

SUMMARY
TRAF1/2 and cIAP1/2 are members of the TNF receptor associated factor (TRAF) and the inhibitor of apoptosis (IAP) families, respectively. They are critical for both the canonical and the noncanonical NF-κB signaling pathways. Here we report the crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes. A TRAF2 trimer interacts with one cIAP2 both in the crystal and in solution. Two chains of the TRAF2 trimer directly contact cIAP2 and key residues at the interface are confirmed by mutagenesis both in vitro and in cells. TRAF1 and TRAF2 preferentially form the TRAF1: (TRAF2)2 heterotrimer, which interacts with cIAP2 more strongly than TRAF2 alone. In contrast, TRAF1 alone interacts very weakly with cIAP2. Surprisingly, TRAF1 and one chain of TRAF2 in the TRAF1: (TRAF2)2: cIAP2 ternary complex mediate interaction with cIAP2. Because TRAF1 is dramatically upregulated by many stimuli, it may modulate the interaction of TRAF2 with cIAP1/2, which explains previously noted regulatory roles of TRAF1 in TNF signaling.
INTRODUCTION
Genetic knockout studies have long established the role of TNF receptor associated factors (TRAFs) in NF-κB and MAP kinase activation pathways in the signaling of multiple receptor families in both innate and adaptive immunity (Yeh et al., 1999). In contrast, the function of cIAP1/2 has remained rather obscure since their original identification as components of the TNFR2 and other TNF receptor family signaling complexes and as TRAF1/2 interacting proteins (Rothe et al., 1995). Recent key discoveries illuminated the essential functions of cIAP1/2 as E3 ligases in both the canonical and the noncanonical NF-κB signaling pathways.
In the noncanonical NF-κB signaling pathway, TRAF2 and cIAP1/2 are critical components of the E3 complex that regulates the stability of NIK, a kinase that phosphorylates IKKα and activates NF-κB (Petersen et al., 2007; Vallabhapurapu et al., 2008; Varfolomeev et al., 2007; Vince et al., 2007; Zarnegar et al., 2008). In unstimulated cells, TRAF2 interacts with TRAF3 which in turn recruits NIK, thus bringing it to the vicinity of cIAP1/2 for K48-linked polyubiquitination and degradation. This suppresses noncanonical NF-κB activation. Upon receptor stimulation, cIAP1/2 switches substrate specificity from NIK to TRAF2 and TRAF3, allowing NIK to accumulate to activate the noncanonical NF-κB pathway.
TRAF2 and cIAP1/2 are also essential for activation of TNFα-induced canonical NF-κB pathway (Mahoney et al., 2008; Vince et al., 2009). Upon interacting with TNFα, TNFR1 recruits the adapter protein TRADD, which in turn recruits TRAF2 and RIP1 (Hsu et al., 1996a; Hsu et al., 1996b). RIP1 becomes modified with K63-linked polyubiquitin chains, a process that is essential for IKKβ activation (Ea et al., 2006; Wu et al., 2006). The ubiquitin ligase for RIP1 polyubiquitination has been speculated to be TRAF2 (Lee et al., 2004b); however, our recent structural analysis suggested that TRAF2 RING does not possess K63-linked ubiquitin ligase activity (Yin et al., 2009b). Consistent with our observation, TNFα induced RIP1 polyubiquitination is dependent on the TRAF2: cIAP1/2 interaction but the TRAF2 RING domain is dispensable (Vince et al., 2009). Instead, when both cIAP1 and cIAP2 were absent, RIP1 polyubiquitination was defective with decreased phosphorylation of IKKβ (Mahoney et al., 2008; Varfolomeev et al., 2008) and reduced cancer cell survival (Bertrand et al., 2008), demonstrating that cIAP1/2 also mediates K63-linked polyubiquitination of RIP1 (Yin et al., 2009b).
Unlike TRAF2, the role of TRAF1 in NF-κB activation is less clear. Accumulating data support its role as both a negative and a positive modulator of signaling by certain TNF family receptors, possibly in a cell-type dependent manner (Lee and Choi, 2007). TRAF1 is upregulated upon TNFα signaling and overexpression of TRAF1 inhibited NF-κB activation by many stimuli (Carpentier and Beyaert, 1999). TRAF1-deficient T cells are hyper-responsive to TNFα with enhanced proliferation and activation of the NF-κB and AP-1 signaling pathways (Tsitsikov et al., 2001). However, TRAF1-deficient dendritic cells show attenuated responses to secondary stimulation by TRAF2-dependent factors (Arron et al., 2002), suggesting a positive regulatory role.
cIAP1 and cIAP2 are often upregulated in many cancers and antagonizing IAPs has enormous promise in specific cancer therapy (LaCasse et al., 2008). Abnormal expressions of TRAF1 and TRAF2 are also present in a large number of lymphocyte malignancies (Zapata et al., 2007). In addition, chromosomal translocation that creates a fusion protein of cIAP2 and MALT1 (cIAP2.MALT1) occurs frequently in mucosa-associated lymphoid tissue (MALT) B-cell lymphoma to mediate constitutive NF-κB activation (Thome, 2004). The ability of cIAP2 to interact with TRAF2 has been shown to be critical for the cIAP2.MALT1 chimera to activate NF-κB (Garrison et al., 2009; Lucas et al., 2007). Collectively, these data suggest the importance of TRAF1/2 and cIAP1/2 in human diseases and the potential in using them as therapeutic targets.
Most TRAF proteins share a common domain organization that contains a RING, several zinc fingers, a TRAF-N domain and a conserved TRAF-C domain (Rothe et al., 1994). TRAF1 does not have an N-terminal RING domain but can form hetero-oligomers with TRAF2. cIAP1 and cIAP2 are composed of three N-terminal BIR domains, a UBA domain, a CARD domain, and a C-terminal RING domain (Rothe et al., 1995). Previous studies have shown that the BIR domains of cIAP1 and cIAP2 directly interact with TRAF1 and TRAF2 (Rothe et al., 1995; Samuel et al., 2006; Varfolomeev et al., 2006),
Despite the importance of the TRAF2: cIAP1/2 interaction, no structural information is currently available. Here we report biochemical, structural and cell biological studies on the interaction between TRAF2 and cIAP2 and on the ability of TRAF1 to modulate this interaction.
RESULTS
Mapping the Interaction Between TRAF2 and cIAP2
Limited biochemical information is available on the interaction between TRAF2 and cIAP2. While the BIR1 domain of cIAP1/2 has been shown to mediate its interaction with TRAF2 (Samuel et al., 2006; Varfolomeev et al., 2006), efforts on mapping the region of TRAF2 for interaction with cIAP1/2 has produced only conflicting results (Rothe et al., 1995; Samuel et al., 2006). To initiate mapping studies on TRAF2, we first co-expressed non-tagged cIAP2 BIR1 domain (residues 26-99) with His-tagged entire C-terminal region of TRAF2 in E. coli. The His-tag pulldown co-purified non-tagged cIAP2 BIR1, confirming that the interaction between TRAF2 and cIAP2 is direct (Supplemental Figure S1). We then trimmed down the TRAF2 construct from both the N- and the C-termini and determined their ability to interact with cIAP2 (Supplemental Figure S1). Surprisingly, the TRAF-C domain of TRAF2 is dispensable for cIAP2 interaction, in contrast to the previous result (Samuel et al., 2006). A number of TRAF2 constructs containing as short as residues 266-329 interacted strongly with cIAP2 BIR1.
Overall Structure of the TRAF2: cIAP2 Complex
The construct of TRAF2 containing residues 266-329 yielded diffracting crystals, both alone and in complex with the cIAP2 BIR1 domain. The structure of the TRAF2: cIAP2 complex was solved at 2.6 Å resolution by multi-wavelength anomalous diffraction (MAD) using the intrinsic zinc atom of the cIAP2 BIR1 (Table 1, Figure 1A, Supplemental Figure S2). The structure of TRAF2 alone was solved by molecular replacement using the TRAF2 structure from the complex as a model (Table 1). The cIAP2-binding region of TRAF2 forms a continuous homotrimeric coiled coil, both alone and in complex with cIAP2. Most surprisingly, despite the trimeric coiled coil structure of TRAF2, each TRAF2 trimer interacts with one, instead of three, cIAP2 molecules in the crystal. The trimeric coiled coil structure of TRAF2 is reminiscent of the previous structure of the TRAF domain of TRAF2 (residues 310-501) (Park et al., 1999). Remarkably, modeling of the full-length TRAF2 structure using the current structure, the TRAF domain structure and structures of the N-terminal region of TRAF2 and TRAF6 (Yin et al., 2009a; Yin et al., 2009b) showed a striking ~300 Å long platform (Figure 1B) onto which many interacting proteins may be docked.
Table 1.
Crystallographic Statistics
| TRAF2: cIAP2 | TRAF2 | TRAF1: TRAF2: cIAP2 | |||
|---|---|---|---|---|---|
| Data Collection | |||||
|
| |||||
| Beamlines | NSLS X25 | NSLS X29 | NSLS X29 | ||
| Space group | I41 | R3 | I41 | ||
| Cell dimensions | |||||
| a, b, c (Å) | 92.1, 92.1, 86.0 | 150.7, 150.7, 86.7 | 91.9, 91.9, 88.7 | ||
| α, β, γ (°) | 90, 90, 90 | 90, 90, 120 | 90, 90, 90 | ||
| Resolution (Å) | 37.0 - 2.6 | 33.4 - 2.6 | 32.5 - 2.8 | ||
| Wavelength (Å) | 1.2822 (peak) | 1.2832 (inflection) | 1.2664 (remote) | 1.0800 | 1.0800 |
| RSYM (%) | 4.9 (39.9) | 4.6 (40.1) | 4.9 (39.3) | 6.8 (37.6) | 9.7 (48.0) |
| I/σI | 29.5 (1.8) | 31.4 (1.8) | 30.5 (2.0) | 24.8 (4.3) | 21.7 (2.7) |
| Completeness (%) | 87.3 (36.9) | 87.7 (37.1) | 88.7 (46.4) | 92.0 (46.0) | 99.8 (99.7) |
| Redundancy | 3.6 (2.4) | 3.6 (2.4) | 3.6 (2.5) | 5.8 (5.1) | 6.1 (4.4) |
|
| |||||
| Refinement | |||||
| Resolution (Å) | 37.0 - 2.6 | 33.4 - 2.6 | 32.5 - 2.8 | ||
| No. Reflections | 9,689 | 19,062 | 8,753 | ||
| Rwork / Rfree | 0.230 / 0.274 | 0.230 / 0.262 | 0.197 / 0.247 | ||
| No. Protein atoms | 1,890 | 2,629 | 2,053 | ||
| No. Zinc atoms | 1 | 0 | 1 | ||
| R.M.S. Deviations | |||||
| Bond lengths (Å) / angles (°) | 0.019 / 1.7 | 0.006 / 1.0 | 0.009 / 1.14 | ||
| Ramachandran plot | |||||
| Most favored / allowed (%) | 95.9 / 100.0 | 95.5 / 100.0 | 96.1 / 98.4 | ||
Values in parenthesis are for the highest-resolution shell.
See also Figure S2.
Figure 1.

Crystal structure of the TRAF2: cIAP2 complex
(A) A ribbon diagram of TRAF2 in complex with cIAP2 BIR1 domain. The three TRAF2 chains are shown in yellow, green and cyan, respectively. cIAP2 is shown in pink.
(B) Atomic model of full-length TRAF2 built from the current structure, the structure of TRAF2 in complex with a receptor peptide (Park et al., 1999) and the N-terminal domain structures of TRAF2 and TRAF6 (Yin et al., 2009a; Yin et al., 2009b). ZF1-ZF4: zinc fingers 1 to 4; CC: coiled coil.
(C) MALS measurement of the TRAF2: cIAP2 complex in line with gel filtration chromatography, showing that the measured molecular mass is consistent with a 3:1 complex of TRAF2 and cIAP2. The horizontal axis shows the elution volume and the vertical axis shows the measured molecular mass.
(D) ITC measurement for the titration of cIAP2 into TRAF2, showing a KD of 1.7 μM and a stoichiometry of 1:3 of cIAP2 and TRAF2.
TRAF2 and cIAP2 Form a 3:1 Complex in Solution
To characterize the interaction between TRAF2 and cIAP2 in solution, we first performed multi-angle light scattering (MALS) measurement in line with gel filtration chromatography using the TRAF2 construct containing residues 266-329. The measurement revealed a molecular mass of the complex of 30.6 kDa, which is consistent with the 33.6 kDa mass calculated for 3 TRAF2 molecules and 1 cIAP2 BIR1 molecule (Figure 1C). We further performed isothermal titration calorimetry (ITC) to independently confirm the interaction stoichiometry and to reveal the interaction thermodynamics (Figure 1D, Supplemental Table S1). Indeed, the same 3:1 stoichiometry was shown with a measured N value of 0.3 (in agreement with the expected N value of 0.333) for the titration of cIAP2 to TRAF2. The interaction exhibits a dissociation constant of 1.7 μM, a low micromolar affinity common for interactions between signaling proteins. The interaction is mainly contributed by favorable enthalpy, which is consistent with the significant hydrophilic content of the interaction (see below).
Interaction Between TRAF2 and cIAP2 and Structure-based Mutagenesis
There are two potential TRAF2: cIAP2 interfaces in the crystal lattice with similar buried surface areas (Figure 2A). The first interface buries a total of 1,200 Å2 surface area and has a sum of 126 direct van der Waals contacts. The second interface is similar in buried surface area but with only 71 direct van der Waals contacts. The shape complementarity (shape correlation statistic) score (Lawrence and Colman, 1993) of the first interface is 0.64, which is excellent, while the second interface gives a good score of 0.61. These analyses suggest that the first interface is somewhat more likely to be the physiological binding interface in solution, which was further confirmed by mutagenesis shown below.
Figure 2.

Interactions between TRAF2 and cIAP2
(A) Two alternative interfaces that cIAP2 makes with a TRAF2 trimer in the crystal lattice. The Site 1 is the correct interface.
(B) A close up view of the interacting region of the TRAF2: cIAP2 complex. TRAF2 residues involved in cIAP2 interaction are colored in red for the yellow chain and in magenta for the green chain. cIAP2 residues involved in TRAF2 interaction are shown in red.
(C) Side chains involved in the TRAF2: cIAP2 interaction. Left: Interfacial TRAF2 residues are shown and labeled. Right: Interfacial cIAP2 residues are shown and labeled.
(D) Detailed interaction between TRAF2 and cIAP2, showing both hydrophobic interactions and hydrogen bonding interactions. For clarity, only side chains of cIAP2 are shown.
(E) Sequence alignment between human cIAP2 and cIAP1. Residues at the correct interface with TRAF2 are shaded in red while those at the incorrect interface are shaded in gray. Residues that coordinate the Zn atom are shaded in yellow. Secondary structures, residues numbers of cIAP2, and mutational effects are shown above the sequence. +: WT-like interaction; −: almost no interaction; w: weak interaction. Residues involved in hydrogen bonding interactions and the surface exposure of the interfacial residues are shown below the sequence. H: hydrogen bonds; e: exposed in the TRAF2: cIAP2 complex; b: buried at the TRAF2: cIAP2 interface.
(F) Sequence alignment between human TRAF2 and TRAF1. Residues involved in cIAP2 interaction are shaded in red and magenta, respectively, for each of the two TRAF2 chains. Residues numbers of TRAF2 and mutational effects are shown above the sequence. +: WT-like interaction; −: almost no interaction; w: weak interaction. Residues involved in hydrogen bonding interactions and the surface exposure of the interfacial residues are shown below the sequence. H: hydrogen bonds; e: exposed in the TRAF2: cIAP2 complex; b: buried at the TRAF2: cIAP2 interface. The a and d positions of the coiled coil are shown.
The interaction between TRAF2 and cIAP2 at the first interface is localized between residues 281 to 295 of TRAF2 and residues 27 to 52 of cIAP2 (Figure 2B, 2C, 2D). Two chains of TRAF2 are in direct contact with cIAP2, with one chain mediating most of the interactions (the yellow chain in Figure 2B) and another chain contributing additional residues (the green chain in Figure 2B). For cIAP2, residues from the α1 and α2 region contact TRAF2. The interaction is both hydrophobic and hydrophilic in nature (Figure 2E, 2F). A hydrophobic patch of TRAF2 involving residues I285 and V288 interacts with residues L30, M33 and S34 of cIAP2. Residue N284 of TRAF2 forms a hydrogen bond with S37 of cIAP2. R291 of TRAF2 forms a salt bridge with E47 of cIAP2. N290 and E292 of TRAF2 and R48 of cIAP2 form a cluster of salt bridges and hydrogen bonds. E294 forms another salt bridge with R52 of cIAP2. The TRAF2-contacting residues are completely identical between cIAP1 and cIAP2 (Figure 2E), suggesting that TRAF2 interacts with cIAP1 in a similar manner and with similar strength. In contrast, there are several differences between the cIAP2-contacting residues of TRAF2 and TRAF1, suggesting that TRAF1 may not have the same energetics of interaction with cIAP2.
Structure based mutagenesis experiments were first performed on cIAP2 to identify the most critical residues for TRAF2 interaction and to confirm the physiological interaction surface (Figure 3A). To assess the mutational effects, we produced His-tagged WT and mutant cIAP2 to pulldown Smt3-fused WT TRAF2. Among the 12 single site mutations, those that caused drastic effects all mapped to the first interface, while mutations on the second interface exhibited minimal effects. Only the Y56A mutation of the second interface had an observable weakening on the interaction. However, this effect may be attributed to a potential local structural alteration from removal of the partially buried large side chain. The mutagenesis results suggest that four residues of cIAP2 buried at the interface, L30, M33, E47 and R48, are energetically most critical for the interaction.
Figure 3.

Structure-based mutagenesis of TRAF2 and cIAP2
(A) Pulldown of Smt3 fusion of TRAF2 (residues 266-329) by His-tagged WT and mutant cIAP2 BIR1 (residues 26-99). The mutations are labeled to indicate whether they belong to Site 1 or Site 2. +: WT-like interaction; −: almost no interaction; w: weak interaction; *: a contaminant band.
(B) Pulldown of cIAP2 BIR1 by His-Smt3 fusion of WT and mutant TRAF2. +: WT-like interaction; −: almost no interaction; w: weak interaction; *: contaminant bands.
(C) Co-immunoprecipitation of lysates from HEK 293T cells co-expressing Myc-TRAF2 and Flag-cIAP2, Flag-cIAP2 L30A, Flag-cIAP2 R48A, or Flag-cIAP2 F67A. Lysates were incubated with anti-Flag M2 beads and TRAF2: cIAP2 complexes were detected by immunoblot analysis against TRAF2.
(D) Co-immunoprecipitation of lysates from HEK 293T cells co-expressing Myc-cIAP2 and Flag-TRAF2 or Flag-TRAF2 E292A. Lysates were incubated with anti-Flag M2 beads and TRAF2: cIAP2 complexes were detected by immunoblot analysis against Myc. All experiments were done at least three times with similar results.
(E) Traf2−/− MEFs were reconstituted with pBABE-Flag vector (EV), pBABE-Flag-TRAF2 WT (WT), or pBABE-Flag-TRAF2 E292A (E292A). Rescue of suppression of constitutive noncanonical NF-κB activation was assessed by immunoblot analysis of NIK.
(F) Traf2−/−Traf5−/− MEFs were reconstituted with pBABE-Flag vector (EV), pBABE-Flag TRAF2 (TRAF2 WT), or pBABE-Flag-TRAF2 E292A (E292A). Rescue of canonical NF-κB activation was assessed by immunoblot analysis of IκBα after TNFα (20 ng/ml) stimulation. Data are representative of three independent experiments.
Conversely, the effects of TRAF2 mutations were determined using WT and mutant His-tagged Smt3 fused TRAF2 and non-tagged WT cIAP2 (Figure 3B). Among the 10 single site mutations of TRAF2, three residues that are buried at the interface, I285, V288 and E292, contribute most to the interaction. Their mutations to Ala radically diminished the interaction with cIAP2. Mutations C287A, N290A and R291A also weakened the interaction with cIAP2. Interestingly, several residues at the peripheral region of the interface did not cause observable effects when mutated, even though they form hydrogen bonding interactions with cIAP2 (Figure 2D). Our mutational results support the “hot spot” theory that central, buried residues at an interface are the major contributors of binding energetics (Clackson and Wells, 1995).
Critical Residues in Full-length TRAF2: cIAP2 Interaction and Signaling
To further determine whether the observed interaction between TRAF2 and cIAP2 in the crystal structure depicted a physiological interaction, we used co-immunoprecipitation to assess the effects of structure-based mutations in full-length cIAP2 and TRAF2 in cells (Figure 3C, 3D). When Flag-tagged WT cIAP2 was immunoprecipitated, it pulled down associated Myc-tagged WT TRAF2. cIAP2 mutants defective in interacting with TRAF2 in vitro, L30A and R48A, failed to pull down TRAF2 (Figure 3C). In contrast, cIAP2 mutant F67A, which was designed based on the alternative interface, did not have an observable effect on TRAF2 interaction. Conversely, Flag-tagged WT TRAF2 also pulled down Myc-tagged WT cIAP2. The TRAF2 mutant E292A defective in cIAP2 interaction in vitro, failed to pull down cIAP2 (Figure 3D). The complete agreement of the mutational effects in full-length TRAF2 and cIAP2 in cells confirmed the structural observation.
To further study the functional role of the TRAF2 mutants, we introduced TRAF2 WT or TRAF2 E292A into Traf2−/− MEFs by the pBABE-Flag retroviral system. Noncanonical NF-κB activation was then examined by immunoblot with anti-NIK. As expected, expression of TRAF2 WT in Traf2−/− MEFs restored NIK to the low normal basal level. However, expression of the cIAP2-binding deficient mutant of TRAF2 failed to do so (Figure 3E). We also examined the canonical NF-κB activation after TNFα stimulation in Traf2−/−Traf5−/− MEFs reconstituted with TRAF2 WT or TRAF2 E292A. Cells reconstituted with the binding deficient mutant of TRAF2 failed to restore TNFα-induced NF-κB activation while cells reconstituted with TRAF2 WT restored TNF-induced NF-κB activation as evidenced by IκBα degradation (Figure 3F).
Asymmetry and Anti-cooperativity in the TRAF2: cIAP2 Interaction
We analyzed the TRAF2 structure in the complex with cIAP2 to understand why only one cIAP2 is bound to trimeric TRAF2. We first wondered if three cIAP2 molecules bound to a TRAF2 trimer would cause steric hindrance among one another, which however was not correct (Supplemental Figure S3A). We then wondered whether the TRAF2 structure in the complex with cIAP2 is somewhat asymmetrical and therefore cannot interact with three cIAP2 molecules simultaneously. Superposition of TRAF2 with its ~120° rotated counterpart showed that they deviate significant from one another, indeed suggesting asymmetry in the TRAF2 structure (Figure 4A). Importantly, the region of TRAF2 in direct contact with cIAP2 does not superimpose well. In addition, structural alignment among the TRAF2 chains in the TRAF2: cIAP2 complex showed that all three chains differ in their conformations (Fig. 4B and 4C). These differences would move the main chain position significantly within the cIAP2-binding region.
Figure 4.

Asymmetry of TRAF2 in complex with cIAP2 and regulation of cIAP2 interaction by TRAF1
(A) Superposition of the TRAF2 structure (shown in yellow, green and cyan) in complex of cIAP2 with its 3-fold rotated counterpart (shown in orange). The cIAP2 bound to the stationary TRAF2 trimer is shown in pink.
(B) Superposition of the three TRAF2 chains using either the N-terminal or C-terminal region sequences.
(C) Superposition of the three TRAF2 chains using the entire sequences. The boxed region is enlarged at the right panel to show the Cα position differences.
(D) Superposition of the TRAF2 structure alone (shown in yellow, green and cyan) with its 3-fold rotated counterpart (shown in orange).
(E) Gel filtration fractions of the TRAF1: cIAP2 complex (top 1), the TRAF2: cIAP2 complex (top 2), the TRAF2 C287A: cIAP2 complex (top 3), the TRAF2 R291K: cIAP2 complex (bottom 3), the TRAF2 C287A/R291K: cIAP2 complex (bottom 2) and the TRAF1: TRAF2: cIAP2 complex (bottom 1).
(F) Preferential formation of a 1:2 complex of TRAF1 and TRAF2. SDS-PAGE lanes of gel filtration fractions from a mixture of His-Smt3-TRAF1 and His-TRAF2 are shown, together with the chromatographic trace at OD280. Each of the peaks was assessed by MALS and the measured molecular mass is shown.
(G) The TRAF1: (TRAF2)2 heterotrimer competed robustly with the TRAF2 trimer for cIAP2 interaction. Gel filtration fractions of a mixture of His-Smt3-TRAF1, His-TRAF2 and a small amount of His-cIAP2 are shown. Most of the cIAP2 proteins co-migrated with the heterotrimer.
To further analyze the consequence of this asymmetry, we placed cIAP2 molecules to the two remaining hypothetical binding sites on a TRAF2 trimer by superposition. Strikingly, these created binding sites were much less optimal than the observed TRAF2: cIAP2 interaction. The buried surface area dropped from 1,200 Å2 total to about 930 Å2 and 950 Å2 respectively with concomitant reduction in the number of van der Waals interactions. Perhaps most drastically, the complementarity measurements decreased from the original score of 0.64 to about half, 0.35 and 0.26, respectively (Supplemental Table S2). It should be noted that calculations from a number of PDB entries showed that most protein complexes have shape complementarity scores above 0.6 (Lawrence and Colman, 1993).
These data suggest that there might be anti-cooperativity in the binding of cIAP2 to TRAF2. Upon binding to one cIAP2 molecule, the TRAF2 trimer undergoes subtle but significant conformational changes to optimize the interaction. This then prohibits the binding of additional cIAP2 molecules to the same TRAF2 trimer. This type of anti-cooperativity has also been proposed for the binding of one linear di-ubiquitin to a dimeric coiled coil region of NEMO (Rahighi et al., 2009). Analysis of the TRAF2 structure in the absence of cIAP2 binding (Table 1) supports this hypothesis. The trimerically related counterpart in this TRAF2 structure superimposes quite well in comparison with the poor degree of superposition for the TRAF2 in complex with cIAP2 (Figure 4D).
TRAF1 Interacts with cIAP2 Weakly
Yeast two hybrid analysis showed that both TRAF1 and TRAF2 interact with cIAP1/2 (Rothe et al., 1995). Despite the high degree of sequence identity between the cIAP2-interacting region of TRAF1 and TRAF2, we noticed that TRAF1 does not interact with cIAP2 as strongly as TRAF2. When co-expressed in E. coli, TRAF1 and cIAP2 could be co-purified by Ni-affinity resin, but this complex dissociated significantly during gel filtration chromatography while the TRAF2: cIAP2 complex mostly stayed intact (Figure 4E, 2 gels at the top). An ITC experiment titrating cIAP2 to TRAF1 yielded a dissociation constant of 111 μM, which is almost two orders of magnitude weaker than the corresponding TRAF2: cIAP2 interaction (Supplemental Figure S3B, Table S3). An equilibrium dialysis experiment showed a similar dissociation constant of 127 μM (Supplemental Figure S3C), confirming the much weaker binding affinity. The ITC measurement also showed that each TRAF1 trimer interacts with three cIAP2 molecules, different from the situation for TRAF2.
There are four amino acid residue differences between TRAF1 and TRAF2 in the cIAP2-interacting sites, T281V, C287A, R291K and R295A (Figure 2F). Because structure-based mutations T281A and R295A of TRAF2 did not cause any observable changes in interacting with cIAP2, while C287A and R291A did reduce the interaction (Figure 3B), we speculated that one reason for the weak interaction of TRAF1 with cIAP2 is due to the changes on C287 and R291. To test this hypothesis, we generated the R291K mutation and the double mutation of C287A and R291K on TRAF2 to see if they affect interactions with cIAP2 (Figure 4E). Gel filtration experiments showed that R291K did not show clear effects on cIAP2 interaction while C287A reduced the interaction (Figure 4E, 2 gels in the middle). The double mutation further reduced the interaction with cIAP2 (Figure 4E, fifth gel from the top), confirming our hypothesis. It is likely that the amino acid difference in TRAF1 weakens the interaction with cIAP2 to such an extent that deformation of the symmetrical TRAF1 trimer cannot occur, which in turn further contributes to the weaker interaction. Although we can measure the TRAF1: cIAP2 interaction in vitro, it is likely that this interaction is too weak to occur in cells.
TRAF1 and TRAF2 Preferentially Form 1: 2 Heterotrimers
To determine whether TRAF1 and TRAF2 form heterotrimers in vitro, we first expressed TRAF1 as a His-Smt3 fusion and TRAF2 as a His-fusion and purified them separately. The smt3-fusion was used to alter the molecular weight of the TRAF1 construct so that it elutes at a different position in gel filtration chromatography from the TRAF2 construct of similar length. We incubated the two proteins overnight at 4°C and performed gel filtration chromatography on the mixture (Figure 4F). Three peaks were seen in the chromatographic profile, with two peaks corresponding to TRAF1 alone and TRAF2 alone, respectively. The third peak is the highest and corresponds to a heterotrimer of TRAF1 and TRAF2. The location of the peak suggested that it was a 1: 2 heterotrimer of TRAF1 and TRAF2.
To confirm this assignment of the heterotrimer peak, we subjected it to MALS measurement in line with gel filtration chromatography, which gave a molecular mass of 39.8 kDa (3% error) (Figure 4F). Because His-Smt3-TRAF1 and His-TRAF2 have molecular weights of 21,109 Da and 9,613 Da, respectively, the calculated mass of a 1:2 heterotrimer of TRAF1 and TRAF2 would be 40.3 kDa, consistent with the measured mass. As controls, both the His-Smt3-TRAF1 trimer and the His-TRAF2 trimer peaks gave measured masses, 61.0 kDa and 23.5 kDa, respectively, that are consistent with their calculated values of 63.3 kDa and 28.8 kDa, respectively. The preferential formation of a 1:2 heterotrimeric complex is surprising because it has been proposed that TRAF1 and TRAF2 can form both 1:2 and 2:1 heterotrimers (Fotin-Mleczek et al., 2004).
We wondered whether the preferred 1:2 heterotrimer of TRAF1 and TRAF2 was formed because TRAF1 was not in overt excess. To determine this, we mixed at least two-fold molar excess of TRAF1-His with His-Smt3-TRAF2 and performed the similar gel filtration experiments. Even in the excess of TRAF1, TRAF2 still preferentially formed 1:2 heterotrimers as shown both by its elution position and the estimated ratio of TRAF1 and TRAF2 coomassie blue staining intensities on the SDS-PAGE (Supplemental Figure S3D). Therefore, in our experiments with or without excess TRAF1, we could not observe a distinct population of the 2:1 heterotrimer of TRAF1 and TRAF2, confirming that only the TRAF1: (TRAF2)2 heterotrimer is preferentially formed.
The TRAF1: (TRAF2)2 Heterotrimer Has an Enhanced Affinity to cIAP2 Relative to TRAF2
To determine the ability of the 1:2 heterotrimer of TRAF1 and TRAF2 to interact with cIAP2, we co-expressed TRAF1, TRAF2 and cIAP2 in E. coli. Only TRAF2 is His-tagged and the complex was co-purified by Ni-affinity and gel filtration chromatography. Consistent with the preference for 1:2 stoichiometry in the heterotrimeric complex, the approximate ratio between the co-purified TRAF1 and the His-tagged TRAF2 appeared to be 1:2. This ratio was maintained even when excess cell lysate of untagged TRAF1 was added to the cell lysate with the co-expressed proteins during Ni-affinity purification (Figure 4E, bottom gel). Based on the coomassie blue staining intensities, one cIAP2 is bound to a heterotrimer of TRAF1 and TRAF2 (Figure 4E, bottom gel), which is similar to the interaction of cIAP2 with the TRAF2 trimer.
We noticed that the TRAF1: (TRAF2)2 heterotrimer in complex with cIAP2 appeared to dissociate the least during gel filtration chromatography, suggesting that the heterotrimer may have an enhanced affinity to cIAP2 (Figure 4E, bottom gel). Because the TRAF1: TRAF2 heterotrimer is in constant equilibrium with the homotrimers, it is unlikely to obtain a pure species to determine the affinity of its interaction with cIAP2 using ITC. Instead, to compare the affinities of TRAF2 and the TRAF1: TRAF2 heterotrimer for cIAP2, we added cIAP2 to the mixture of His-Smt3-TRAF1 and excess His-TRAF2 and performed gel filtration chromatography. Despite having apparently less heterotrimer than the TRAF2 trimer in the gel filtration profile when TRAF2 was in excess, most cIAP2 co-migrated with the heterotrimer peak (Figure 4G), confirming that the heterotrimer indeed possesses higher affinity than the TRAF2 trimer for cIAP2. Two additional gel filtration runs of somewhat different ratio mixtures of TRAF1, TRAF2 and cIAP2 further showed that the relative affinities for cIAP2 from low to high are TRAF1, TRAF2, and the TRAF1: (TRAF2)2 heterotrimer (Supplemental Figure S3E).
Crystal Structure of the TRAF1: (TRAF2)2 Heterotrimer in Complex with cIAP2
We crystallized the TRAF1: (TRAF2)2: cIAP2 ternary complex and determined its crystal structure at 2.8 Å resolution (Table 1). Because TRAF1 only interacts weakly with cIAP2, we anticipated that most likely the cIAP2-binding site in the heterotrimer is composed of two TRAF2 chains. To our surprise, the cIAP2 binding site in the heterotrimer consists of one TRAF1 and one TRAF2 chains (Supplemental Figure S4A). In addition, it is TRAF1 that contributes the majority of residues to the cIAP2 interaction, not TRAF2.
Although the structure of the TRAF1: (TRAF2)2: cIAP2 complex is very similar to that of the TRAF2: cIAP2 complex, when the cIAP2 molecules are superimposed, the TRAF trimers show a 8.9° rotation relative to each other (Figure 5A). Conversely, when the TRAF trimers are superimposed, the cIAP2 molecules exhibit a similar degree of orientational difference (Supplemental Figure 4B). In this superposition mode, the TRAF side chains at the interface with cIAP2 have similar conformations (Figure 5B). In contrast, the cIAP2 side chains at the interface with the TRAF trimer show significant differences due to the orientational difference (Figure 5C).
Figure 5.

The TRAF1: TRAF2: cIAP2 ternary complex
(A) Superposition of the cIAP2 molecule in the ternary complex structure with the cIAP2 molecule in the TRAF2: cIAP2 binary complex structure, showing the resulting orientational difference in the TRAF trimers. The TRAF2: cIAP2 complex is shown in magenta.
(B) A ribbon diagram of the TRAF1: TRAF2 heterotrimer shown with side chains involved in cIAP2 interaction. Side chains at the heterotrimer are shown with carbon atoms colored according to the ribbon colors. Equivalent side chains at the TRAF2 homotrimer are also shown, with carbon atoms in yellow. The yellow circle indicates the residue only involved in the TRAF2: cIAP2 interaction; the black circle indicates the residue only involved in the cIAP2 interaction in the heterotrimer.
(C) A ribbon diagram of the cIAP2 BIR1 domain, shown with side chains involved in the heterotrimer interaction. Side chains are shown with carbon atoms colored according to the ribbon colors. Equivalent side chains of cIAP2 involved in the TRAF2 homotrimer interaction are also shown, with carbon atoms in yellow. The yellow circle indicates the residue only involved in the TRAF2: cIAP2 interaction; the black circle indicates the residue only involved in the heterotrimer interaction.
(D) Detailed interaction between the TRAF1: TRAF2 heterotrimer and cIAP2. For clarity, only side chains of cIAP2 are shown.
(E) Mutational effects of the TRAF1: TRAF2 complex in cIAP2 interaction. The defective phenotype of the V288A mutation in TRAF2 can be rescued by WT TRAF1.
(F) Pulldown of TRAF1 and TRAF2 by NIK. NIK−/− MEFs were reconstituted with an empty vector (EV) or tandem affinity purification tag (TAP, with a CBP tag and a FLAG tag) fused NIK (TAP-NIK). The cells were lysed and immunoprecipitated with anti-Flag M2 beads followed by immunoblotting with the indicated antibodies.
(G) Immunoassays on lysates from wild-type (WT), Traf1−/−, and Traf3−/− murine transformed B cells. Basal NIK level and p100 to p52 processing in whole cell extracts were detected by immunoblotting.
(H) Co-immunoprecipitation of lysates from HEK 293T cell co-expressing Myc-cIAP2 and Flag-TRAF2, Flag-TRAF2 E292A or Flag-TRAF2 V288A in the presence or absence of His-TRAF1. Lysates were incubated with anti-Flag M2 beads and TRAF2: cIAP2 and TRAF2: TRAF1 complexes were detected by immunoblot analysis against Myc and TRAF1 respectively. All experiments were done at least three times with similar results.
The orientational difference between the TRAF trimer and cIAP2 in the TRAF1: TRAF2: cIAP2 complex generates a tilt that allows more contacts at the lower part of the interface on both the TRAF trimer and cIAP2 (Figure 5B, 5C, 5D). Particularly, TRAF1 residue V196 (equivalent to TRAF2 residue T281) extends the hydrophobic patch formed by I200 and V203 (equivalent to TRAF2 residues I285 and V288), and directly interacts with Y31 from cIAP2. TRAF2 residue E283 interacts with L26 of cIAP2 in the ternary complex, both of which are not involved in the interaction in the TRAF2: cIAP2 complex. In contrast, the interactions at the TRAF2 residue R295 (equivalent to A210 of TRAF1) and at the cIAP2 residue R52 are no longer present in the ternary complex. The replacement of R291 of TRAF2 to K206 of TRAF1 eliminated the hydrogen bonding interaction mediated by R291. Instead, the K206 side chain of TRAF1 forms hydrogen bonds with the main chain carbonyl oxygen atoms of S37 and F39 of cIAP2.
We performed structure-based mutagenesis on TRAF1 to confirm its critical role in recognition of cIAP2 by the TRAF1: TRAF2 heterotrimer (Figure 5E). Mutations on TRAF1 residues I200, V203, K206 and E207, which are equivalent to TRAF2 residues I285, V288, R291 and E292 respectively, drastically or moderately reduced the interaction of the heterotrimer with cIAP2. In addition, when V288 is mutated to Ala in the TRAF2: cIAP2 complex, the interaction was drastically compromised. However, when V288A is generated in the heterotrimer, cIAP2 interaction is mostly maintained, consistent with the structural observation that V288 of TRAF2 is not critical in cIAP2 interaction in the heterotrimer.
Overall, the interaction between the TRAF1: TRAF2 heterotrimer and cIAP2 buries a similar extent of surface area and has a similar number of van der Waals contacts in comparison with the TRAF2: cIAP2 complex. However, there seems to be a better shape complementarity (Supplemental Table S4) and a higher number of hydrophobic contacts, which could contribute to the higher affinity. It is also possible that the apparent higher stability of the TRAF1: (TRAF2)2 heterotrimer facilitates cIAP2 interaction.
TRAF1 is a Component of the E3 complex for NIK
The ability of the TRAF1: (TRAF2)2 complex to interact with cIAP2 robustly suggests that TRAF1 may also be a physiological component of the ubiquitin ligase complex of cIAP1/2. To test this hypothesis, we reconstituted NIK−/− MEFs with an empty vector (EV) or tandem affinity purification (TAP) tag fused NIK. Immunoprecipitation using anti-Flag M2 beads showed that both TRAF2 and TRAF1 were pulled down by NIK, confirming that TRAF1 is a component of the E3 for NIK (Figure 5F).
TRAF1 Enhances the TRAF2: cIAP2 Interaction in Cells
To elucidate the biological relevance of TRAF1 in cIAP1/2 interaction, we first used Traf1−/− B cells to determine whether TRAF1 is required for NIK degradation and noncanonical NF-κB activation. The experiment showed that TRAF1 is not critical for this activity (Figure 5G), consistent with a role of TRAF1 as a regulatory, rather than obligatory molecule in cIAP1/2 function. We then used co-transfection to determine whether TRAF1 has any effect in the TRAF2: cIAP2 interaction. We showed consistently that indeed TRAF1 enhanced the pulldown of cIAP2 by TRAF2 significantly (Figure 5H). In addition, the V288A mutant of TRAF2 is rescued for interaction with cIAP2 by TRAF1. In contrast, the E292A mutant of TRAF2 did not interact with TRAF1 and was not rescued for interaction with cIAP2 (Figure 5H). Collectively, these studies suggest that TRAF1 and TRAF2 do form a complex in cells to modulate cIAP2 recruitment.
DISCUSSION
The Molecular Mechanisms of TRAF1/2: cIAP1/2 Interactions
Our current study elucidated the mode of interaction between TRAF1/2 and cIAP1/2 and confirmed their interactions in cells. Interestingly, the surface of the BIR1 domain of cIAP1/2 for TRAF interaction is equivalent to that of the BIR1 domain of XIAP for TAB1 interaction (Lu et al., 2007) (Figure 6A), identifying this surface of the BIR domains as a common interaction surface. The opposite side of this surface defines another common interaction surface of the BIR domains, which interacts with Smac in the BIR2 and BIR3 domains of cIAP1/2 and XIAP, and dimerizes in the BIR1 domain of XIAP (LaCasse et al., 2008; Lu et al., 2007) (Figure 6A, 6B).
Figure 6.
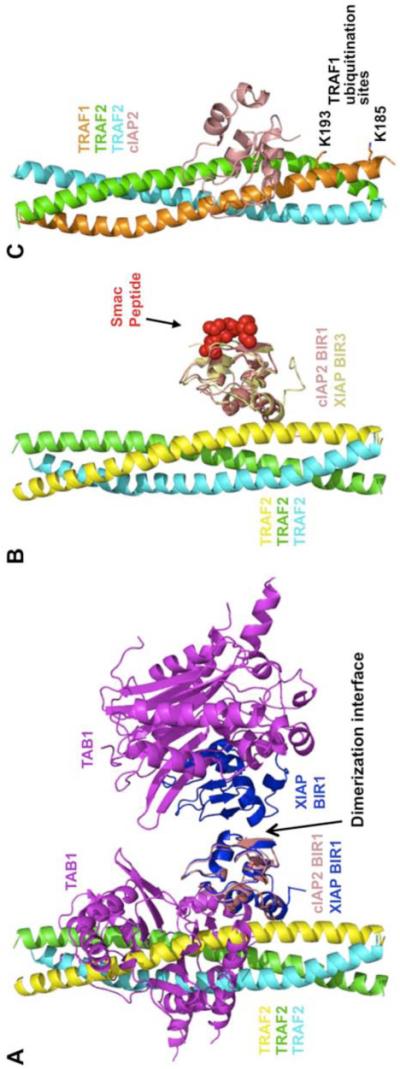
Implications of the TRAF1/2: cIAP1/2 interaction
(A) Comparison of the TRAF2: cIAP2 interaction with the dimeric XIAP BIR1: TAB1 interaction. The TAB1-interacting surface of XIAP BIR1 overlaps with the TRAF2-interacting surface of cIAP BIR1. The dimerization interface of XIAP BIR1 is opposite of the above interface.
(B) Comparison of the TRAF2: cIAP2 interaction with the XIAP BIR3: Smac peptide interaction. Smac peptide is bound to the opposite side of the TRAF2: cIAP interface.
(C) The heterotrimer of TRAF1 and TRAF2 in complex with cIAP2, showing the ubiquitination sites on TRAF1.
One unexpected finding is that TRAF1 and TRAF2 are not created equal with regards to interaction with cIAP1/2. TRAF2 interacts with cIAP2 at an affinity that is about two orders of magnitude higher than that of TRAF1. In contrast, the 1:2 heterotrimer of TRAF1 and TRAF2, the preferred oligomerization form when both TRAF1 and TRAF2 are present, interacts with cIAP2 at a stronger affinity than TRAF2.
Another unexpected finding is that both TRAF2 trimer and the TRAF1: TRAF2 heterotrimer interact with one, instead of three, cIAP2 molecules. This may be attributed to generation of asymmetry in the TRAF trimer upon cIAP2 binding and therefore prohibition of interacting with more cIAP2 molecules. In contrast, TRAF1 trimer can interact with three cIAP2 molecules simultaneously. It is likely that the amino acid difference in TRAF1 weakens the interaction with cIAP2 to such an extent that deformation of the symmetrical TRAF1 trimer cannot occur, which in turn further contributes to the weaker interaction. The mode of TRAF2 binding for cIAP2 uncovered here should also be true for cIAP1, which has identical binding site residues. Existing mutagenesis data have implicated cIAP1 residues equivalent to L30, E47 and R48 of cIAP2 as important for TRAF2 interaction (Samuel et al., 2006; Varfolomeev et al., 2006).
Multiple Mechanisms of TRAF2 Regulation by TRAF1
Accumulating data support a role for TRAF1 as both a negative and a positive modulator of signaling by certain TNF family receptors, possibly in a cell-type dependent manner (Fotin-Mleczek et al., 2004; Lee and Choi, 2007). Our study revealed that the TRAF1: (TRAF2)2 heterotrimer has an enhanced affinity to cIAP2 in comparison with the TRAF2 trimer. We confirmed this observation in cells, providing one possible mechanism for regulation of TRAF2 function by TRAF1. We further showed that TRAF1 rescued a cIAP2-binding defective mutant of TRAF2 in cIAP2 interaction through heterotrimerization. Because TRAF2 alone also interacts with cIAP1/2 robustly, TRAF1−/− cells may not exhibit dramatic phenotypes. However, it has been shown that physiological expression of TRAF1 enhances NF-κB activation and interleukin-8 production induced by TNFR2 (Wicovsky et al., 2009), consistent with the role of TRAF1: (TRAF2)2 heterotrimer in promoting signal-dependent NF-κB activation.
The structure of the TRAF1: TRAF2: cIAP2 may also explain how TRAF1 can inhibit TNFR2-induced proteasomal degradation of TRAF2. Both TRAF1 and TRAF2 are targets of polyubiquitination and degradation by cIAP1/2 upon recruitment to TNF family receptors (Dupoux et al., 2009). The major sites of ubiquitination of TRAF2 by cIAP1/2 are not known. However, in the structure of the heterotrimer: cIAP2 complex, the major ubiquitination sites of TRAF1, K185 and K193 (Lee et al., 2004a), are immediately adjacent to the binding site for cIAP2 (Figure 6C). This provides a potential mechanism for the preference of TRAF1 as a ubiquitination target of cIAP1/2 and therefore for inhibiting TRAF2 degradation.
The TRAF1/2: cIAP1/2 Interaction as a Potential Therapeutic Target
The TRAF1/2: cIAP1/2 interaction appears to be a critical switch between cell survival and cell death. It has long been observed that TRAF1, TRAF2 and cIAP1/2 suppress caspase-8 activation and apoptosis induction (Wang et al., 1998). Recent studies have revealed the molecular basis for this observation. When cIAP1/2 are recruited by TRAF1/2 to a TNF family signaling complex, RIP1 is modified with K63-linked polyubiquitin chains, leading to IKKβ recruitment and NF-κB activation. When cIAP1/2 are induced to auto-ubiquitinate and to degrade, e.g. by Smac mimetics, RIP1 is de-ubiquitinated and leave the NF-κB signaling complex to form alternative complexes for cell death induction (Wang et al., 2008).
A similar induction of cell death may be elicited by inhibiting the interaction between TRAF2 and cIAP1/2 because releasing cIAP1/2 from a TNF family signaling complex would prohibit K63-linked polyubiquitination of RIP1. This would switch the cell fate from NF-κB activation and cell survival to cell death. This induction of cell death is also supported by the observation that when the TRAF2: cIAP1/2 complex is competitively recruited to other TNF receptor family members, induction of caspase-8 activation by TNFα is accelerated (Fotin-Mleczek et al., 2002). Therefore, our current study offers both the rational and the potential applicability for yet another way to inhibit IAP family members to combat cancer.
EXPERIMENTAL PROCEDURES
Crystallization and Structure Determination
The TRAF2: cIAP2 complex was crystallized by seeding using 8 % PEG3350 and 6% Tacsimate at pH 5.0. The TRAF1: (TRAF2)2: cIAP2 complex was crystallized using 0.15 M Ammonium sulfate, 0.1 M MES pH 6 and 15 % (w/v) PEG 4000 in a sitting drop vapor diffusion system at 4 °C.
The structure of the TRAF2: cIAP2 complex was determined by multi-wavelength anomalous diffraction (MAD) of the intrinsic zinc atoms. The structures of TRAF2 alone and of the TRAF1: TRAF2: cIAP2 complex were solved by molecular replacement.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Miao Lu for his earlier work on the project, PXRR staff for data collection at X29 and X25 of NSLS, and Jin Wu for maintaining X-ray and computer equipment. This work was supported by National Institute of Health (RO1 AI045937 to HW).
ACCESSION NUMBERS
The atomic coordinates have been deposited in the RCSB Protein Data Bank with the PDB codes of 3M06, 3M0A, and 3M0D for TRAF2 alone, the TRAF2: cIAP2 complex and the TRAF1: TRAF2: cIAP2 complex, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Arron JR, Pewzner-Jung Y, Walsh MC, Kobayashi T, Choi Y. Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J Exp Med. 2002;196:923–934. doi: 10.1084/jem.20020774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- Carpentier I, Beyaert R. TRAF1 is a TNF inducible regulator of NF-kappaB activation. FEBS Lett. 1999;460:246–250. doi: 10.1016/s0014-5793(99)01356-3. [DOI] [PubMed] [Google Scholar]
- Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- Dupoux A, Cartier J, Cathelin S, Filomenko R, Solary E, Dubrez-Daloz L. cIAP1-dependent TRAF2 degradation regulates the differentiation of monocytes into macrophages and their response to CD40 ligand. Blood. 2009;113:175–185. doi: 10.1182/blood-2008-02-137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Fotin-Mleczek M, Henkler F, Hausser A, Glauner H, Samel D, Graness A, Scheurich P, Mauri D, Wajant H. Tumor necrosis factor receptor-associated factor (TRAF) 1 regulates CD40-induced TRAF2-mediated NF-kappaB activation. J Biol Chem. 2004;279:677–685. doi: 10.1074/jbc.M310969200. [DOI] [PubMed] [Google Scholar]
- Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, Scheurich P, Schmid JA, Wajant H. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J Cell Sci. 2002;115:2757–2770. doi: 10.1242/jcs.115.13.2757. [DOI] [PubMed] [Google Scholar]
- Garrison JB, Samuel T, Reed JC. TRAF2-binding BIR1 domain of c-IAP2/MALT1 fusion protein is essential for activation of NF-kappaB. Oncogene. 2009;28:1584–1593. doi: 10.1038/onc.2009.17. [DOI] [PubMed] [Google Scholar]
- Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996a;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu H-B, Pan M-G, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996b;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27:6252–6275. doi: 10.1038/onc.2008.302. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- Lee JS, Hong US, Lee TH, Yoon SK, Yoon JB. Mass spectrometric analysis of tumor necrosis factor receptor-associated factor 1 ubiquitination mediated by cellular inhibitor of apoptosis 2. Proteomics. 2004a;4:3376–3382. doi: 10.1002/pmic.200401000. [DOI] [PubMed] [Google Scholar]
- Lee SY, Choi Y. TRAF1 and its biological functions. Adv Exp Med Biol. 2007;597:25–31. doi: 10.1007/978-0-387-70630-6_2. [DOI] [PubMed] [Google Scholar]
- Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004b;279:33185–33191. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
- Lu M, Lin SC, Huang Y, Kang YJ, Rich R, Lo YC, Myszka D, Han J, Wu H. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol Cell. 2007;26:689–702. doi: 10.1016/j.molcel.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas PC, Kuffa P, Gu S, Kohrt D, Kim DS, Siu K, Jin X, Swenson J, McAllister-Lucas LM. A dual role for the API2 moiety in API2-MALT1-dependent NF-kappaB activation: heterotypic oligomerization and TRAF2 recruitment. Oncogene. 2007;26:5643–5654. doi: 10.1038/sj.onc.1210342. [DOI] [PubMed] [Google Scholar]
- Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YC, Burkitt V, Villa AR, Tong L, Wu H. Structural basis for self-association and receptor recognition of human TRAF2. Nature. 1999;398:533–538. doi: 10.1038/19110. [DOI] [PubMed] [Google Scholar]
- Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, Harran P, Wang X. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. 2007;12:445–456. doi: 10.1016/j.ccr.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- Samuel T, Welsh K, Lober T, Togo SH, Zapata JM, Reed JC. Distinct BIR domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem. 2006;281:1080–1090. doi: 10.1074/jbc.M509381200. [DOI] [PubMed] [Google Scholar]
- Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol. 2004;4:348–359. doi: 10.1038/nri1352. [DOI] [PubMed] [Google Scholar]
- Tsitsikov EN, Laouini D, Dunn IF, Sannikova TY, Davidson L, Alt FW, Geha RS. TRAF1 is a negative regulator of TNF signaling. enhanced TNF signaling in TRAF1-deficient mice. Immunity. 2001;15:647–657. doi: 10.1016/s1074-7613(01)00207-2. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev E, Wayson SM, Dixit VM, Fairbrother WJ, Vucic D. The inhibitor of apoptosis protein fusion c-IAP2.MALT1 stimulates NF-kappaB activation independently of TRAF1 AND TRAF2. J Biol Chem. 2006;281:29022–29029. doi: 10.1074/jbc.M605116200. [DOI] [PubMed] [Google Scholar]
- Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC, Davidson AJ, Callus BA, Wong WW, Gentle IE, et al. TRAF2 must bind to cIAPs for TNF to efficiently activate NF-{kappa}B and to prevent TNF-induced apoptosis. J Biol Chem. 2009 doi: 10.1074/jbc.M109.072256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c- IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- Wicovsky A, Henkler F, Salzmann S, Scheurich P, Kneitz C, Wajant H. Tumor necrosis factor receptor-associated factor-1 enhances proinflammatory TNF receptor-2 signaling and modifies TNFR1-TNFR2 cooperation. Oncogene. 2009;28:1769–1781. doi: 10.1038/onc.2009.29. [DOI] [PubMed] [Google Scholar]
- Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Hakem R, Woo M, Mak TW. Gene targeting in the analysis of mammalian apoptosis and TNF receptor superfamily signaling. Immunol Rev. 1999;169:283–302. doi: 10.1111/j.1600-065x.1999.tb01323.x. [DOI] [PubMed] [Google Scholar]
- Yin Q, Lamothe B, Darnay BG, Wu H. Structural Basis for the Lack of E2 Interaction in the RING Domain of TRAF2. Biochemistry. 2009a doi: 10.1021/bi901462e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, Lin SC, Lamothe B, Lu M, Lo YC, Hura G, Zheng L, Rich R, Campos AD, Myszka DG, et al. E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol. 2009b;16:658–666. doi: 10.1038/nsmb.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata JM, Lefebvre S, Reed JC. Targeting TRAfs for therapeutic intervention. Adv Exp Med Biol. 2007;597:188–201. doi: 10.1007/978-0-387-70630-6_15. [DOI] [PubMed] [Google Scholar]
- Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.