (2–3 months) expressing eGFP (enhanced green fluorescent protein) (Clontech) under the control of the GnRH promoter were used to facilitate identification of GnRH neurons (Suter et al., 2000a). Mice were maintained under a 14/10 h light/dark photoperiod with Harlan 2916 chow (Harlan) and water available ad libitum. Mice were ovariectomized (OVX) under isoflurane (Abbott Laboratories) anesthesia to remove ovarian steroid feedback; some OVX mice received steroid implants containing 0.625 μg of 17β-estradiol in sesame oil (OVX+E), which produce physiological levels of estradiol in the circulation as previously described (Christian et al., 2005). 17β-Estradiol was administered in vivo and was not present in any recording solutions except for specific studies of the rapid effects of estradiol, in which case the steroid was applied acutely in vitro as described below. Postoperative analgesia was provided by a long-acting local anesthetic (0.25% bupivicaine; 7.5 μl/site; Abbott Laboratories). Recordings were made 2–5 d after surgery and steroid replacement. All animal procedures were approved by the University of Virginia Animal Care and Use Committee.
Brain slice preparation.
All chemicals were obtained from Sigma-Aldrich unless noted. Brain slices were prepared as previously described (Nunemaker et al., 2002; Chu and Moenter, 2005). Briefly, mice were killed at times that corresponded to negative feedback (8:30–9:30 A.M.; referred to as A.M.) or surge peak (positive feedback; 1:30–2:30 P.M.; referred to as P.M.) in estradiol-treated animals. The brain was rapidly removed and placed in ice-cold high-sucrose saline solution containing the following (in mm): 250 sucrose, 3.5 KCl, 26 NaHCO3, 10 d-glucose, 1.25 Na2HPO4, 1.2 MgSO4, 2.5 MgCl2. Coronal (300 μm) slices were cut with a Vibratome 3000 (Ted Pella). Slices were incubated for 30 min at 30–32°C in 50% high-sucrose saline and 50% artificial CSF (ACSF) solution, containing the following (in mm): 135 NaCl, 3.5 KCl, 26 NaHCO3, 10 d-glucose, 1.25 Na2HPO4, 1.2 MgSO4, 2.5 CaCl2, pH 7.4. Slices were then transferred to 100% ACSF solution at room temperature (∼21–23°C) for 0.5–2.5 h. For recording, slices were placed in a recording chamber on the stage of an Olympus BX51WI upright fluorescent microscope and continuously superfused at 5–6 ml/min with oxygenated ACSF at 25 or 32°C as described below. Slices were stabilized in the chamber for ≥5 min before recording.
Electrophysiological recording.
GFP-GnRH neurons in the preoptic area were identified by brief illumination at 470 nm. Macroscopic Ca2+ currents from GnRH neurons were recorded using the whole-cell configuration of the patch-clamp technique. Patch pipettes (2.5–3.5 MΩ) were drawn from borosilicate glass capillaries (1.65 mm outer diameter; 1.12 mm inner diameter; World Precision Instruments) using a Sutter P97 pipette puller. Electrode capacitance was electronically compensated. Liquid junction potential (less than −4 mV for voltage clamp and approximately −13 mV for current clamp) was not corrected (Barry, 1994). Currents and voltages were recorded with an Axopatch-700B amplifier (Molecular Devices) and filtered at 10 kHz. Voltage and current command pulses were generated using pCLAMP9.2 software (Molecular Devices). Neuron membrane potential was held at −60 mV between protocols during voltage-clamp recordings. During whole-cell recording, input resistance (Rin), series resistance (Rs), and membrane capacitance (Cm) were continually monitored. Only recordings with Rin > 500 MΩ, Rs < 20 MΩ, and Cm > 10 pF, and holding current between 0 and −50 pA were included for analysis. There were no differences among groups in any passive recording properties or series resistance attributable to steroid treatment or time of day or response to treatment. Cells with bad clamping (identified as sluggish, incomplete current response to pulse protocol, and/or 10–90% rise time > 2.5 ms) were discarded. Recordings were performed from 1 to 3 h after preparation of brain slices was complete. No more than four cells per animal were recorded. Recorded cells were mapped to an atlas (Paxinos and Franklin, 2001) to determine whether any trends based on anatomical location emerged; no such trends were apparent in these data sets (data not shown).
Drugs and solutions.
For voltage-clamp recording of ICa, the bath solution consisted of the following (in mm): 120 NaCl, 10 glucose, 26 NaHCO3, 1.25 Na2HPO4, 1.2 MgSO4, 2.5 CaCl2, 5 4-aminopyridine (4AP), 5 CsCl, 10 tetraethylammonium (TEA)-Cl, and 0.0005 TTX, pH 7.4 (gassed with 95% O2 and 5% CO2); the pipette solution contained the following (in mm): 120 Cs-gluconate, 10 HEPES, 10 EGTA, 0.5 CaCl2, 4 Mg-ATP, 0.4 NaGTP, and 20 TEA-Cl, pH 7.3 (titrated with CsOH, 310 mOsm). For current-clamp recording, ACSF was used for bath solution; the pipette solution contained the following (in mm): 125 K-gluconate, 20 KCl, 10 HEPES, 5 EGTA, 4.0 MgATP, 0.4 NaGTP, 0.1 CaCl2, pH 7.3, 290 mOsm. Bicuculline (20 μm), APV [d-(−)-2-amino-5-phosphonovaleric acid] (20 μm), and CNQX (6-cyano-7-nitroquinoxaline) (10 μm) were included in the bath solution to block ionotropic GABAergic and glutamatergic currents. Nitrendipine (50 μm), agatoxin IVA (166 nm), conotoxin GVIA (700 nm), estradiol 17β (100 pm to 100 nm), estradiol 17α (100 nm), the estrogen α receptor agonist 1,3,5-tris(4-hydroxyphenyl)-4-propyl-1H-pyrazole (PPT) (10 nm), β receptor agonist 2,3-bis(4-hydroxyphenyl)propionitrile (DPN) (10 nm), the estrogen receptor (ER) antagonist 7α,17β-[9[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol (ICI 182780) (1 μm), GPR30 agonist G1 (100 nm), cadmium chloride (200 μm), and nickel chloride (100 μm or 1 mm) were bath applied; SNX-482 (1 μm) was locally applied by pressure micropipette.
Voltage-clamp protocols.
All currents were corrected for leak and capacitive currents on-line by a P/-6 protocol. To generate Ca2+ channel current–voltage (I–V) activation curves, currents were elicited by a voltage protocol of a 250 ms prepulse at −120 mV to remove inactivation, followed by current measurement at test potentials (250 ms) from −80 to +60 mV at 10 mV increments. To determine the steady-state inactivation of calcium currents, the membrane potential was initially hyperpolarized to −100 mV for 500 ms to remove inactivation, followed by a 1 s prepulse of −100 to 10 mV in 10 mV increments, and then a test pulse of 10 mV for 500 ms; current was quantified during the test pulse. Both of these protocols were done at 32°C and at 25°C in separate slices to determine the effect of temperature. Several different voltage protocols were used to examine LVA currents, which were recorded at 32°C to maximize amplitude. To isolate LVA currents by subtraction, separate voltage protocols of a 250 ms prepulse at either −100 or −50 mV, were followed by test potentials (250 ms) from −80 to +60 mV at 10 mV increments. To isolate LVA currents by selective activation, a 1 s prepulse at −100 mV was followed by a 250 ms test pulse at −50 mV. To examine tail currents, channels were activated by a step from prepulse at −100 mV (250 ms) to 10 mV for 10 ms, followed by a step to −60 mV for determination of tail current kinetics.
To examine subtypes of HVA currents contributing to the macroscopic current and the effects of in vivo and in vitro estradiol treatment on these currents, recordings were done at 25°C to minimize rundown. After 3–5 min stabilization, a voltage protocol consisting of a prepulse at −100 mV for 250 ms followed by a step for 250 ms to +10 mV (peak of IV response curve) was repeated every 30 s to determine peak and sustained current. These measures were made during a 5 min control period, followed by a 10–15 min bath application of specific VGCC blockers, vehicle, 17α-estradiol, 17β-estradiol, or estradiol receptor antagonists and agonists, followed by a 10 min washout period. Nitredipine was applied for 20 min with no washout.
Analysis.
The peak amplitude and the sustained amplitude 200 ms after the beginning of test potential were calculated. Current waveforms were fitted with the Clampfit program (Molecular Devices) or GraphPad Prism program (GraphPad Software). The voltage dependencies of activation and steady-state inactivation were described with a single Boltzmann distribution: I(V) = Imax/(1 + exp[(V1/2 − V)/k]), where Imax is the maximal current elicited, V1/2 is the half-maximal voltage, and k is the voltage dependence (slope) of the distribution. The time course of tail current was fit by two exponentials function with Igor Pro (Wavemetrics). For all current–voltage (I–V) curves and steady-state inactivation curves, fitted values were typically reported with 95% linear confidence limits.
To isolate LVA currents, the current recorded after a −50 mV prepulse was subtracted from that recorded after the −100 mV prepulse, or repeated 50 times to reduce noise level with the 1 s −100 mV prepulse protocol. To isolate different subtype VGCCs, the first 5 min of recording under control conditions from each cell was fitted to determine the rundown rate; average effects of blockers were subtracted from the rundown rate at 11–15 min (in vivo) or 16–20 min (in vitro) after applications from the theoretical rundown line. The R-type component (in vivo) was corrected for a small (6–7%) artifact that was observed in the macroscopic current after pressure applying bath solution. Control ICa variations was <4% 15–20 min into the recording. Cells were considered to respond to estradiol if ICa changed by ≥20%.
Parametric and nonparametric analyses were performed in Prism as dictated by data distribution. Statistical comparisons of in vivo treatments were done with ANOVA followed by Bonferroni's post hoc test. Two-tailed paired comparisons of current before and after in vitro treatments were done with each cell serving as its own control. Statistical significance was set at p < 0.05. The data shown represent mean ± SEM.
Results
Adult GnRH neurons in brain slice express a small LVA macroscopic Ca current, which is not affected by estradiol in vivo
Electrophysiological assessment of calcium currents in various GnRH neuronal model systems has led to different conclusions as to the expression of LVA versus HVA currents. We thus first characterized the macroscopic calcium current in acutely prepared brain slices from adult OVX mice. Under our recording conditions, all GnRH neurons displayed prominent HVA currents, activating positive to a membrane potential of −40 mV, and reaching maximum amplitude around +10 mV (Fig. 1A,C,D); this current was blocked with the nonspecific Ca channel blocker cadmium (data not shown). However, none of the 121 neurons recorded from OVX mice had measurable currents activating negative to −40 mV using this protocol. In contrast, non-GnRH neurons recorded in the same slices (n = 17) displayed prominent currents activated at −60 mV (Fig. 1B–D) (n = 17), which were rapidly inactivated as is characteristic of T-type LVA currents. Thus, prominent LVA currents could be detected by this protocol under these recording conditions.
Figure 1.
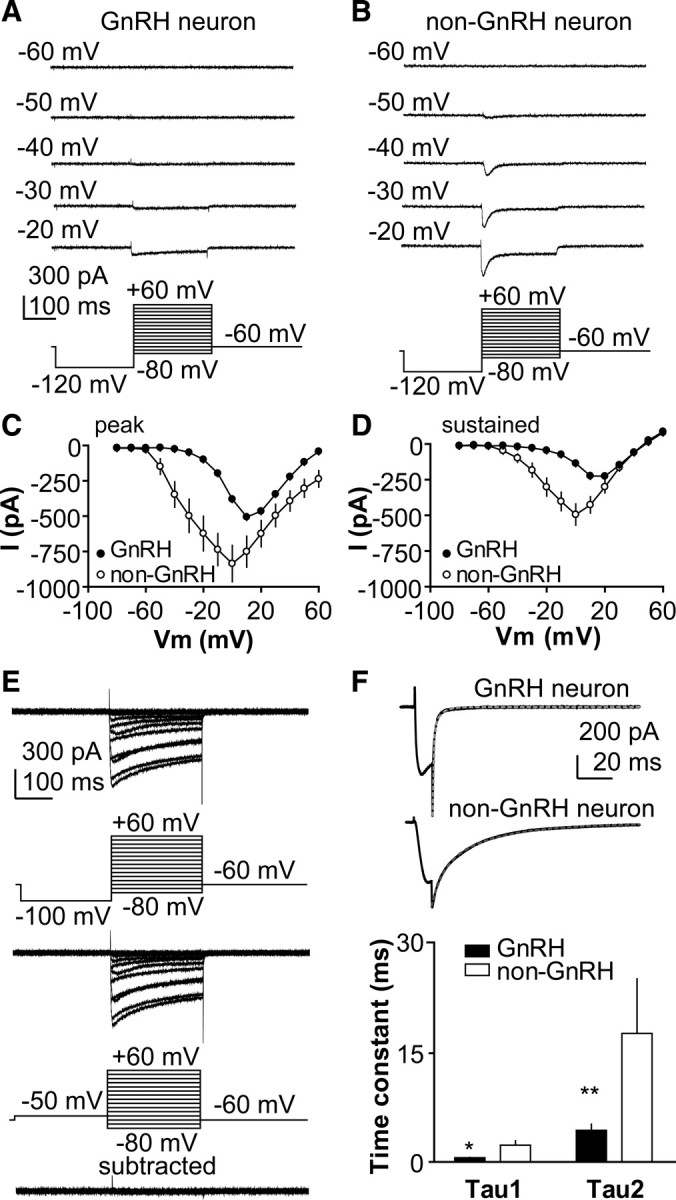
Adult GnRH neurons exhibit limited LVA calcium current. A, B, Representative of calcium current recorded at different membrane potentials in GnRH and non-GnRH neuron, respectively. C, D, Average current–voltage curves of peak and sustained current from GnRH and non-GnRH neurons. E, Subtraction of calcium current recorded with a −50 mV prepulse from current recorded with a −100 mV prepulse does not reveal LVA-mediated current. F, Representative tail current and time course of tail current (τ1 and τ2) from GnRH neuron (top) and non-GnRH neuron (middle), and mean ± SEM time constants (bottom). *p < 0.05; **p < 0.01.
We next attempted to reveal LVA-mediated currents in GnRH neurons using a subtraction protocol. Current recorded after a −50 mV prepulse, which should inactivate LVA channels, was subtracted from current recorded after a −100 mV prepulse, which should remove inactivation from LVA channels. Again, minimal LVA current is observed in GnRH neurons using this approach (Fig. 1E) (n = 8). We next examined tail currents, which are of longer duration for LVA than HVA channels. Tail currents in GnRH neurons were of short duration (τ1, 0.56 ± 0.05 ms; τ2, 4.31 ± 0.84 ms; n = 12), consistent with a predominance of HVA channels. In contrast, tail currents in random non-GnRH neurons were prolonged (τ1, 2.28 ± 0.60 ms, n = 17, p < 0.05; τ2, 17.54 ± 2.79 ms, n = 17, p < 0.01) (Fig. 1F), indicating the presence of LVA channels.
Finally, we used averaging to increase the signal-to-noise ratio. A prolonged (1 s) prepulse at −100 mV was given to provide a strong signal to remove inactivation, and then the membrane potential was stepped to −50 mV; this protocol was repeated 50 times at 30 s intervals and the resulting current traces averaged. Under these conditions, 41% of GnRH neurons exhibit a small amplitude LVA-mediated current (6.5 ± 0.2 pA in cells from OVX mice; n = 15) (Fig. 2A,B) that was blocked by 100 μm Ni2+ (n = 13 cells) (Fig. 2A). It is possible that LVA currents activated by this protocol or other protocols would be masked by large outward A-type potassium currents that are typical of GnRH neurons (DeFazio and Moenter, 2002; Zhang et al., 2007, 2009b). Neither the percentage of GnRH neurons exhibiting LVA current nor amplitude of this current was altered by the A-type potassium current blocker 4AP (n = 7) (data not shown), suggesting this compound did not contribute to the failure to detect LVA current in the previous attempts. Cadmium (200 μm) reduced the percentage of GnRH neurons exhibiting this current but did not eliminate it (n = 10) (data not shown). The ability of cadmium to block channels is voltage dependent, with blocking ability decreasing at more hyperpolarized potentials (Swandulla and Armstrong, 1989). Together, these data suggest a subpopulation of adult GnRH neuron expresses small LVA currents as measurable by recordings at the cell soma.
Figure 2.

LVA-mediated currents function in GnRH neurons but are not altered in a diurnal model of estradiol positive and negative feedback. A, Representative average of 50 repeats of a voltage protocol to reveal LVA current (top) and its blockade by Ni2+. B, LVA-mediated current does not change with estradiol or time of day in this model. Error bars indicate SEM. C–G, Differentiation of fast and slow rebound potential and current in GnRH neurons. C, D, Representative current-clamp recordings of the slow (C) and fast (D) rebound potential generated by termination of a 50 pA hyperpolarizing current injection. E, ZD7288 (right) inhibits the slow but not the fast rebound potential (p < 0.05). F, Voltage-clamp recording of a cell exhibiting both fast and slow rebound current. Cell was stepped from a holding potential of −50 mV to the potentials indicated on the left for 1.2 s, and then returned to −50 mV. G, Voltage dependence of the slow (open symbols) and fast (closed symbols) rebound current. H, Representative current-clamp recording of GnRH neuron with a resting potential near the average and that did not exhibit rebound depolarization. I, Representative example of a GnRH neuron with a depolarized resting potential that did not exhibit rebound depolarization. J, Left, Representative current-clamp recording of a GnRH neuron with a depolarized resting potential that exhibited a rebound potential after termination of hyperpolarizing current injections. In this example, the rebound potential contributes to action potential generation, which is blocked by TTX (right). K, Nickel (100 μm) completely blocked the rebound potential in GnRH neurons. L, Depolarizing GnRH neurons (right) that exhibit hyperpolarized resting membrane potential does not induce rebound potential.
To study the effect of estradiol on LVA channels, we compared the current measured using the averaging protocol (Fig. 2A) among GnRH neurons from OVX and OVX+E mice recorded at different times of day (Fig. 2B). The peak amplitude of LVA current was not different among groups (OVX A.M., n = 7; OVX P.M., n = 8; OVX+E A.M., n = 10; OVX+E P.M., n = 10; p > 0.1). These data suggest estradiol in vivo does not alter LVA-mediated currents in GnRH neurons in this model of steroid replacement.
GnRH neurons typically have input resistance near 1 GΩ; thus, even relatively small currents such as the LVA currents measured here can impact the physiology of these cells. To assess whether LVA-mediated current contributes to GnRH neuron activity, we used current clamp to investigate the rebound in membrane potential after hyperpolarization. Rebound depolarizations can be generated by activation of either LVA-mediated or hyperpolarization-activated nonspecific cation (HCN)-mediated currents (Ih) during a preceding membrane hyperpolarization. To differentiate between rebound potentials mediated by LVA versus HCN channels, latency to peak of the response and sensitivity to the Ih blocker 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288) (50 μm) were determined. After hyperpolarization in current clamp, GnRH neurons exhibit two types of rebound potentials, slow (Fig. 2C) and fast (Fig. 2D). ZD7288 has no effect on the fast rebound but eliminates the slow rebound (n = 11, p < 0.05) (Fig. 2C–E). In voltage clamp, a greater degree of hyperpolarization is required to activate the slow current (latency to peak of current, 97.4 ± 4.2 ms) on return to −50 mV than the fast current (latency to peak of current, 22.5 ± 2.8 ms) (Fig. 2F,G). Furthermore, the activation of the fast current reaches a plateau by approximately −100 mV, whereas greater hyperpolarization further increases the slow current. Finally, ∼50% of cells exhibit the slow current, which is blocked by the specific Ih blocker ZD7288 (Z. Chu, H. Takagi, and S. M. Moenter, unpublished data), and thus distinguished from the LVA-mediated rebound discussed below. Together, these data demonstrate that we can distinguish between fast and slow rebound currents that are likely mediated by LVA and HCN channels, respectively.
To reveal rebound potentials, hyperpolarizing current steps (10–40 pA; 1 s) were injected, and depolarization relative to initial membrane potential (“rebound”) was quantified. A total of 73 cells with an average resting potential (Vrest) of −57.6 ± 1.0 mV was examined. Most cells (n = 55) exhibited Vrest that was more hyperpolarized than −50 mV and none of these cells exhibited a depolarizing rebound of membrane potential after termination of a hyperpolarizing current injection (Fig. 2H). A smaller subpopulation exhibited Vrest that was depolarized to −50 mV (46.3 ± 1.4 mV; n = 18). Of these depolarized cells, approximately one-half (8 of 18) were similar to the more hyperpolarized cells as they exhibited no rebound depolarization (Fig. 2I). In contrast, the other one-half (10 of 18 cells) exhibited a rebound depolarization (10.4 ± 1.0 mV) after a 40 pA hyperpolarizing current injection (bringing membrane potential to −85.2 ± 1.0 mV) that could result in action potential generation that was blocked by TTX (Fig. 2J). The time to peak of the rebound potential from the end of hyperpolarizing current injection was 110.7 ± 2.4 ms, and the decay time was 130.1 ± 5.5 ms. This rebound depolarization was blocked by Ni2+ (100 μm), suggesting it may be mediated by LVA channels (Fig. 2K). Hyperpolarization of these cells to −54.0 ± 0.8 mV by DC current injection (5 pA) reduced the amplitude of the rebound (4.9 ± 0.6 mV; n = 6; p < 0.05) (data not shown). In contrast, depolarization of the majority of GnRH neurons that exhibited more hyperpolarized Vrest to 43.1 ± 1.3 mV did not generate a rebound depolarization (n = 20) (Fig. 2L). These results suggest that, in GnRH neurons, rebound current is dependent on the resting membrane potential, and LVA can contribute to the function of a subpopulation of GnRH neurons.
Estradiol alters HVA-mediated current of GnRH neurons in a diurnal manner
To study whether changes in calcium current (ICa) mediated via HVA channels are altered by estradiol feedback, ICa was recorded from GnRH neurons in slices made from OVX and OVX+E mice during the time of negative (A.M.) or positive (P.M.) feedback. Recordings were made at 32°C, near the physiological temperature. In OVX mice, neither peak nor sustained components of the HVA-mediated current were altered by time of day (Fig. 3A,B) (A.M., n = 16; P.M., n = 16). In contrast, peak and sustained ICa density varied with time of day in cells from OVX+E mice, being significantly higher in the P.M. (n = 17) than in the A.M. (n = 17) at most membrane test potentials between −10 and +50 mV (p < 0.01). Furthermore, the peak ICa current density of GnRH neurons from OVX+E mice was lower than that from OVX mice in the A.M., when estradiol exerts negative feedback (membrane potentials from −10 to +40 mV; p < 0.01; n = 16 OVX cells from 4 mice; n = 17 OVX+E cells from 5 mice), and was higher than that from OVX mice in the P.M., when estradiol exerts positive feedback (membrane potentials, 0 and 10 mV; p < 0.05; n = 16 OVX cells from 4 mice; n = 17 OVX+E cells from 5 mice). The activation (Fig. 3C,E), steady-state inactivation (Fig. 3D,F), rise time (10–90%) (Fig. 3G), and membrane capacitance (data not shown) of GnRH neurons did not vary among the four groups. Interestingly, decay time (90–50%) (Fig. 3H) of GnRH neurons from OVX+E mice was higher in P.M. than in A.M. (p < 0.05); the prolonged current could contribute to increased excitability of GnRH neurons at this time. These results suggest that estradiol modulates HVA currents in GnRH neurons in a diurnal manner consistent with previously observed changes in GnRH neuronal activity (Christian et al., 2005).
Figure 3.
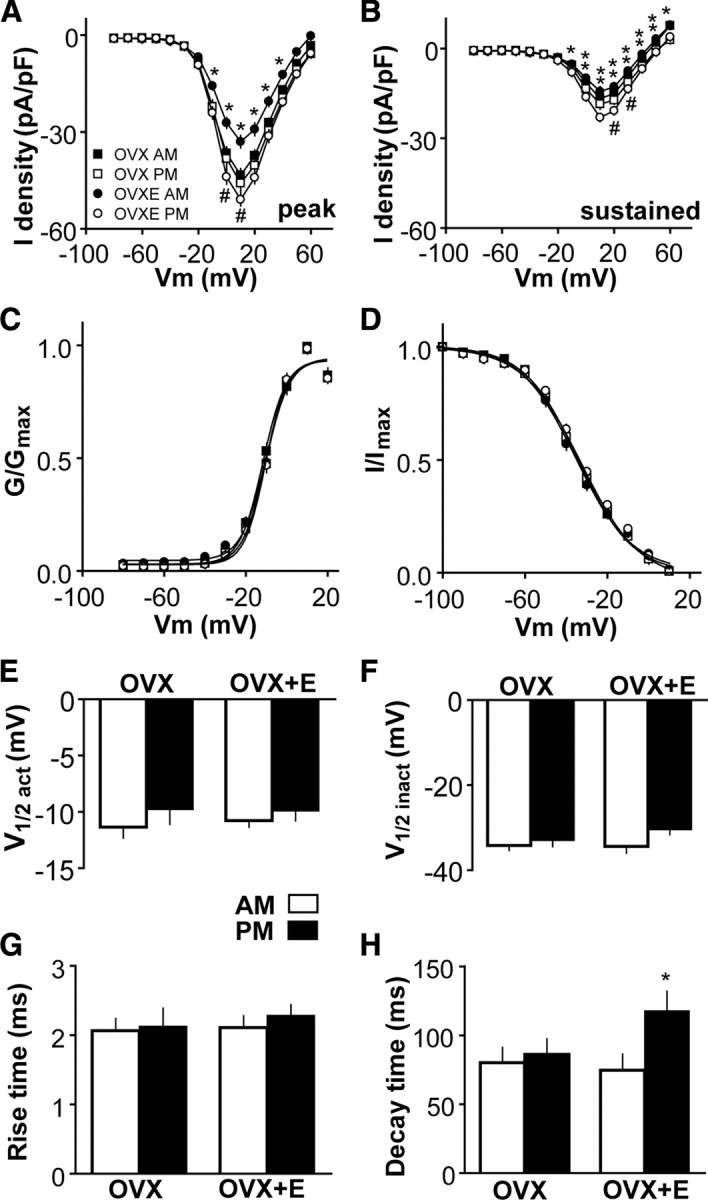
HVA-mediated ICa in GnRH neurons is modulated in an estradiol-dependent diurnal manner. A, B, Current–voltage plots of peak and sustained current density from OVX and OVX+E mice in the A.M. and P.M. [*p < 0.05, OVX vs OVX+E in the A.M.; #p < 0.05, OVX vs OVX+E in the P.M.; **p < 0.01, OVX+E A.M. vs OVX+E P.M. (not marked in A as all values are significant)]. C, D, Activation and steady-state inactivation curves from OVX and OVX+E mice in the A.M. and P.M. E–H, Mean ± SEM V1/2act (E), V1/2inact (F), rise time (10–90%) (G), and decay time (90–50%) (H) from OVX and OVX+E mice in the A.M. and P.M.
Estradiol modulates specific subtypes of HVA in GnRH neurons
We next determined the subtypes of HVA altered by in vivo estradiol feedback. It is important in these longer duration recordings to minimize rundown, which we accomplished by recording at room temperature rather than more physiological temperatures. Most electrophysiological studies of VGCCs are done at room temperature. However, recent reports suggest temperature can substantially modify ICa in cardiac myocytes (Tsien et al., 1987), neurons (Acerbo and Nobile, 1994), muscle (Klöckner et al., 1990), and pituitary cells (Rosen, 1996). We thus first compared the properties of HVA-mediated currents at 25°C (OVX A.M., n = 58 cells from 15 animals; OVX P.M., n = 44 cells from 14 animals; OVX+E A.M., n = 57 cells from 16 animals; OVX+E P.M., n = 44 cells from 15 animals) with measures made at 32°C (Fig. 3). Averaged traces of ICa recorded during a step to +10 mV (peak activation) from −100 mV are shown in Figure 4A–C for cells from OVX mice, OVX+E mice in the A.M., and OVX+E mice in the P.M., respectively. In all models, recording at the lower room temperature reduced peak and sustained current amplitude (Fig. 4D) and current density (data not shown). A small but significant (p < 0.05) difference appeared between cells from OVX and OVX+E mice in V1/2act; at 25°C, estradiol depolarized this potential but had no effect at 32°C (Fig. 4E). Because this effect disappeared as physiological temperatures were approached, it is likely of little impact on GnRH neurons in vivo. Importantly, the membrane potential at which current amplitude was maximum (data not shown), and the main effects of estradiol to reduce macroscopic HVA-current in the A.M. during negative feedback relative to that in cells from OVX mice and to increase current in the P.M. during positive feedback were reproduced at 25°C (p < 0.05 for both at 10 mV step) (Fig. 4D) (p < 0.05).
Figure 4.

Temperature alters decay time and peak current but does not alter in vivo estradiol-dependent diurnal modulation of calcium current. A–C, Average maximal current (at +10 mV) curve of cells from OVX mice, OVX+E mice in A.M. and OVX+E mice in P.M., respectively, at 25 and 32°C (gray) and 25°C scaled to compare kinetics (scaled 25). D, The amplitude of ICa is greater at 32°C than 25°C in cells from both OVX and OVX+E mice, but estradiol-dependent effects persist at both temperatures. E, F, Mean ± SEM effects of temperature on V1/2act and V1/2inact in cells from OVX and OVX+E mice in the A.M. and (*p < 0.05, OVX vs OVX+E; #p < 0.05, OVX+E A.M. vs OVX+E P.M.). G, H, Mean ± SEM effects of temperature on rise time (10–90%) and decay time (90–50%) in cells from OVX and OVX+E mice in the A.M. and P.M. (*p < 0.05, OVX vs OVX+E; #p < 0.05, OVX+E A.M. vs OVX+E P.M.).
We thus made recordings at 25°C to study which subtypes of VGCCs are modulated by estradiol using a pharmacological approach with specific channel blockers. To minimize recording duration and further reduce rundown, current was recorded only at +10 mV test pulse; membrane potential was held at −60 mV between trials, which consisted of a −100 mV prepulse for 250 ms and followed by a test pulse at +10 mV for 250 ms at 30 s intervals. The current was stabilized within ∼5 min of achieving the whole-cell configuration, after which it ran down ∼30% over 30 min (Fig. 5A). A representative experiment using SNX-482 to block R-type channels is shown in Figure 5B. Response to each blocker was calculated from the linear fit of rundown as shown. In cells from OVX mice (Fig. 5C,D, left), the peak and sustained components of macroscopic ICa contributed by channel subtypes were not different between A.M. and P.M. (from 28 animals; nitrendipine, L-type antagonist, n = 7 in A.M., n = 6 in P.M.), conotoxin GVIA (N-type antagonist, n = 6 each in A.M. and P.M.), agatoxin IVA (P/Q-type antagonist, n = 6 each in A.M. and P.M.), and SNX-482 (R-type antagonist, n = 8 in A.M., n = 7 in P.M.). In contrast, in cells from OVX+E mice (Fig. 5C,D, right), the peak and sustained components of macroscopic ICa conducted via L- and N-type channels were greater in the P.M. during positive feedback than in the A.M. during negative feedback (p < 0.05; n = 6 in both A.M. and P.M. for both channel subtypes). There was no difference in the proportion of current conducted via P/Q- (n = 6 in both A.M. and P.M.) or R-type (n = 9 in A.M., n = 8 in P.M.) channels between A.M. and P.M. These results suggest in vivo treatment with estradiol modulates specific subtypes of VGCCs in GnRH neurons.
Figure 5.

In vivo treatment with estradiol modulates specific subtypes of HVA calcium channels. A, Example of rundown of ICa under control conditions. B, SNX-482 was locally applied for 10 min after a 5 min control period. The proportion of HVA subtype blocked by specific agents was assessed by the linear fit of the rundown during the first 5 min of control recording. C, D, The proportions of peak and sustained current of different subtype HVA from OVX and OVX+E mice in the A.M. and P.M. (*p < 0.05 A.M. vs P.M. in OVX+E mice). Error bars indicate SEM.
Rapid effects of estradiol on ICa in GnRH neurons
There is increasing evidence that estradiol can also rapidly modulate neuronal function by changing ion channel activity (Mermelstein et al., 1996; Lee et al., 2002; Ullrich et al., 2007; Zhao and Brinton, 2007; Sarkar et al., 2008), including in GnRH neurons (Temple et al., 2004; Abe and Terasawa, 2005; Abe et al., 2008; Romanò et al., 2008; Chu et al., 2009). We thus studied whether there are rapid, nongenomic effects of estradiol on HVA-mediated currents in GnRH neurons. We used a similar design to that used for the study of HVA subtypes above (Fig. 5). After stabilization of ICa for 5–10 min, and 5 min of control recording, 17β-estradiol was bath-applied for 15 min, followed by washout for 10 min. 17β-Estradiol rapidly (within 5 min) and reversibly (washout within 5 min) potentiated ICa in both A.M. and P.M.; there was no difference based on time of day, and data were pooled for analysis (Fig. 6A,B). Cells were classified as responding if there was a ≥20% change in peak ICa. Vehicle had no effect (0.1% ethanol; n = 8) (Fig. 6C). The percentage of responsive neurons increased with increasing estradiol concentration (100 pm, n = 13; 1 nm, n = 14 cells; 100 nm, n = 6 cells) (Fig. 6C). The stereoisomer 17α (100 nm) elicited a 20% change in peak ICa in only one cell, indicating stereo-specificity (Mermelstein et al., 1996; Lee et al., 2002) and suggesting the increase in response was not attributable to changes in membrane fluidity induced by intercolation of estradiol into the membrane (data not shown) (n = 6). Although the percentage of responding cells increased with greater estradiol concentrations, the percentage change in ICa did not vary with concentration of estradiol (range, 20–24% increase in peak current, 32–37% increase in sustained current). This suggests the ability of GnRH neurons to respond to acutely applied estradiol may be “all or none” (the exception to this is the response to G1 discussed below). Because the percentage of cells responding to estradiol is the primary factor affected, we focused on this measure.
Figure 6.

In vitro treatment with estradiol rapidly increases ICa through classical receptor ERβ and GPR30. A, Representative traces show estradiol (1 nm; gray) rapidly increases ICa in GnRH neurons from OVX mice in both the A.M. and P.M. B, Time course of the rapid effect of estradiol (1 nm) on ICa. C, The percentage of cells responding to estradiol with a rapid change in ICa. Black bars, Cells from OVX mice treated with vehicle or estradiol 17β; white bar, cells from OVX+E mice treated with 1 nm estradiol 17β; gray bars, cells from OVX mice treated with specific receptor agonists and antagonists.
The above studies were done in OVX mice, raising the question of whether or not acutely applied estradiol alters ICa in cells that were recently exposed to estradiol in vivo. To test this, we repeated aspects of the above study and found that 1 nm estradiol has a similar effect in GnRH neurons in cells from OVX+E mice (n = 10) (Fig. 6C) as that observed in slices from OVX mice in terms of percentage of cells responding, magnitude of response, and the lack of effect of time of day on the response.
We next investigated the type of receptor mediating the rapid potentiation of ICa in GnRH neurons (Fig. 6C). The effect of estradiol was mostly mimicked by the ERβ agonist DPN (10 nm; n = 10), but not by the ERα agonist PPT (10 nm; n = 9). Interestingly, the classical receptor antagonist ICI 182780 (1 μm) reduced (from 57 to 38%) but did not eliminate the ability to respond to 1 nm estradiol (n = 10 cells), suggesting a second pathway may exist in addition to ERβ. ICI itself had no effect on ICa (n = 6) (data not shown). With regard to alternative pathways, we studied the effect of G1, an agonist for the putative G-protein-coupled estrogen receptor GPR30 (Prossnitz et al., 2008). G1 (100 nm) increased ICa in 33% of cells (n = 4 of 12 cells), similar to the percentage that responded to 17β-estradiol in the presence of ICI 182780. Of note, the increase in ICa in response to G1 was more robust than for estradiol, suggesting it may be a superagonist or have additional targets not related to estradiol signaling. These data suggest that 17β-estradiol increases ICa through classical estradiol receptor ERβ and membrane GPR30.
We next examined the subtypes of HVA channels that respond to acutely applied 17β-estradiol in GnRH neurons (Fig. 7A). Preincubation with the L-type VGCC blocker nitrendipine (50 μm) reduced the percentage of cells responding to 1 nm 17β-estradiol (n = 10 cells). Addition of the N-type VGCC blocker conotoxin GIVA (1 μm) was not different from nitrendipine itself (n = 7 cells), suggesting 17β-estradiol has no acute effect on N-type VGCCs. Furthermore, nitrendipine blocked the ability of DPN to increase ICa in GnRH neurons (n = 9 cells). These data suggest that 17β-estradiol potentiates L-type VGCCs through ERβ. Next, we studied the subtypes of HVA activated by G1 in OVX mice. Since 17β-estradiol has no acute effect on N-type VGCCs (Fig. 7A), we tried to block the effects of G1 with L-, P/Q-, and R-type VGCCs blockers. Nitrendipine (n = 11 cells), SNX-482 (1 μm; n = 10 cells), and agatoxin IVA (200 μm; n = 10) did not change the percentage of GnRH neurons responding to G1 (Fig. 7B). Interestingly, unlike other agents tested, in which the percentage increase in ICa was consistently in the same range, the R-type blocker SNX-482 reduced the percentage increase in ICa that was induced by G1 (Fig. 7C, percentage increase shown relative to control values so that no change in ICa is shown as 0%) (n = 4 of 12 responding cells; p < 0.05). Because the response to G1 was greater than that to estradiol, we confirmed no current was induced by G1 in the presence of cadmium (400 μm) (Fig. 7B) (n = 8), indicating the G1-activated current was indeed mediated by calcium channels or calcium entry-sensitive mechanisms. These data suggest that 17β-estradiol potentiates R-type VGCCs through GPR30.
Figure 7.

In vitro treatment with estradiol potentiates different subtypes of HVA through different receptors. A, Nitrendipine (50 μm) decreased the percentage of cells responding to estradiol (1 nm). Additional treatment with conotoxin GVIA (700 nm) did not change percentage of cells responding. Nitrendipine (50 μm) blocked the effect of DPN (10 nm). B, Nitrendipine (50 μm), SNX (1 μm), or agatoxin IVA (166 nm) did not block the effect of G1 (100 nm). SNX-482 (1 μm) did not change the percentage of cells responding to G1 (100 nm). C, SNX-482 decreased the percentage increase in ICa in response to G1 (*p < 0.05; 0% indicates no difference from control conditions). D, The combined application of nitrendipine and SNX-482 blocked the effect of 17β-estradiol in both OVX and OVX+E mice. Error bars indicate SEM.
Finally, we applied both nitrendipine and SNX-482 to brain slices from OVX and OVX+E mice to confirm the above results. In both animal models, the combination of nitrendipine and SNX-482 essentially blocked the effect of 17β-estradiol with both the percentage of responding cells and the percentage change in current reduced below 10% (n = 10 OVX; n = 11 OVX+E) (Fig. 7D). Together, these data suggest 17β-estradiol rapidly potentiates ICa via multiple pathways initiated by ligand binding to ERβ and/or GPR30, and that L-type and R-type calcium channels are the respective targets of this acute modulation.
Discussion
Estradiol feedback regulation of GnRH release is a key component generating the female reproductive cycle. Hormone release and neuronal excitability are calcium dependent, and VGCCs are a main pathway for calcium influx. Here, we report estradiol modulates different HVA subtypes of VGCCs in GnRH neurons via different estrogen receptors and mechanisms.
Reports of LVA-mediated currents in GnRH neurons have varied with species, model, and developmental stage (Bosma, 1993). Several explanations may account for these differences. First, CaV3 subunits that comprise LVA channels may be expressed by only some GnRH neurons (Zhang et al., 2009b). GnRH neurons are often divided into subpopulations by both gene expression (Smith et al., 2000) and function (Christian and Moenter, 2007; Chen and Moenter, 2009). Second, localization of CaV3 message and protein can be somewhat different; this may be methodological but may indicate not all cells with abundant message translate it to similar levels of protein (McKay et al., 2006). Third, current from LVA channels in distal processes may dampen before arrival at the cell body, precluding accurate measurements at the soma. Such currents may impact on neuronal excitability; recent physiological and anatomical work suggests GnRH neurons may couple via dendrodendritic bundling (Roberts et al., 2008; Campbell et al., 2009). Fourth, LVA channels may be regulated as a critical part of steroid feedback. An estradiol replacement paradigm different from that used in the present study increased LVA-mediated T-type currents in GnRH neurons in models of both negative and positive feedback; however, increased excitability was only observed during positive feedback (Zhang et al., 2009b). That estradiol regimen involves an injection to mimic the proestrous increase, which, although more physiological with regard to pattern of steroid, makes diurnal changes difficult to interpret as they may be attributable to steroid level or time of day. Here, in a model exhibiting daily switches between estradiol negative and positive feedback in a constant estradiol milieu, no changes in LVA-mediated current with estradiol or time of day were observed, indicating the higher estradiol level is needed for this regulation.
LVA channels can play an important role in regulating firing patterns and oscillatory behavior because they require only a small depolarization from rest to open (Perez-Reyes, 2003; Molineux et al., 2006). In brain slices, individual GnRH neurons exhibit burst firing and episodic activity (Suter et al., 2000b; Kuehl-Kovarik et al., 2002; Nunemaker et al., 2003a) that could underlie the pulsatile secretion observed in vivo that is critical for fertility (Knobil et al., 1980). Although the amplitude of the current was low in the present study, LVA-mediated currents generated rebound spikes and may contribute to burst firing. GnRH neurons have high input resistance; thus, even small currents can substantially alter membrane potential (Suter et al., 2000a; Sim et al., 2001; DeFazio and Moenter, 2002; Chu and Moenter, 2006). Furthermore, cells expressing this current could play a role in initiating burst activity and communicating it to the balance of the GnRH neuronal network.
In this model, there was no effect of estradiol or time of day on LVA current. In contrast, HVA currents may play a role in mediating estradiol-dependent diurnal changes in GnRH neuron activity. Specifically, HVA-mediated current did not change with time of day in OVX mice, whereas in vivo estradiol treatment reduced HVA-mediated currents during estradiol negative feedback but increased these currents during positive feedback. These results reveal a novel mechanism for feedback modulation of GnRH neurons by estradiol in that an intrinsic property of these neurons is targeted, in addition to synaptic transmission (Mahesh et al., 1999; Han et al., 2004; Christian and Moenter, 2007, 2008a,b; Clarkson et al., 2008; Roseweir et al., 2009). Although these changes may be subsequent to alterations in estradiol-sensitive neuromodulation of GnRH neurons, for example by VIP (Harney et al., 1996; Smith et al., 2000, 2005; Christian and Moenter, 2008a) or kisspeptin (Smith et al., 2005; Pielecka-Fortuna et al., 2008) rather than estradiol action directly at the GnRH neuron, they suggest that multiple aspects of GnRH neuron physiology are targeted by the normal cyclical switch between estradiol negative and positive feedback.
The effects of in vivo estradiol on HVA-mediated currents specifically altered L- and N-type channels. L-type channels are typically found on cell bodies where they contribute to calcium-dependent gene transcription (Gomez-Ospina et al., 2006). In contrast, N-type channels are found in dendrites, soma, and nerve terminals, and their opening mediates calcium-dependent release of neurotransmitters (Westenbroek et al., 1992; Reid et al., 2003; Catterall et al., 2005).
In addition to long-term changes in cell physiology brought about by the action of estradiol as a modulator of transcription, estradiol can alter the intrinsic and synaptic physiology of neurons within minutes (Kelly et al., 1976, 1977; Mermelstein et al., 1996; Lee et al., 2002; Zhao and Brinton, 2007; Sarkar et al., 2008). In GnRH neurons, estradiol at high physiological or pharmacological levels acutely increases firing activity (Abe and Terasawa, 2005; Chu et al., 2009) and increases frequency of oscillations in intracellular calcium concentration (Temple et al., 2004; Abe et al., 2008; Romanò et al., 2008).
In the present study, estradiol rapidly (<5 min) increased HVA-mediated calcium currents in GnRH neurons regardless of time of day and regardless of the estradiol condition of the animal. Rapid estradiol effects were mimicked by the ERβ agonist DPN, consistent with the expression of ERβ in GnRH neurons (Herbison and Pape, 2001) and other rapid effects of estradiol in GnRH neurons (Abrahám et al., 2003; Chu et al., 2009). The pure classical estrogen receptor antagonist ICI 182780, however, only partially blocked the acute effects of estradiol, suggesting other receptors may participate (Toran-Allerand et al., 2002; Roepke et al., 2009). GPR30, a candidate estrogen receptor (Prossnitz et al., 2008), is expressed in primate GnRH neurons and is involved in the rapid effect of estradiol on GnRH release and intracellular calcium oscillations (Noel et al., 2009). Here, the GPR30 agonist G1 rapidly increased HVA currents in one-third of GnRH neurons, suggesting a subpopulation may express GPR30. Of interest, G1 elicited an even greater response than estradiol. One explanation is that supraphysiological levels of estradiol are needed to fully activate GPR30; such concentrations may be locally available within the brain (Corpéchot et al., 1981; Toran-Allerand et al., 2005). As the G1-induced change in current is eliminated by cadmium, the ultimate target does appear to be calcium channels, or mechanisms dependent on an influx via these channels. Acute actions of estradiol were mediated by ERβ and GPR30, which increased L- and R-type currents, respectively.
The different effects of estradiol on HVA subtypes when given in vivo versus in vitro suggest different mechanisms are involved. Importantly, because the rapid actions of estradiol appear similar in both OVX and OVX+E animals, these changes may be additive in their regulation of GnRH neurons. In this regard, the longer time course of in vivo estradiol treatment enables mechanisms including changes in message and protein levels. Estradiol-sensitive afferents may be engaged (Wintermantel et al., 2006; Heldring et al., 2007). For example, estradiol might interact with the circadian pacemaker in the suprachiasmatic nuclei, changing calcium channel function of GnRH neurons via VIP (Harney et al., 1996; Smith et al., 2000; Christian and Moenter, 2008b) or vasopressin (Palm et al., 1999). In vivo estradiol might also change HVA-mediated current through changes in signaling via metabotropic receptors for GABA or glutamate (Chu and Moenter, 2005; Dumalska et al., 2008; Zhang et al., 2009a). GABAergic (Christian and Moenter, 2007) and glutamatergic (Christian et al., 2009) transmission indeed change with time of day in this model in an estradiol-dependent manner; thus, the afferent signal is present. In contrast to in vivo estradiol, acutely applied estradiol is thought to reveal nongenomic mechanisms. Physiologically, an acute change in estradiol may come from a burst of local synthesis, but such mechanisms remain speculative (Woolley, 2007). Acute estradiol likely acts directly on the GnRH neuron itself or via rapid changes in neuromodulation. Because both action potential generation and ionotropic receptors for GABA and glutamate were blocked in the present studies, these acute effects of estradiol are likely directly on the GnRH neuron. This interpretation is supported by the lack of any diurnal changes in response to acutely applied estradiol, as opposed to the well documented time-of-day-dependent changes that occur in response to estradiol in vivo (Moenter et al., 2009). This latter observation suggests the acute estradiol signal is not interacting with the circadian pacemaker in the suprachiasmatic nucleus under these experimental conditions.
The present study shows two novel pathways by which estradiol can modulate VGCCs in GnRH neurons. The effects of both in vivo and in vitro suggest that estradiol has multiple effects on VGCCs. These data provide a new link between estradiol and GnRH neuron function. Estrogen changes intrinsic properties in addition to synaptic transmission and other neuromodulators to regulate GnRH neuron activity for female reproductive success. Determining the underlying signal pathways of estradiol modulation on VGCCs will be important to understand the central neural control of reproduction.
Footnotes
This work was supported by National Institutes of Health–Eunice Kennedy Shriver National Institute of Child Health and Human Development Grants R01 HD41469 and R01 HD34860. We thank Debra Fisher for expert technical assistance, Dr. Yi Zhang from Baylor College of Medicine for scientific advice, and Peilin Chen, Alison Roland, Pei-San Tsai, and R. Anthony DeFazio for helpful editorial comments.
References
- Abe H, Terasawa E. Firing pattern and rapid modulation of activity by estrogen in primate luteinizing hormone releasing hormone-1 neurons. Endocrinology. 2005;146:4312–4320. doi: 10.1210/en.2005-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H, Keen KL, Terasawa E. Rapid action of estrogens on intracellular calcium oscillations in primate luteinizing hormone-releasing hormone-1 neurons. Endocrinology. 2008;149:1155–1162. doi: 10.1210/en.2007-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahám IM, Han SK, Todman MG, Korach KS, Herbison AE. Estrogen receptor β mediates rapid estrogen actions on gonadotropin-releasing hormone neurons in vivo. J Neurosci. 2003;23:5771–5777. doi: 10.1523/JNEUROSCI.23-13-05771.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acerbo P, Nobile M. Temperature dependence of multiple high voltage activated Ca2+ channels in chick sensory neurones. Eur Biophys J. 1994;23:189–195. doi: 10.1007/BF01007610. [DOI] [PubMed] [Google Scholar]
- Barry PH. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J Neurosci Methods. 1994;51:107–116. doi: 10.1016/0165-0270(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Batra S. Increase by oestrogen of calcium entry and calcium channel density in uterine smooth muscle. Br J Pharmacol. 1987;92:389–392. doi: 10.1111/j.1476-5381.1987.tb11335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma MM. Ion channel properties and episodic activity in isolated immortalized gonadotropin-releasing hormone (GnRH) neurons. J Membr Biol. 1993;136:85–96. doi: 10.1007/BF00241492. [DOI] [PubMed] [Google Scholar]
- Bourguignon JP, Gerard A, Debougnoux G, Rose J, Franchimont P. Pulsatile release of gonadotropin-releasing hormone (GnRH) from the rat hypothalamus in vitro: calcium and glucose dependency and inhibition by superactive GnRH analogs. Endocrinology. 1987;121:993–999. doi: 10.1210/endo-121-3-993. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Gaidamaka G, Han SK, Herbison AE. Dendro-dendritic bundling and shared synapses between gonadotropin-releasing hormone neurons. Proc Natl Acad Sci U S A. 2009;106:10835–10840. doi: 10.1073/pnas.0903463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- Chen P, Moenter SM. GABAergic transmission to gonadotropin-releasing hormone (GnRH) neurons is regulated by GnRH in a concentration-dependent manner engaging multiple signaling pathways. J Neurosci. 2009;29:9809–9818. doi: 10.1523/JNEUROSCI.2509-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Moenter SM. Estradiol induces diurnal shifts in GABA transmission to gonadotropin-releasing hormone neurons to provide a neural signal for ovulation. J Neurosci. 2007;27:1913–1921. doi: 10.1523/JNEUROSCI.4738-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Moenter SM. Vasoactive intestinal polypeptide can excite gonadotropin-releasing hormone neurons in a manner dependent on estradiol and gated by time of day. Endocrinology. 2008a;149:3130–3136. doi: 10.1210/en.2007-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Moenter SM. Critical roles for fast synaptic transmission in mediating estradiol negative and positive feedback in the neural control of ovulation. Endocrinology. 2008b;149:5500–5508. doi: 10.1210/en.2008-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Mobley JL, Moenter SM. Diurnal and estradiol-dependent changes in gonadotropin-releasing hormone neuron firing activity. Proc Natl Acad Sci U S A. 2005;102:15682–15687. doi: 10.1073/pnas.0504270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian CA, Pielecka-Fortuna J, Moenter SM. Estradiol suppresses glutamatergic transmission to gonadotropin-releasing hormone neurons in a model of negative feedback in mice. Biol Reprod. 2009;80:1128–1135. doi: 10.1095/biolreprod.108.075077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Moenter SM. Endogenous activation of metabotropic glutamate receptors modulates GABAergic transmission to gonadotropin-releasing hormone neurons and alters their firing rate: a possible local feedback circuit. J Neurosci. 2005;25:5740–5749. doi: 10.1523/JNEUROSCI.0913-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Moenter SM. Physiologic regulation of a tetrodotoxin-sensitive sodium influx that mediates a slow afterdepolarization potential in gonadotropin-releasing hormone neurons: possible implications for the central regulation of fertility. J Neurosci. 2006;26:11961–11973. doi: 10.1523/JNEUROSCI.3171-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Andrade J, Shupnik MA, Moenter SM. Differential regulation of gonadotropin-releasing hormone neuron activity and membrane properties by acutely applied estradiol: dependence on dose and estrogen receptor subtype. J Neurosci. 2009;29:5616–5627. doi: 10.1523/JNEUROSCI.0352-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson J, d'Anglemont de Tassigny X, Moreno AS, Colledge WH, Herbison AE. Kisspeptin-GPR54 signaling is essential for preovulatory gonadotropin-releasing hormone neuron activation and the luteinizing hormone surge. J Neurosci. 2008;28:8691–8697. doi: 10.1523/JNEUROSCI.1775-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpéchot C, Robel P, Axelson M, Sjövall J, Baulieu EE. Characterization and measurement of dehydroepiandrosterone sulfate in rat brain. Proc Natl Acad Sci U S A. 1981;78:4704–4707. doi: 10.1073/pnas.78.8.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFazio RA, Moenter SM. Estradiol feedback alters potassium currents and firing properties of gonadotropin-releasing hormone neurons. Mol Endocrinol. 2002;16:2255–2265. doi: 10.1210/me.2002-0155. [DOI] [PubMed] [Google Scholar]
- Dumalska I, Wu M, Morozova E, Liu R, van den Pol A, Alreja M. Excitatory effects of the puberty-initiating peptide kisspeptin and group I metabotropic glutamate receptor agonists differentiate two distinct subpopulations of gonadotropin-releasing hormone neurons. J Neurosci. 2008;28:8003–8013. doi: 10.1523/JNEUROSCI.1225-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungan HM, Gottsch ML, Zeng H, Gragerov A, Bergmann JE, Vassilatis DK, Clifton DK, Steiner RA. The role of kisspeptin GPR54 signaling in the tonic regulation and surge release of gonadotropin-releasing hormone/luteinizing hormone. J Neurosci. 2007;27:12088–12095. doi: 10.1523/JNEUROSCI.2748-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–376. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]
- Fukushima A, Sano A, Aiba S, Kimura F. Role of Na+ and Ca2+ channels in the preoptic LH surge generating mechanism in proestrous rats. Endocr J. 2003;20:145–153. doi: 10.1507/endocrj.50.145. [DOI] [PubMed] [Google Scholar]
- Giri M, Kaufman JM. In vitro GnRH release from the isolated medial basal hypothalamus of the male guinea pig: evidence for the existence of two pools of releasable GnRH. Brain Res. 1994;648:270–280. doi: 10.1016/0006-8993(94)91127-4. [DOI] [PubMed] [Google Scholar]
- Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel CaV1.2 encodes a transcription factor. Cell. 2006;127:591–606. doi: 10.1016/j.cell.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Todman MG, Herbison AE. Endogenous GABA release inhibits the firing of adult gonadotropin-releasing hormone neurons. Endocrinology. 2004;145:495–499. doi: 10.1210/en.2003-1333. [DOI] [PubMed] [Google Scholar]
- Haneda K, Oka Y. Selective modulation of voltage-gated calcium channels in the terminal nerve gonadotropin-releasing hormone neurons of a teleost, the dwarf gourami (Colisa lalia) Endocrinology. 2004;145:4489–4499. doi: 10.1210/en.2004-0353. [DOI] [PubMed] [Google Scholar]
- Harney JP, Scarbrough K, Rosewell KL, Wise PM. In vivo antisense antagonism of vasoactive intestinal peptide in the suprachiasmatic nuclei causes aging-like changes in the estradiol-induced luteinizing hormone and prolactin surges. Endocrinology. 1996;137:3696–3701. doi: 10.1210/endo.137.9.8756535. [DOI] [PubMed] [Google Scholar]
- Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: how do they signal and what are their targets? Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- Herbison AE, Pape JR. New evidence for estrogen receptors in gonadotropin-releasing hormone neurons. Front Neuroendocrinol. 2001;22:292–308. doi: 10.1006/frne.2001.0219. [DOI] [PubMed] [Google Scholar]
- Hiruma H, Uemura T, Kimura F. Neuronal synchronization and ionic mechanisms for propagation of excitation in the functional network of immortalized GT1-7 neurons: optical imaging with a voltage-sensitive dye. J Neuroendocrinol. 1997;9:835–840. doi: 10.1046/j.1365-2826.1997.00645.x. [DOI] [PubMed] [Google Scholar]
- Jarvis SE, Zamponi GW. Trafficking and regulation of neuronal voltage-gated calcium channels. Curr Opin Cell Biol. 2007;19:474–482. doi: 10.1016/j.ceb.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Joëls M, Karst H. Effects of estradiol and progesterone on voltage-gated calcium and potassium conductances in rat CA1 hippocampal neurons. J Neurosci. 1995;15:4289–4297. doi: 10.1523/JNEUROSCI.15-06-04289.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BD, Zheng W, Korach KS, Scheuer T, Catterall WA, Rubanyi GM. Increased expression of the cardiac L-type calcium channel in estrogen receptor-deficient mice. J Gen Physiol. 1997;110:135–140. doi: 10.1085/jgp.110.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsch FJ, Moenter SM, Caraty A. The neuroendocrine signal for ovulation. Anim Reprod Sci. 1992;28:329–341. [Google Scholar]
- Kato M, Ui-Tei K, Watanabe M, Sakuma Y. Characterization of voltage-gated calcium currents in gonadotropin-releasing hormone neurons tagged with green fluorescent protein in rats. Endocrinology. 2003;144:5118–5125. doi: 10.1210/en.2003-0213. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Moss RL, Dudley CA. Differential sensitivity of preoptic-septal neurons to microelectrophoresed estrogen during the estrous cycle. Brain Res. 1976;114:152–157. doi: 10.1016/0006-8993(76)91017-9. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Moss RL, Dudley CA, Fawcett CP. The specificity of the response of preoptic-septal area neurons to estrogen: 17alpha-estradiol versus 17beta-estradiol and the response of extrahypothalamic neurons. Exp Brain Res. 1977;30:43–52. doi: 10.1007/BF00237857. [DOI] [PubMed] [Google Scholar]
- Klöckner U, Schiefer A, Isenberg G. L-type Ca-channels: similar Q10 of Ca-, Ba- and Na-conductance points to the importance of ion-channel interaction. Pflugers Arch. 1990;415:638–641. doi: 10.1007/BF02583518. [DOI] [PubMed] [Google Scholar]
- Knobil E, Plant TM, Wildt L, Belchetz PE, Marshall G. Control of the rhesus monkey menstrual cycle: permissive role of hypothalamic gonadotropin-releasing hormone. Science. 1980;207:1371–1373. doi: 10.1126/science.6766566. [DOI] [PubMed] [Google Scholar]
- Krsmanović LZ, Stojilković SS, Merelli F, Dufour SM, Virmani MA, Catt KJ. Calcium signaling and episodic secretion of gonadotropin-releasing hormone in hypothalamic neurons. Proc Natl Acad Sci U S A. 1992;89:8462–8466. doi: 10.1073/pnas.89.18.8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krsmanović LZ, Mores N, Navarro CE, Tomić M, Catt KJ. Regulation of Ca2+-sensitive adenylyl cyclase in gonadotropin-releasing hormone neurons. Mol Endocrinol. 2001;15:429–440. doi: 10.1210/mend.15.3.0610. [DOI] [PubMed] [Google Scholar]
- Kuehl-Kovarik MC, Pouliot WA, Halterman GL, Handa RJ, Dudek FE, Partin KM. Episodic bursting activity and response to excitatory amino acids in acutely dissociated gonadotropin-releasing hormone neurons genetically targeted with green fluorescent protein. J Neurosci. 2002;22:2313–2322. doi: 10.1523/JNEUROSCI.22-06-02313.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata K, Takebayashi M, Kagaya A, Morinobu S, Yamawaki S. Effect of beta-estradiol on voltage-gated Ca2+ channels in rat hippocampal neurons: a comparison with dehydroepiandrosterone. Eur J Pharmacol. 2001;416:203–212. doi: 10.1016/s0014-2999(01)00880-9. [DOI] [PubMed] [Google Scholar]
- Kusano K, Fueshko S, Gainer H, Wray S. Electrical and synaptic properties of embryonic luteinizing hormone-releasing hormone neurons in explant cultures. Proc Natl Acad Sci U S A. 1995;92:3918–3922. doi: 10.1073/pnas.92.9.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Chai YG, Lee EB, Kim KW, Nah SY, Oh TH, Rhim H. 17Beta-estradiol inhibits high-voltage-activated calcium channel currents in rat sensory neurons via a non-genomic mechanism. Life Sci. 2002;70:2047–2059. doi: 10.1016/s0024-3205(01)01534-x. [DOI] [PubMed] [Google Scholar]
- Legan SJ, Karsch FJ. A daily signal for the LH surge in the rat. Endocrinology. 1975;96:57–62. doi: 10.1210/endo-96-1-57. [DOI] [PubMed] [Google Scholar]
- Mahesh VB, Zamorano P, De Sevilla L, Lewis D, Brann DW. Characterization of ionotropic glutamate receptors in rat hypothalamus, pituitary and immortalized gonadotropin-releasing hormone (GnRH) neurons (GT1-7 cells) Neuroendocrinology. 1999;69:397–407. doi: 10.1159/000054442. [DOI] [PubMed] [Google Scholar]
- McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW, Turner RW. Cav3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur J Neurosci. 2006;24:2581–2594. doi: 10.1111/j.1460-9568.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- Mermelstein PG, Becker JB, Surmeier DJ. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J Neurosci. 1996;16:595–604. doi: 10.1523/JNEUROSCI.16-02-00595.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BH, Olson SL, Levine JE, Turek FW, Horton TH, Takahashi JS. Vasopressin regulation of the proestrous luteinizing hormone surge in wild-type and Clock mutant mice. Biol Reprod. 2006;75:778–784. doi: 10.1095/biolreprod.106.052845. [DOI] [PubMed] [Google Scholar]
- Moenter SM, Chu Z, Christian CA. Neurobiological mechanisms underlying oestradiol negative and positive feedback regulation of gonadotrophin-releasing hormone neurones. J Neuroendocrinol. 2009;21:327–333. doi: 10.1111/j.1365-2826.2009.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineux ML, McRory JE, McKay BE, Hamid J, Mehaffey WH, Rehak R, Snutch TP, Zamponi GW, Turner RW. Specific T-type calcium channel isoforms are associated with distinct burst phenotypes in deep cerebellar nuclear neurons. Proc Natl Acad Sci U S A. 2006;103:5555–5560. doi: 10.1073/pnas.0601261103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- Noel SD, Keen KL, Baumann DI, Filardo EJ, Terasawa E. Involvement of G protein-coupled receptor 30 (GPR30) in rapid action of estrogen in primate LHRH neurons. Mol Endocrinol. 2009;23:349–359. doi: 10.1210/me.2008-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman RL, Blake CA, Sawyer CH. Estrogen-dependent 24-hour periodicity in pituitary LH release in the female hamster. Endocrinology. 1973;93:965–970. doi: 10.1210/endo-93-4-965. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Moenter SM. Estradiol-sensitive afferents modulate long-term episodic firing patterns of GnRH neurons. Endocrinology. 2002;143:2284–2292. doi: 10.1210/endo.143.6.8869. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, Straume M, DeFazio RA, Moenter SM. Gonadotropin-releasing hormone neurons generate interacting rhythms in multiple time domains. Endocrinology. 2003a;144:823–831. doi: 10.1210/en.2002-220585. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Moenter SM. Calcium current subtypes in GnRH neurons. Biol Reprod. 2003b;69:1914–1922. doi: 10.1095/biolreprod.103.019265. [DOI] [PubMed] [Google Scholar]
- Palm IF, Van Der Beek EM, Wiegant VM, Buijs RM, Kalsbeek A. Vasopressin induces a luteinizing hormone surge in ovariectomized, estradiol-treated rats with lesions of the suprachiasmatic nucleus. Neuroscience. 1999;93:659–666. doi: 10.1016/s0306-4522(99)00106-2. [DOI] [PubMed] [Google Scholar]
- Patterson E, Ma L, Szabo B, Robinson CP, Thadani U. Ovariectomy and estrogen-induced alterations in myocardial contractility in female rabbits: role of the L-type calcium channel. J Pharmacol Exp Ther. 1998;284:586–591. [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. Ed 2. New York: Academic; 2001. [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Pielecka-Fortuna J, Chu Z, Moenter SM. Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology. 2008;149:1979–1986. doi: 10.1210/en.2007-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. 2008;70:165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- Reid CA, Bekkers JM, Clements JD. Presynaptic Ca2+ channels: a functional patchwork. Trends Neurosci. 2003;26:683–687. doi: 10.1016/j.tins.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Roberts CB, Hemond P, Suter KJ. Synaptic integration in hypothalamic gonadotropin releasing hormone (GnRH) neurons. Neuroscience. 2008;154:1337–1351. doi: 10.1016/j.neuroscience.2008.04.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TA, Qiu J, Bosch MA, Rønnekleiv OK, Kelly MJ. Cross-talk between membrane-initiated and nuclear-initiated oestrogen signalling in the hypothalamus. J Neuroendocrinol. 2009;21:263–270. doi: 10.1111/j.1365-2826.2009.01846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanò N, Lee K, Abrahám IM, Jasoni CL, Herbison AE. Nonclassical estrogen modulation of presynaptic GABA terminals modulates calcium dynamics in gonadotropin-releasing hormone neurons. Endocrinology. 2008;149:5335–5344. doi: 10.1210/en.2008-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen AD. Temperature modulation of calcium channel function in GH3 cells. Am J Physiol. 1996;271:C863–C868. doi: 10.1152/ajpcell.1996.271.3.C863. [DOI] [PubMed] [Google Scholar]
- Roseweir AK, Kauffman AS, Smith JT, Guerriero KA, Morgan K, Pielecka-Fortuna J, Pineda R, Gottsch ML, Tena-Sempere M, Moenter SM, Terasawa E, Clarke IJ, Steiner RA, Millar RP. Discovery of potent kisspeptin antagonists delineate physiological mechanisms of gonadotropin regulation. J Neurosci. 2009;29:3920–3929. doi: 10.1523/JNEUROSCI.5740-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SN, Huang RQ, Logan SM, Yi KD, Dillon GH, Simpkins JW. Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc Natl Acad Sci U S A. 2008;105:15148–15153. doi: 10.1073/pnas.0802379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim JA, Skynner MJ, Herbison AE. Heterogeneity in the basic membrane properties of postnatal gonadotropin-releasing hormone neurons in the mouse. J Neurosci. 2001;21:1067–1075. doi: 10.1523/JNEUROSCI.21-03-01067.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology. 2005;146:3686–3692. doi: 10.1210/en.2005-0488. [DOI] [PubMed] [Google Scholar]
- Smith MJ, Jiennes L, Wise PM. Localization of the VIP2 receptor protein on GnRH neurons in the female rat. Endocrinology. 2000;141:4317–4320. doi: 10.1210/endo.141.11.7876. [DOI] [PubMed] [Google Scholar]
- Spergel DJ. Calcium and small-conductance calcium-activated potassium channels in gonadotropin-releasing hormone neurons before, during and after puberty. Endocrinology. 2007;148:2383–2390. doi: 10.1210/en.2006-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter KJ, Song WJ, Sampson TL, Wuarin JP, Saunders JT, Dudek FE, Moenter SM. Genetic targeting of green fluorescent protein to gonadotropin-releasing hormone neurons: characterization of whole-cell electrophysiological properties and morphology. Endocrinology. 2000a;141:412–419. doi: 10.1210/endo.141.1.7279. [DOI] [PubMed] [Google Scholar]
- Suter KJ, Wuarin JP, Smith BN, Dudek FE, Moenter SM. Whole-cell recordings from preoptic/hypothalamic slices reveal burst firing in gonadotropin-releasing hormone neurons identified with green fluorescent protein in transgenic mice. Endocrinology. 2000b;141:3731–3736. doi: 10.1210/endo.141.10.7690. [DOI] [PubMed] [Google Scholar]
- Swandulla D, Armstrong CM. Calcium channel block by cadmium in chicken sensory neurons. Proc Natl Acad Sci U S A. 1989;86:1736–1740. doi: 10.1073/pnas.86.5.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple JL, Laing E, Sunder A, Wray S. Direct action of estradiol on gonadotropin-releasing hormone-1 neuronal activity via a transcription-dependent mechanism. J Neurosci. 2004;24:6326–6333. doi: 10.1523/JNEUROSCI.1006-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly ES, Jr, Nethrapalli IS, Tinnikov AA. ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci. 2002;22:8391–8401. doi: 10.1523/JNEUROSCI.22-19-08391.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toran-Allerand CD, Tinnikov AA, Singh RJ, Nethrapalli IS. 17α-Estradiol: a brain-active estrogen? Endocrinology. 2005;146:3843–3850. doi: 10.1210/en.2004-1616. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Fox AP, Hess P, McCleskey EW, Nilius B, Nowycky MC, Rosenberg RL. Multiple types of calcium channel in excitable cells. Soc Gen Physiol Ser. 1987;41:167–187. [PubMed] [Google Scholar]
- Ullrich ND, Koschak A, MacLeod KT. Oestrogen directly inhibits the cardiovascular L-type Ca2+ channel Cav1.2. Biochem Biophys Res Commun. 2007;361:522–527. doi: 10.1016/j.bbrc.2007.07.054. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Krsmanović LZ, Catt KJ, Stojilkovic SS. Control of action potential-driven calcium influx in GT1 neurons by the activation status of sodium and calcium channels. Mol Endocrinol. 1999;13:587–603. doi: 10.1210/mend.13.4.0261. [DOI] [PubMed] [Google Scholar]
- Van Goor F, LeBeau AP, Krsmanović LZ, Sherman A, Catt KJ, Stojilkovic SS. Amplitude-dependent spike-broadening and enhanced Ca2+ signaling in GnRH-secreting neurons. Biophys J. 2000;79:1310–1323. doi: 10.1016/S0006-3495(00)76384-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Sakuma Y, Kato M. High expression of the R-type voltage-gated Ca2+ channel and its involvement in Ca2+-dependent gonadotropin-releasing hormone release in GT1-7 cells. Endocrinology. 2004;145:2375–2383. doi: 10.1210/en.2003-1257. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- Wildt L, Marshall G, Knobil E. Experimental induction of puberty in the infantile female rhesus monkey. Science. 1980;207:1373–1375. doi: 10.1126/science.6986658. [DOI] [PubMed] [Google Scholar]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Gröne HJ, Todman MG, Korach KS, Greiner E, Pérez CA, Schütz G, Herbison AE. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006;52:271–280. doi: 10.1016/j.neuron.2006.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS. Acute effects of estrogen on neuronal physiology. Annu Rev Pharmacol Toxicol. 2007;47:657–680. doi: 10.1146/annurev.pharmtox.47.120505.105219. [DOI] [PubMed] [Google Scholar]
- Zhang C, Bosch MA, Levine JE, Rønnekleiv OK, Kelly MJ. Gonadotropin-releasing hormone neurons express KATP channels that are regulated by estrogen and responsive to glucose and metabolic inhibition. J Neurosci. 2007;27:10153–10164. doi: 10.1523/JNEUROSCI.1657-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Bosch MA, Rønnekleiv OK, Kelly MJ. γ-Aminobutyric acid B receptor mediated inhibition of gonadotropin-releasing hormone neurons is suppressed by kisspeptin-G protein-coupled receptor 54 signaling. Endocrinology. 2009a;150:2388–2394. doi: 10.1210/en.2008-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Bosch MA, Rick EA, Kelly MJ, Ronnekleiv OK. 17β-Estradiol regulation of T-type calcium channels in gonadotropin-releasing hormone neurons. J Neurosci. 2009b;29:10552–10562. doi: 10.1523/JNEUROSCI.2962-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Brinton RD. Estrogen receptor alpha and beta differentially regulate intracellular Ca2+ dynamics leading to ERK phosphorylation and estrogen neuroprotection in hippocampal neurons. Brain Res. 2007;1172:48–59. doi: 10.1016/j.brainres.2007.06.092. [DOI] [PubMed] [Google Scholar]