

Prostaglandin H2 Synthase (PGHS) is responsible for the conversion of arachidonic acid (AA) to prostaglandin H2, the first committed step in the biosynthesis of the prostanoids.1a Two different isoforms of PGHS have been identified. PGHS-1 is constitutively expressed and appears to function as a housekeeping enzyme, while PGHS-2 can be induced by a wide variety of stimulants and is thought to participate in such diverse physiological and pathological processes as reproduction, cardiovascular function, atherosclerosis, tumorigenesis, and inflammation.1b The two isoforms have approximately 60% sequence identity and very similar three-dimensional structures, as well as similar kinetic profiles, indicating that they probably share the same basic mechanism. PGHS has two distinct active sites, which are functionally connected by the generation of a radical on Y385 (PGHS-1 numbering). The radical is generated by the heme-containing peroxidase site and is used to initiate the catalytic cycle of the cyclooxygenase (COX) site via abstraction of the 13 pro-S hydrogen of AA.
The Y385 radical (Y385•), which forms in ovine PGHS-1 (oPGHS-1) within ~10 s after reacting the substrate-free enzyme with hydroperoxides, initially produces a characteristic “wide doublet” EPR spectrum (WD1, 19 G splitting, 35 G overall peak-to-trough width) when measured at X-band frequencies (9 GHz). On the order of seconds, the WD1 spectrum converts to a “wide singlet” (WS1, 35 G width) and finally to a “narrow singlet” (NS1b, 25–28 G width) before decaying to zero intensity.2 Preincubating the enzyme with COX inhibitor produces a narrow singlet spectrum (NS1a) which has better resolved hyperfine features than those of NS1b.3 The human PGHS-2 isoform displays a similar set of EPR spectra, but with different time scales (e.g., the initial WD2 signal converts to a WS2 in ~50 ms,1 and no NS1b was observed).
The structural characterization of these various radical species remains a crucial aspect of determining the mechanisms of catalysis and self-inactivation of PGHS. All of the species described above have been identified as tyrosyl radicals.2,3a However, identification of the specific tyrosine residue and/or the structural details of that residue, and thus the explanation for the evolution of the EPR spectrum, remain unresolved. One possibility for conversion of the WD to WS to NS is the migration of the radical from Y385• to another tyrosine residue. The second residue would have different phenyl ring orientation with respect to the methylene β-protons, leading to a smaller hyperfine coupling and narrower EPR spectrum. An alternative mechanism is that the WD tyrosyl radical undergoes a ring rotation itself, and that all spectra arise from the Y385•. The WS spectra have been shown to be arithmetic sums of WD and NS,2e,4,5 but this does not differentiate between the radical hopping and ring-rotation mechanisms.
To further characterize PGHS-1 tyrosyl radical structure, two studies have been conducted to detect a hydrogen bond to Y385• in ovine PGHS-1 (oPGHS-1), with conflicting results. In the first, continuous wave (CW) EPR of WD at 9 GHz gave a gX value consistent with that of a tyrosyl radical lacking a hydrogen bond.4 Also, 1H CW ENDOR showed no features affected by solvent exchange with 2H2O, and thus, it was concluded that no hydrogen bond was present in the WD.4 The second study, however, examined WD using high-frequency (HF) EPR and determined that the gX value was in the range associated with a hydrogen-bonded tyrosyl radical, and that a distribution in gX values was required to simulate the data.5 In this study, we directly detect the presence of a hydrogen bond to the WD radical, as well as the distribution in gX values. We also determine the geometry of the hydrogen bond, propose a hydrogen bond partner, and show that there is sufficient disorder in the hydrogen bond to account for the distribution in gX observed in the HFEPR spectrum.
The X-band CW EPR spectrum of the oPGHS-1 WD is shown in Figure 1 (inset). The spectrum can be simulated using a gX value of 2.0066, which indicates the presence of a hydrogen bond to the tyrosyl radical.6 At 9 GHz, the spectrum is dominated by hyperfine interactions, and it is not possible to distinguish between the simulations with and without a gX distribution. Because the hyperfine interactions are, to first order, independent of applied field and the Zeeman interactions are proportional to the field, the g values and any distribution in gX will be more resolved at higher frequencies. Figure 1 shows the HF echo-detected EPR spectrum of a WD sample exchanged in 2H2O. The same g tensor was used for both the X-band and HF simulations. However, the HF simulation could only be fit to the data if a distribution wasincorporated in gX. This distribution was assumed to be Gaussian in shape, centered at 2.0066, with a full-width at half-height (fwhh) of 0.0020.
Figure 1.
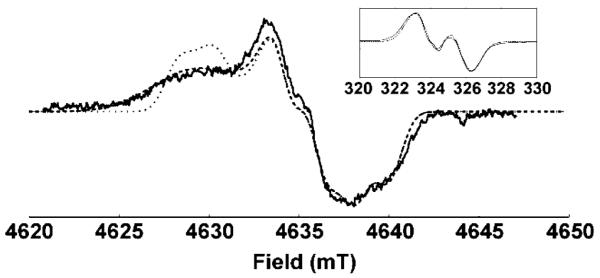
Derivative of the HF echo-detected EPR absorption spectrum of 2H-exchanged WD1 (–), and simulations with (- - -) and without (···) a distribution in gX. Experimental parameters: 9 K; EPR frequency = 130.00 GHz; pulse widths ) 50 ns; tau = 120 ns; repetition rate = 100 Hz; shots per scan = 100; total scans = 7. Inset: X-band EPR spectrum (–) and simulations with (- - -) and without (···) a distribution in gX. Experimental parameters: 77 K; EPR frequency = 9.101 GHz; power = 0.5 mW; modulation amplitude = 0.25 mT; time constant = 1 s; scan time = 4 min; total scans = 4.
To directly examine the hydrogen bond and to determine the position of the hydrogen bond proton, HF 2H Mims ENDOR was carried out on the same sample used for the HF EPR spectrum. The resulting ENDOR spectrum is shown in Figure 2. Simulations of the ENDOR spectrum yielded a significantly better fit when a distribution was included in the hydrogen bond length. On the basis of these simulations, the proton is located in the plane of the aromatic ring of Y385, forming a 120° bond angle with the C–O bond. The distribution of O···H distances was centered around 1.5 Å, with a fwhh of 0.2 Å. It is notable that a recent X-band ENDOR study of Mn-depleted PSII also detected disorder in the hydrogen-bonding environment of the tyrosyl radical Yz.7
Figure 2.
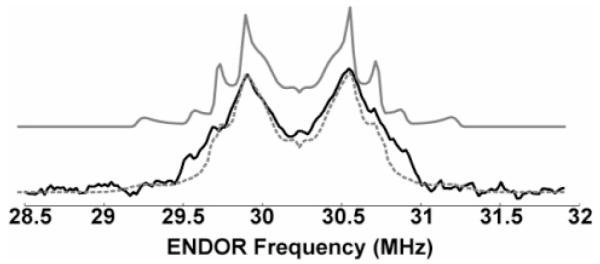
Mims HFENDOR of 2H WD1. Experimental spectrum (–) and simulations with (- - -, gray) and without (–, gray) a distribution in hydrogen bond length (see text). Experimental parameters: 7 K; EPR frequency = 130.00 GHz; magnetic field = 4635.8 mT; EPR pulse widths = 40 ns; ENDOR pulse width = 20 ms; tau = 100 ns; T = 21 μs; repetition rate = 100 Hz; shots per scan = 30; total scans = 1320; RF power output ~1 kW.
It is valuable to use the HFENDOR results presented here to determine if this H-bond distribution can explain the observed gX distribution. This can be done using a simple empirical formula that has been proposed to relate the gX values of tyrosyl radicals to hydrogen bond lengths: gX = 2.0094–0.0033/(r – 0.5)2, where r is the O···H distance in angstroms.6a,8 From our data, we have independently obtained values for gX and r (from the HFEPR and HFENDOR spectra, respectively). The r values predict a gX distribution whose fwhh is 2.0053–2.0067; the central r value corresponds to a gX value of 2.0061. These results agree well with the observed gX values, and thus, we propose that the distribution in gX arises primarily from the structural disorder in the H bond.
The crystal structure of oPGHS-1 has been solved with AA bound in the COX active site.9 In this structure, Y385 is located at the edge of the hydrophobic channel that forms the cyclooxygenase active site. Its side chain is directed into the channel, such that the phenoxyl oxygen is 2.7 Å from C13 of the AA, a finding consistent with the proposed catalytic mechanism. Y348 is found on the opposite side of the channel, with its side chain oriented toward that of Y385. The oxygen–oxygen distance is 2.4 Å, and the angle between the line connecting the two oxygen atoms and the Y385 C—O bond is 128°. This geometry is highly favorable for hydrogen bonding, and Y348 has, therefore, been proposed as a hydrogen bond partner for the Y385•.9,10 The ENDOR data in this work have located the hydrogen bond proton to be in a position consistent with the presumed location of the phenoxyl proton of Y348, based on the crystal structure. It is important to note that while the crystal structure was not determined from the WD form of oPGHS-1, the dihedral angles between the β-carbon protons and the ring of Y385 are consistent with angles predicted on the basis of the observed hyperfine couplings arising from these protons. A recent crystal structure of oPGHS-1 in a complex with the cyclooxygenase inhibitor, α-methyl-4-biphenylacetic acid, reveals a water molecule in a nearly symmetrical arrangement on the other side of Y385, at a slightly longer distance from the phenoxyl O of Y385 (2.84 Å, as opposed to 2.66 Å for Y348 in this structure).11 The ENDOR simulation cannot determine to which side of the symmetric tyrosyl residue the hydrogen bond is directed, but the determination of the O···H distance as 1.5 Å is more consistent with a hydrogen bond to Y348.
Hydrogen-bonding networks have been shown to provide pathways for proton-coupled electron transfer in enzymes.12 The demonstrated ability of Y385• to participate in a hydrogen bond suggests that such a network may be involved in the mechanism of self-inactivation and/or regeneration of neutral Y385. Recent EPR studies using site-directed mutants have provided evidence for the radical migration mechanism to explain the EPR spectral changes that occur upon inactivation of PGHS-2.13 However, the data presented here cannot rule out tyrosine ring rotation as an explanation for the evolution of the EPR spectra. Further study of wild type and mutants will help clarify the role of the tyrosine residues and the hydrogen bond to Y385• in PGHS inactivation.
Acknowledgment
We thank Vladimir Krymov for technical assistance, and Professors Brian Hoffman and Robert Bittl for helpful discussions. J.C.W. is supported by the Medical Scientist Training Program at the AECOM. This work was supported by NIH Grants GM44911 (A.L.T.) and GM60609 (G.J.G.).
Footnotes
Supporting Information Available: A description of the simulations, a full parameter set, and details of sample preparation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1)(a).Tsai AL, Kulmacz RJ. Prostaglandins Other Lipid Mediators. 2000;62:231–254. doi: 10.1016/s0090-6980(00)00083-6. [DOI] [PubMed] [Google Scholar]; (b) Warner TD, Mitchell JA. FASEB J. 2004;18:790–804. doi: 10.1096/fj.03-0645rev. [DOI] [PubMed] [Google Scholar]
- (2)(a).Karthein R, Dietz R, Nastainczyk W, Ruf HH. Eur. J. Biochem. 1988;171:313–320. doi: 10.1111/j.1432-1033.1988.tb13792.x. [DOI] [PubMed] [Google Scholar]; (b) Kulmacz RJ, Ren Y, Tsai AL, Palmer G. Biochemistry. 1990;29:8760–8671. doi: 10.1021/bi00489a037. [DOI] [PubMed] [Google Scholar]; (c) Lassmann G, Odenwaller R, Curtis JF, DeGray JA, Mason RP, Marnett LJ, Eling TE. J. Biol. Chem. 1991;266:20045–20055. [PubMed] [Google Scholar]; (d) Tsai AL, Palmer G, Kulmacz RJ. J. Biol. Chem. 1992;267:17753–17759. [PubMed] [Google Scholar]; (e) DeGray JA, Lassmann G, Curtis JF, Kennedy TA, Marnett LJ, Eling TE, Mason RP. J. Biol. Chem. 1992;267:23583–23588. [PubMed] [Google Scholar]
- (3)(a).Tsai AL, Hsi LC, Kulmacz RJ, Palmer G, Smith WL. J. Biol. Chem. 1994;269:5085–5091. [PubMed] [Google Scholar]; (b) Kulmacz RJ, Palmer G, Tsai AL. Mol. Pharmacol. 1991;40:833–837. [PubMed] [Google Scholar]; (c) Shimokawa T, Kulmacz RJ, DeWitt DL, Smith WL. J. Biol. Chem. 1990;265:20073–20076. [PubMed] [Google Scholar]
- (4).Shi W, Hoganson CW, Espe M, Bender CJ, Babcock GT, Palmer G, Kulmacz RJ, Tsai AL. Biochemistry. 2000;39:4112–4121. doi: 10.1021/bi992561c. [DOI] [PubMed] [Google Scholar]
- (5).Dorlet P, Seibold SA, Babcock GT, Gerfen GJ, Smith WL, Tsai AL, Un S. Biochemistry. 2002;41:6107–6114. doi: 10.1021/bi015871f. [DOI] [PubMed] [Google Scholar]
- (6)(a).Un S, Atta M, Fontecave M, Rutherford AW. J. Am. Chem. Soc. 1995;117:10713–10719. [Google Scholar]; (b) Ivancich A, Mattioli TA, Un S. J. Am. Chem. Soc. 1999;121:5743–5753. [Google Scholar]; (c) Un S, Gerez C, Elleingand E, Fontecave M. J. Am. Chem. Soc. 2001;123:3048–3054. doi: 10.1021/ja003650b. [DOI] [PubMed] [Google Scholar]
- (7).Force DA, Randall DW, Britt RD, Tang XS, Diner BA. J. Am. Chem. Soc. 1995;117:12643–12644. [Google Scholar]
- (8).Un S, Tang XS, Diner BA. Biochemistry. 1996;35:679–684. doi: 10.1021/bi9523769. [DOI] [PubMed] [Google Scholar]
- (9).Malkowski MG, Ginell SL, Smith WL, Garavito RM. Science. 2000;289:1933–1937. doi: 10.1126/science.289.5486.1933. [DOI] [PubMed] [Google Scholar]
- (10)(a).Picot D, Loll PJ, Garavito RM. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]; (b) Loll PJ, Picot D, Garavito RM. Nat. Struct. Biol. 1995;2:637–643. doi: 10.1038/nsb0895-637. [DOI] [PubMed] [Google Scholar]; (c) Loll PJ, Picot D, Ekabo O, Garavito RM. Biochemistry. 1996;35:7330–7340. doi: 10.1021/bi952776w. [DOI] [PubMed] [Google Scholar]; (d) Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Biochemistry. 2001;40:5172–5180. doi: 10.1021/bi010045s. [DOI] [PubMed] [Google Scholar]
- (11).Gupta K, Selinsky BS, Kaub CJ, Katz AK, Loll PJ. J. Mol. Biol. 2004;335:503–518. doi: 10.1016/j.jmb.2003.10.073. [DOI] [PubMed] [Google Scholar]
- (12).Ekberg M, Sahlin M, Eriksson M, Sjöberg BM. J. Biol. Chem. 1996;271:20655–20659. doi: 10.1074/jbc.271.34.20655. [DOI] [PubMed] [Google Scholar]
- (13).Rogge CE, Liu W, Wu G, Wang L-H, Kulmacz RJ, Tsai AL. Biochemistry. 2004;43:1560–1568. doi: 10.1021/bi035717o. [DOI] [PubMed] [Google Scholar]