

Abstract
Daxx is a regulatory protein for apoptosis signal-regulating kinase 1 (ASK1) which activates Jun N-terminal kinase (JNK) and p38 pathways in response to stressors such as tumor necrosis factor α (TNFα). Here we show that TNFα treatment induces the accumulation of Daxx protein through ASK1 activation by preventing its proteasome-dependent degradation. ASK1 directly phosphorylates Daxx at Ser176 and Ser184 and Daxx is required for the sustained activation of JNK. Tumorigenic mutant p53, which binds to Daxx and inhibits Daxx-dependent activation of ASK1, prevents Daxx phosphorylation and stabilization. When mutant p53 was depleted in cancer cells, Daxx was accumulated and the cell killing effect of TNFα was restored. Our results indicate that Daxx not only activates ASK1 but also is a downstream target of ASK1 and that accumulated Daxx further activates ASK1. Thus, the Daxx-ASK1 positive feedback loop amplifying JNK/p38 signaling plays an important role for cell killing effects of stressors, such as TNFα. Tumorigenic mutant p53 disrupts this circuit and makes cells more tolerable to stresses, as its gain-of-function mechanism.
Keywords: mutant p53, Daxx, ASK1, phosphorylation, protein stabilization
Introduction
Daxx was identified as a Fas death-domain interacting protein and shown to enhance Fas-induced apoptosis through the activation of c-Jun N-terminal kinase (JNK) (1). Daxx binds to and activates apoptosis signal-regulating kinase 1 (ASK1), a member of stress-responsive mitogen-activated protein kinase kinase kinase (MAP3K) family (2, 3). ASK1 activates the MKK4/JNK and MKK6/p38 MAPK pathways through their phosphorylation. ASK1 displays pro-apoptotic activity in responding to diverse stimuli such as tumor necrosis factor (TNFα), anti-Fas antibody and oxidative stress (3–5).
Daxx is a ubiquitously expressed gene (6) and has an essential developmental function (7). Based on a recent study, Daxx-silencing in cells compels a resistance to cell death induced by UV irradiation and oxidative stress, and an impaired JNK activation (8), suggesting that Daxx is a regulatory upstream factor of JNK to induce apoptosis. In addition, ASK1−/− and JNK−/− cells are resistant to TNFα- and H2O2-induced apoptosis (9, 10). Nevertheless, controversial results have been reported that Daxx has an anti-apoptotic effect (11) or sensitizes cells to Fas-induced apoptosis (12). Blocking of the JNK activation and the apoptotic induction has been observed in cells overexpressing a dominant-negative kinase-inactive mutant of ASK1 (K709M) (3, 4). Furthermore, ASK1 is required for sustained activation of JNK/p38 and apoptosis (10). These findings suggest a physiological significance in the network of Daxx-ASK1-JNK signaling for apoptotic cell death.
Translocation of Daxx shuttling between the nucleus and cytoplasm has been implicated in its apoptotic function (13). Daxx is mainly located in the nucleus, whereas it is a pro-apoptotic signal mediator in the cytoplasm when ASK1 is over-expressed (14). This seems to be consistent with the report describing the cytoplasmic localization of Daxx upon exposure to stresses such as Fas crosslinking (15). Homeodomain-interacting protein kinases (HIPKs) have been found to phosphorylate Daxx. HIPK1 phosphorylates Daxx at Ser 667 and consequently relocalizes Daxx from promyelocytic leukemia protein-nuclear bodies (PML-NBs). The relocalization of Daxx by HIPK1 is dependent on the HIPK1 active-kinase domain, however, independent of Daxx phosphorylation (16). The translocated Daxx binds to ASK1 and leads to ASK1 oligomerization (17). HIPK2 also binds to Daxx and induces its phosphorylation, which in turn leads to JNK activation.
Here we report that TNFα treatment induces phosphorylation and stabilization of Daxx protein through ASK1 activation, which is essential for sustained JNK activation and resultant apoptosis. This positive feedback loop mechanism is inhibited by tumorigenic mutant p53.
Materials and Methods
Plasmids construction and siRNA synthesis
Various Daxx substitution mutants were generated by PCR with the primers; T174A/F (5′-CAAATGCTGAAAACGCTGCCTCTCAG-3′), T174A/R (5′-CTGAGAGGCAGCGTTTTCAGCATTTG-3′), S176A/F (5′-GAAAACACTGCCGCTCAGTCTCCAAG-3′), S176A/R (5′-CTTGGAGACTGAGCGGCAGTGTTTTC-3′), T181A/F (5′-GTCTCCAAGGGCCCGTGGTTCCCGGCGGCAG-3′), T181A/R (5′-CTGCCGCCGGGAACCACGGGCCCTTGGAGAC-′ 3), S184A/F (5′-GTCTCCAAGGACCCGTGGTGCCCGGCGGCAG-3′ ′), S184A/R (5′-CTGCCGCCGGGCACCACGGGTCCTTGGAGAC-3′), T174AS176AS178AT181AS184A/F (5′-GAAAACGCTGCCGCTCAGGCGCCAAGGGCCCGTGGTGCC-3′), T174AS176AS178AT181AS184A/R (5′-GGCACCACGGGCCCTTGGCGCCTGAGCGGCAGCGTTTTC-3′). For generating GST-Daxx 70–216 WT and its substitution mutants, the N-terminal region of Daxx was amplified using primers (5′-GGAGGAGGATCCCCTTCCTTGAACTTTGTAAGATGCAG-3′) and (5′-GGAGGAGAATTCCTAATCCAATTCTGAGAGATCCAACTC-3′). The siRNAs for Daxx and negative control were obtained from Invitrogen, The siRNA sequence for Daxx-1 was described previously (11): 5′-GGAAAAGGAGUUGGAUCUCUCAGAA-3′. The siRNA sequence for Daxx-2 is 5′-CCCUCCCACACACCUCUCC-3′.
Cell culture and transfection
HeLa, ME180, A431, A2058 and 293 cells were cultured in Dulbecco’s modified Eagles medium (HyClone) supplemented with 10% fetal calf serum (FCS). HT29 cells were maintained in McCoy’s5A (HyClone) supplemented with 10% FCS. For transfection with plasmid DNA, 293 cells were transfected by using FuGene 6 (Roche) and transfection into HeLa and ME180 cells were performed by electroporation with Nucleofector (Amaxa biosystems). Duplex siRNAs were transfected by using oligofectamine (Invitrogen). HT29 cells (~2 × 105) were infected with control or p53 shRNA (IMGENEX) adenoviruses (MOI = 200) for 5 days.
Production of anti-phospho-Daxx-specific antibody
Anti-rabbit phospho-Daxx specific antibody was generated by Open Biosystems with a synthetic phospho-peptide (CNTApSQSPRTRGpSRR), which corresponded to pSer176 and pSer184-containing Daxx.
Immunoprecipitation and western blotting
Cell lysate preparations, western blotting, and immunoprecipitation are described previously (18). For immunoprecipitation of phosphorylated Daxx at Ser176 and Ser184, 1 mg of cell lysates were incubated with 5 μg of anti-phospho-specific Daxx antibody or control rabbit immunoglobulin for 2 h at 4°C. Immunoprecipitated materials were separated by SDS-PAGE. The primary antibodies are anti-FLAG (M5, Sigma), anti-HA (3F10, Roche), anti-EGFP (Santa Cruz), anti-phospho-JNK (Cell Signaling), anti-JNK (Cell Signaling), anti-p53 (FL393, Santa Cruz), anti-Daxx (Sigma), anti-actin (Santa Cruz), anti-PARP (Affinity Bioreagents Inc.), anti-p53 (DO-1, Santa Cruz), and anti-GST (Santa Cruz).
IP-kinase assay
IP-kinase assay were performed as described (18). Briefly, cells were lysed and subjected to immunoprecipitation with the 3F10 anti-HA antibody (Roche) for 2 h at 4°C. The beads were washed and then incubated with 0.2 μg of substrate for 10 min at 30°C in the kinase reaction buffer supplemented with 100 μM ATP and 0.3 μCi of [γ-32P] ATP. The reactions were terminated by addition of SDS-sample buffer. The phosphorylated proteins were resolved by SDS-PAGE and measured by PhosphorImager (BioRad).
Results
Accumulation of cellular Daxx protein in response to TNFα treatment coincides with the activation of ASK1
TNFα exposure induces two types of JNK activation, namely early and sustained induction, mediated by a mammalian MAP3K ASK1 (3, 4, 19). Daxx enhances apoptosis on cells by interacting with and activating ASK1, which activates the JNK/p38 pathway. Interestingly, we observed that the cellular protein level of Daxx was significantly increased in TNFα treated cells (Fig 1A and Supplemental Figs s1). Therefore, we investigated the effect of TNFα on cellular Daxx at the protein level. Treatment with TNFα increased endogenous Daxx expression levels in a dose-dependent manner. These results indicate that there is a TNFα-dependent mechanism to regulate Daxx levels in cells.
Fig 1. ASK1 increases the Daxx expression by preventing proteasome-dependent degradation.
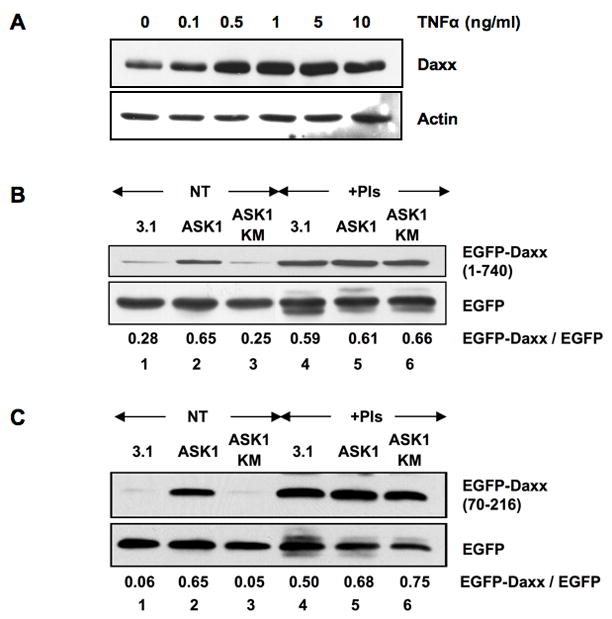
(a) TNFα treatment increases the expression level of Daxx. HeLa cells were treated with TNFα at the indicated concentrations for 18 h. The cell extracts were subjected to western blotting for Daxx and actin. (b, c) ASK1 stabilizes Daxx. 293 cells were transfected with expression plasmids for EGFP-Daxx, either full length (b) or a deletion mutant (70–216) (c), and EGFP together with control vector (3.1), ASK1-HA or kinase inactive mutant K709M (KM)-HA, and then cultivated for 2 days. Twelve hours after the treatment with or without proteasome inhibitors (PIs), cell extracts were prepared and subjected to western blotting with anti-EGFP antibody. The relative EGFP-Daxx protein levels are shown at the bottom as a ratio over the EGFP protein levels. NT: no treatment. Note that the expression level of EGFP serves as an internal reference of ectopic EGFP fusion protein expression.
Since ASK1 is activated by TNFα (4), the ASK1 activity is likely to be critical in the cellular Daxx accumulation. 293 cells were transfected with either control vector or expression plasmids for ASK1-HA or a dominant negative kinase-dead mutant, ASK1 K709M (KM)-HA (4), together with EGFP-Daxx and control EGFP to test the effect of ASK1 activity on the amount of cellular Daxx. The cellular level of EGFP-Daxx was evaluated by western blotting with EGFP as a reference (EGFP-Daxx/EGFP ratio). Daxx accumulated in cells only when transfected with ASK1-HA wild type (WT) (Fig 1B, lanes 1–3) but not with control vector or ASK1 KM-HA, and this was due to Daxx stabilization since proteasome inhibitors eliminated this effect (Fig 1B, lanes 4–6). Importantly, ASK1-dependent stabilization on Daxx level was also observed in three of its deletion mutants (amino acids 1–625, 1–418, and 70–216), but not other mutants (amino acids 1–69, 625–740, 419–740, 279–740 and 217–740) (Fig 1C and Supplemental Fig s2). These results suggest that the N-terminal region, amino acid 70–216, of Daxx is the only domain required for Daxx stabilization. The ASK1-dependent phosphorylation in this region may lead to Daxx accumulation by preventing proteasomal degradation.
Therefore, it is reasonable to assume that the same region of Daxx directly interacts with ASK1. ASK1-Daxx interaction was studied by immunoprecipitation of ASK1-HA from 293 cell lysates expressing both ASK1-HA and the various truncated forms of FLAG-Daxx (Fig 2A). ASK1-HA efficiently coimmunoprecipitated with FLAG-Daxx 1–216, 1–493 and 1–625 (Fig 2B, lanes 1, 3, and 5), but not with 217–740, 494–740 and 625–740 (lanes 2, 4, and 6) of Daxx lacking the N-terminal region. Thus, the N-terminal 216 amino acids of Daxx are sufficient for the interaction with ASK1. We were also able to detect an association between deleted region 70–216 of Daxx and ASK1, regardless of the ASK1 activity (Fig 2C). These results indicate that amino acids 70 to 216 of Daxx are responsible for the interaction with ASK1. The kinase activity of ASK1 is not required for the interaction.
Fig 2. ASK1 binds to the N-terminal region of Daxx.
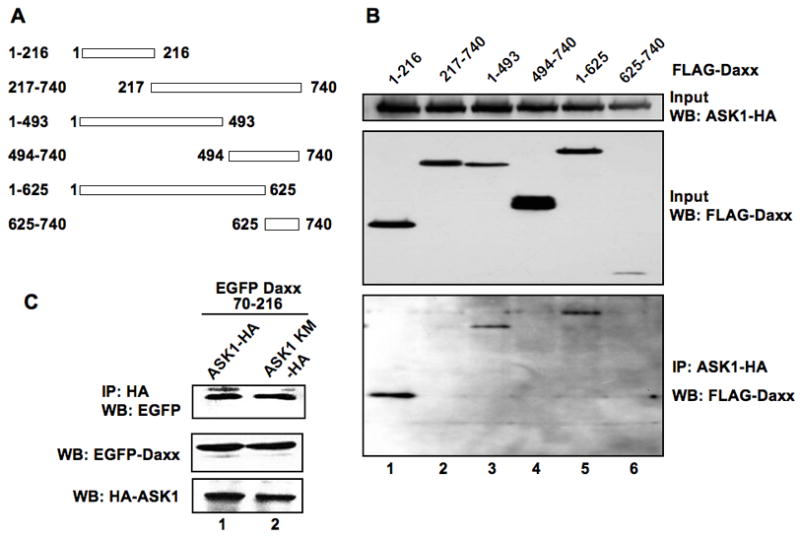
(a) Schematic diagram of deletion mutants of Daxx. Numbers indicate amino acid residues. (b) Mapping of the ASK1-interacting region of Daxx. 293 cells were transfected with the plasmids expressing ASK1-HA and various deletion mutants of FLAG-Daxx, as indicated in (a). Two days post-transfection, cell extracts were subjected to immunoprecipitation with anti-HA antibody, and the precipitated materials were analyzed by western blotting with anti-FLAG antibody. The 20% inputs for immunoprecipitation were analyzed for FLAG-Daxx and ASK1-HA proteins by western blotting as shown (Input WB). (c) ASK1 kinase activity is dispensable for the interaction of ASK1 with Daxx. Cell extracts from 293 cells expressing EGFP-Daxx 70–216 and either ASK1-HA or ASK1 KM-HA were prepared and the expression levels of EGFP-Daxx 70–216 and ASK1-HA were determined by anti-EGFP and -HA antibodies, respectively. Since the expression level of EGFP-Daxx 70–216 was higher in cells when expressed with ASK1-HA than with ASK1 KM-HA, the amounts of lysates for immunoprecipitation were adjusted based on the western blotting data for EGFP-Daxx 70–216 and ASK1. Cell lysates were immunoprecipitated with anti-HA antibody followed by western blotting for EGFP. IP: immunoprecipitation. WB: western blotting.
ASK1 phosphorylates Daxx on Ser176 and Ser184
We next asked whether Daxx was a direct substrate for ASK1 kinase. An in vitro ASK1 kinase assay was performed with bacterially produced GST-Daxx 70–216 as a substrate. Wild type ASK1 immunopurified from 293 cell extracts clearly phosphorylated GST-Daxx 70–216 (Supplemental Fig s3).
There are 21 serine/threonine residues within amino acid positions 70–216 of Daxx (Fig 3A). We generated various substitution mutants of 21 serine (S)/threonine (T) to alanine (A) within amino acids 70–216 of Daxx to determine the phosphorylation sites (Fig 3B and Supplemental Figs s4 and s5). The in vitro phosphorylation of GST-Daxx 70–216 WT was evident by ASK1-ΔN, a constitutively active mutant of ASK1 (Fig 3C, lanes 2 and 5) (5). Among mutants, GST-Daxx ST0, which lacks five serine/threonine residues within amino acids 176 to 184 (Figs 3B), nearly diminished its phosphorylation (Fig 3C, lanes 4 and 6). Likewise, GST-Daxx S176/184A, which is a double substituted mutant at Ser176 and Ser184, also drastically reduced its phosphorylation level (Fig 3C, lane 3 and Supplemental Fig s5). Since these phosphorylation deficient mutants still interact with ASK1-ΔN, reduced phosphorylation is not due to the luck of interaction with ASK1 (Supplemental Fig s6). Conversely, GST-Daxx ST0-176/184S, which restored both Ala176 and Ala184 residues to serines in ST0, regained its phosphorylation level. Again, there was no difference between ST0-176/184S and wild type in the interaction with ASK1 (Supplemental Fig s6). Single revertant mutants ST0-176S and ST0-184S, carrying restored residues Ala176 and Ala184 to serine of ST0, respectively, displayed partial restoration of phosphorylation (Fig 3C, lanes 8 and 9).
Fig 3. ASK1 phosphorylates on Ser 176 and Ser 184 in Daxx.
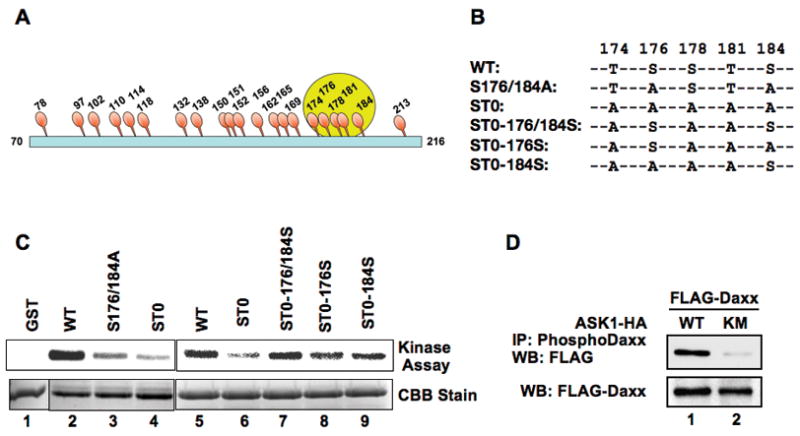
(a) Schematic diagram of serine (Ser)/threonine (Thr) residues within amino acids 70 to 216 of Daxx. Numbers indicate the position of amino acid residues. (b) Five Ser and Thr residues between 174–184 (circled in (a)) were characterized in detail. Substitution mutants between amino acids 174 and 184 are shown. (c) ASK1 phosphorylates Daxx on Ser 176 and Ser 184 in vitro. Recombinant mutant Daxx GST-fusion proteins were incubated with a constitutively active mutant ASK1 ΔN-HA in the presence of [γ-32P] ATP as described in Materials and Methods. The reaction mixtures were separated in SDS-PAGE, and phosphorylated GST-fusion proteins were determined by PhosphorImager. The protein levels of GST-Daxx WT and its substitution mutants in the kinase reaction were visualized by Coomassie brilliant blue (CBB) staining. (d) Phosphorylation of Daxx at Ser 176 and Ser 184 in vivo. FLAG-Daxx was expressed with full-length WT ASK1-HA or ASK1 KM-HA and the phosphorylation status of FLAG-Daxx was determined. The cell extracts were immunoprecipitated with anti-phospho-Daxx specific antibody followed by western blotting with anti-Daxx or FLAG antibodies. Since the expression level of FLAG-Daxx was increased by the presence of ASK1, the amounts of cell lysate in the immunoprecipitation reactions were adjusted to contain equal amount of Daxx protein (WB: FLAG-Daxx).
ASK1-dependent Daxx phosphorylation was monitored in vivo with an anti-phospho-Daxx specific antibody raised against a synthetic phosphopeptide encompassing the pS176 and pS184 of Daxx. Western blotting of precipitated materials with anti-phospho Daxx antibody showed that FLAG-Daxx Daxx was phosphorylated at Ser176 and Ser184 by ASK1 WT but not by kinase-dead ASK1 KM (Fig 3D). Similar results were obtained for endogenous Daxx when constitutively active ASK1 ΔN or ASK1 KM was expressed (Supplemental Fig s7A). Daxx was phosphorylated at Ser176 and Ser184 by endogenous ASK1 when cells were treated with TNFα (Supplemental Fig s7B). Similar experiments were performed with a phosphorylation deficient Daxx mutant FLAG-Daxx S176/184A. The amount of immunoprecipitated Daxx was not increased either by ASK1 WT or TNFα-treatment indicating that the anti-phospho Daxx antibody recognizes phosphorylated Daxx on Ser176 and Ser184 (Supplemental Fig s7C, D). These in vitro and in vivo results demonstrate that Daxx is a direct substrate of ASK1 and the Ser176 and Ser184 of Daxx are phosphorylated. Furthermore, ASK1-dependent phosphorylation of Daxx is induced by TNFα treatment, indicating that Daxx is a physiological substrate of ASK1 in cells.
ASK1-dependent phosphorylation of Daxx is critical for its stabilization
We investigated the effect of Daxx phosphorylation by ASK1 on its accumulation. The null-phosphorylation mutant FLAG-Daxx ST0 failed to be accumulated by ASK1-ΔN (Fig 4, lane 8), whereas the revertant mutant FLAG-Daxx ST0-176/184S, phosphorylatable by ASK1-ΔN, restored its ASK1-dependent accumulation to the similar extent of that of wild type FLAG-Daxx (Fig 4, lanes 5 and 2). These results indicate that ASK1-dependent phosphorylation of Daxx at Ser176 and Ser184 is required for its accumulation in cells.
Fig 4. Phosphorylation on Ser 176 and Ser 184 in Daxx is critical for Daxx stabilization.
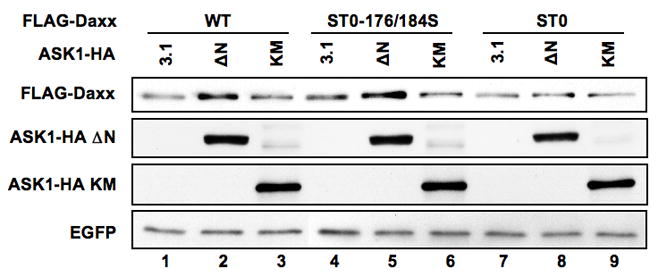
293 cells were transfected with expression plasmids for FLAG-Daxx (WT, ST0-176/184S, or ST0) and EGFP together with either empty vector (3.1), or expression plasmid for ASK1 ΔN-HA or ASK1 KM-HA. Two days after transfection, cell extracts were prepared and FLAG-Daxx levels were compared by western blotting with anti-FLAG antibodies using the expression level of EGFP as a reference. The expression levels of ASK-HA ΔN and ASK1-HA KM are also shown by western blotting with anti-HA antibody.
We next determined the functional significance of ASK1-dependent Daxx phosphorylation in JNK activation. Ectopic expression of ASK1 together with phosphorylation-deficient FLAG-Daxx S176/184A failed to activate JNK (Fig 5A, lane 4). Moreover, JNK was not activated with mutant FLAG-Daxx S176/184A expression by TNFα treatment (Fig 5B, lane 4). Since sustained late phase JNK activation mediated through ASK1 enhances apoptosis on mouse embryonic fibroblasts (10), it is likely that Daxx also plays a critical role on the ASK1-dependent sustained JNK activation and apoptosis in human cells. We determined the role of Daxx on JNK activation induced by TNFα in HeLa cells. Cells were transfected with Daxx-1, Daxx-2, or control siRNA and the activation of JNK was determined (Fig 5C). JNK was effectively activated in response to an increasing concentration of TNFα. When Daxx protein was reduced more than 90% by siRNA, the activation of JNK by TNFα was completely abolished, and the JNK activation became even lower than the basal level. Since the total JNK protein level was not changed during TNFα treatment, the reduction of the phosphorylated form of JNK was due to the inhibition of the ASK1-dependent JNK activation. These results indicate that Daxx is required for the TNFα-dependent sustained activation of JNK. We also determined the requirement of Daxx in the apoptosis induced by TNFα. When Daxx expression was depleted by siRNA, both the induction level of PUMAα (20) and the cleavage of PARP (11) were clearly reduced (Supplemental Fig s8). These results indicate that Daxx plays a critical role in TNFα-dependent apoptotic cell death. The biological significance of the phosphorylation of Daxx in TNFα-dependent apoptosis was also addressed. When S176/184A mutant Daxx was expressed, the cleavage of PARP was not enhanced by TNFα treatment (Supplemental Fig s9). These results indicate that the phosphorylation of Daxx on Ser176 and Ser184 plays a critical role in cellular response induced by TNFα treatment.
Fig 5. The activation of JNK by TNFα treatment requires Daxx and the phosphorylation on Ser 176 and Ser 184 in Daxx is critical.
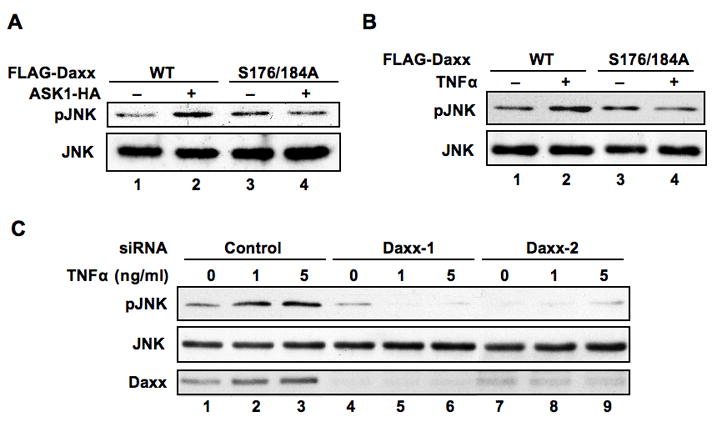
(a) Phosphorylation on Ser 176 and Ser 184 in Daxx is essential for ASK1-dependent sustained JNK activation. HeLa cells were transfected with the indicated expression plasmids. Cell extracts were prepared and analyzed for expression levels of phospho-JNK and total JNK by western blotting. (b) Phosphorylation on Ser 176 and Ser 184 in Daxx is essential for TNFα-induced sustained JNK activation. Either empty vector, FLAG-Daxx WT or FLAG-Daxx S176/184A mutant expression plasmid was transfected into HeLa cells. Twelve hours post transfection, cells incubated in the presence or absence of TNFα (1 ng/ml) for 18 h. The cellular levels of phospho-JNK and total JNK were determined by western blotting. (c) Daxx is essential for sustained activation of JNK. siRNA for control, Daxx-1 or Daxx-2 were transfected into HT29 cells. Three days after transfection, cells were incubated with TNFα (0, 1 or 5 ng/ml) for 18 h. The activation of JNK was determined by western blotting with anti-phospho-JNK antibody. Total JNK and Daxx were determined by western blotting using anti-JNK and Daxx antibodies, respectively.
Tumorigenic mutant p53 inhibits ASK1-dependent phosphorylation of Daxx and prevents TNFα-dependent accumulation of Daxx
We have previously reported that the inhibition of Daxx-dependent ASK1 activation is a mechanism for mutant p53 gain-of-function (18). We therefore tested the effect of mutant p53 on the ASK1-dependent phosphorylation of Daxx. We observed that mutant p53 inhibits the ASK1-dependent phosphorylation of Daxx in a dose-dependent manner (Fig 6A, lanes 3–5). The wild type p53, however, failed to inhibit Daxx phosphorylation (Fig 6A, lanes 6–8). These results indicated that the inhibitory effect is mutant p53 specific. Furthermore, the Daxx-ASK1 association was not dependent on ASK1 kinase activity and tumorigenic mutant p53 inhibits the complex formation between Daxx and ASK1 (Supplemental Fig s10).
Fig 6. Mutant p53 inhibits ASK1-dependent phosphorylation of Daxx and prevents Daxx accumulation induced by TNFα.
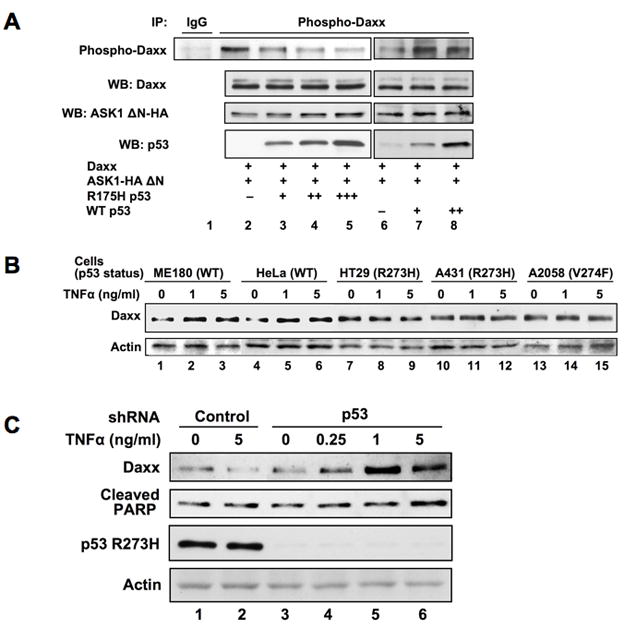
(a) Mutant p53 inhibits ASK1-dependent phosphorylation of Daxx. HeLa cells were transfected with expression plasmids, including wild type and mutant p53 plasmids, in the indicated combinations. Thirty hours after transfection, cell extracts were subjected to immunoprecipitation with anti-phospho-Daxx specific antibody followed by western blotting with anti-Daxx antibody. Since the expression level of Daxx was reduced by the presence of mutant p53 (18), the amounts of cell lysate in the immunoprecipitation reactions were adjusted to contain equal amount of Daxx protein (WB: Daxx). (b) TNFα does not induce the accumulation of Daxx in cells expressing natural tumorigenic mutant p53. Two cell lines with wild type p53 (ME180 and HeLa) and three cell lines with expressing mutant p53 (HT29, A431, and A2058) were examined for the accumulation of Daxx in response to TNFα treatment. Cells were incubated with TNFα (0, 1, 5 ng/ml) for 18 h and the cell extracts were subjected to western blotting for Daxx and actin. (c) Depletion of mutant p53 restores TNFα-induced accumulation of Daxx and apoptosis. HT29 cells were infected (m.o.i. = 200) with adenoviruses encoding control or p53 shRNA for 5 days followed by the treatment with TNFα at the indicated concentration for 18 h. The cell extracts were analyzed by western blotting with indicated antibodies.
The TNFα-dependent accumulation of Daxx was determined in two cell lines (ME180, HeLa) with wild type p53 and three cell lines (HT29, A431, A2058) carrying endogenous mutant p53. Daxx accumulation was observed in ME180 and HeLa cells (Fig 6B, lanes 1–6) but not in HT29, A431, or A2058 (Fig 6B, lanes 7–15) suggesting that the expression of mutant p53 interferes with Daxx from TNFα-dependent accumulation.
If this is the case, depletion of mutant p53 should restore the TNFα-dependent accumulation of Daxx and induces apoptosis in cells. HT29 cells which express R273H (Arg to His substitution mutant at amino acid 273) were infected with adenoviruses expressing shRNA for p53 knockdown followed by the treatment with TNFα. The amount of Daxx was not increased by TNFα treatment when control adenoviruses were infected (Fig 6C, lanes 1 and 2). However, the treatment with TNFα induced the accumulation of Daxx when mutant p53 was depleted (Fig 6C, lanes 3–6). Furthermore, when the accumulation of Daxx was restored, TNFα-dependent apoptosis became evident judged by the cleaved PARP. These results suggest that HT29 cells fail to undergo apoptosis by TNFα treatment because mutant p53 blocks JNK activation by preventing Daxx-ASK1 interaction and the accumulation of Daxx in cells.
Discussion
We demonstrated that the expression level of Daxx in cells was upregulated by ASK1 and the ASK1 kinase activity is required for this effect. Indeed, Daxx serves as a direct substrate for ASK1 and is phosphorylated at Ser 176 and Ser 184 by ASK1. The serine residues at 176 and 184 in human Daxx are well conserved in mammals, including chimpanzee, monkey, dog, cow, mouse, and rat, suggesting the importance of these residues and thus, the regulation of Daxx function through the phosphorylation of these serines may be also conserved. The molecular mechanisms for TNFα-dependent sustained JNK activation have been proposed involving caspase-dependent mammalian sterile 20-like kinase 1 (MST1) cleavage or reactive oxygen species (ROS) production (21, 22). We found that Daxx is essential for the sustained activation of JNK and apoptosis induced by TNFα in cells and Daxx phosphorylations are necessary for TNFα-responsive cellular Daxx accumulation as well as JNK activation. These results are consistent with what was shown in ASK1−/− mouse embryonic fibroblasts, where ASK1 was essential for TNFα-induced sustained JNK activation (10). In our Daxx knockdown experiment, TNFα treatment further reduced the level of phospho-JNK when the protein level of cellular Daxx was largely undetectable. This could be due to the activation of MAP kinase phosphatases (MKPs) by TNFα as reported previously (23).
Previously, it has been reported that ASK1 interacts with the C-terminal region of Daxx during glucose deprivation (17). We found, however, that ASK1 interacted with the N-terminal region of Daxx and the interaction was independent from the ASK1 kinase activity since both the kinase-inactive mutant of ASK1 (ASK1 KM) and wild type ASK1 bind Daxx equally well. Most likely the mode of ASK1-Daxx interaction is different from the ASK1-MKKs complexation since Daxx is missing the DVD docking domain present at the C-terminus of MKK3/MKK6 and MKK4/MKK7 (24). The sequence surrounding the phosphorylation sites, Ser176 and Ser184, in Daxx differs from the previously identified sequences in MKKs or the autophosphorylation sites on ASK1 (25). Mutations on both serines did not affect Daxx-ASK1 interaction indicating that these residues are dispensable for the interaction. Furthermore, these results also confirm that the interaction between ASK1 and Daxx is not affected by the Daxx phosphorylation status ofboth serines.
The involvement of ubiquitination in regulating MAPKs has been recently illustrated (26). Since proteasome inhibitors abolish ASK1 supported accumulation of Daxx, it suggests that Daxx accumulation or degradation is proteasome-dependent. We and others found that Daxx degradation is ubiquitin-dependent (27, and unpublished). However, a ubiquitin-independent proteasome-dependent Daxx degradation initiated by human cytomegalovirus pp71 tegument protein was also reported (28). It is reasonable to speculate that the phosphorylation of Daxx by ASK1 affects the ubiquitin status of Daxx.
Phosphorylation of proteins often plays a critical role in ubiquitinating target proteins and the consequent protein degradation (29, 30). Recently, phosphorylation of Daxx has been shown to induce its cellular degradation. Peptidyl-prolyl isomerase (Pin1) binds to the phosphorylated Ser178-Pro179 motif, although this is only present in human, in the Daxx protein and induces the rapid degradation of Daxx via the ubiquitin-proteasome pathway (27). Pin1 catalyzes the cis-trans isomerization of phosphorylated Ser/Thr-Pro motifs within its specific target substrates. ASK1 phosphorylates two serines close to each other in Daxx, thus if Pin1 twists the Daxx molecule at Pro179 flipping Ser176 and Ser184 in trans position, it may prevent ASK1 from phosphorylating Daxx. The phosphorylation on Ser178 and Ser176/Ser184 could be a tag-of-war situation to regulate Daxx metabolism.
It has been recently reported that in response to DNA damage Daxx undergoes ubiquitination and subsequent proteasomal degradation. The Daxx degradation promotes the translocation of RASSF1C from the nucleus to the cytoplasm (31). Cellular inhibitor of apoptosis protein 1 (c-IAP1), a regulator of TNF signaling, is a ubiquitin ligase for ASK1, and p38 and JNK phosphorylations are prolonged in c-IAP1−/− B cells (32). Since p38/JNK activation by Daxx depends on ASK1, c-IAP1 is also a key factor in modulating Daxx activity and stability in stress signaling.
Mutations in p53 have been found in more than 50% of all human cancer cells. Mutant p53s escape from the MDM2/HDM2-dependent degradation with the help of p16INK4a inactivation (33) and accumulate to induce the malignant transformation (34, 35). Tumorigenic mutant p53 inhibits the interaction between Daxx and ASK1 and thus, inhibits the ASK1-dependent phosphorylation of Daxx. As a result, mutant p53 inhibits the Daxx-dependent JNK activation (18). We propose a mechanism that mutant p53 inhibits Daxx accumulation by preventing phosphorylation of Daxx in response to stress signals. Heat shock protein 27 (HSP27) has been reported to block apoptosis by inhibiting the interaction between Daxx and ASK1 (15), providing another example for the biological significance of the cooperative activation between Daxx and ASK1 in JNK signaling cascade. Therefore, blocking Daxx-ASK1 circuit could be one molecular mechanism for the gain-of-function of mutant p53 which contributes to tumorigenesis.
Based on our results, we propose a positive feedback loop mechanism between Daxx and ASK1 (Supplemental Fig. s11). When ASK1 is activated by stresses, such as oxidative stress, UV and cytotoxic cytokines like TNFα, ASK1 phosphorylates Daxx and stabilizes it. This accumulated Daxx, in turn, further activates ASK1 which reinforces the amplification of downstream kinase activations including JNK and p38. Tumorigenic mutant p53 and possibly HSP27 may break the circuit between Daxx and ASK1 and thus cells will become more tolerant to stresses.
Supplementary Material
Acknowledgments
The authors thank Dr. H Ichijo for ASK1 WT, K709M and ΔN expression plasmids and Dr. H Kasahara for adenoviruses. This work was supported by Department of Radiation Oncology, Washington University School of Medicine and National Institute of Health R01CA98666 (N. Horikoshi).
References
- 1.Yang DD, Kuan CY, Whitmarsh AJ, et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–70. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 2.Wang XS, Diener K, Jannuzzi D, et al. Molecular cloning and characterization of a novel protein kinase with a catalytic domain homologous to mitogen-activated protein kinase kinase kinase. J Biol Chem. 1996;271:31607–11. doi: 10.1074/jbc.271.49.31607. [DOI] [PubMed] [Google Scholar]
- 3.Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998;281:1860–3. doi: 10.1126/science.281.5384.1860. [DOI] [PubMed] [Google Scholar]
- 4.Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–4. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 5.Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal- regulating kinase (ASK) 1. Embo J. 1998;17:2596–606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiriakidou M, Driscoll DA, Lopez-Guisa JM, Strauss JF., 3rd Cloning and expression of primate Daxx cDNAs and mapping of the human gene to chromosome 6p21.3 in the MHC region. DNA Cell Biol. 1997;16:1289–98. doi: 10.1089/dna.1997.16.1289. [DOI] [PubMed] [Google Scholar]
- 7.Michaelson JS, Bader D, Kuo F, Kozak C, Leder P. Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev. 1999;13:1918–23. doi: 10.1101/gad.13.15.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khelifi AF, D’Alcontres MS, Salomoni P. Daxx is required for stress-induced cell death and JNK activation. Cell Death Differ. 2005;12:724–33. doi: 10.1038/sj.cdd.4401559. [DOI] [PubMed] [Google Scholar]
- 9.Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c- mediated death pathway. Science. 2000;288:870–4. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 10.Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–8. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michaelson JS, Leder P. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J Cell Sci. 2003;116:345–52. doi: 10.1242/jcs.00234. [DOI] [PubMed] [Google Scholar]
- 12.Chen LY, Chen JD. Daxx silencing sensitizes cells to multiple apoptotic pathways. Mol Cell Biol. 2003;23:7108–21. doi: 10.1128/MCB.23.20.7108-7121.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salomoni P, Khelifi AF. Daxx: death or survival protein? Trends Cell Biol. 2006;16:97–104. doi: 10.1016/j.tcb.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Ko YG, Kang YS, Park H, et al. Apoptosis signal-regulating kinase 1 controls the proapoptotic function of death-associated protein (Daxx) in the cytoplasm. J Biol Chem. 2001;276:39103–6. doi: 10.1074/jbc.M105928200. [DOI] [PubMed] [Google Scholar]
- 15.Charette SJ, Lavoie JN, Lambert H, Landry J. Inhibition of Daxx-Mediated Apoptosis by Heat Shock Protein 27. Mol Cell Biol. 2000;20:7602–12. doi: 10.1128/mcb.20.20.7602-7612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ecsedy JA, Michaelson JS, Leder P. Homeodomain-interacting protein kinase 1 modulates Daxx localization, phosphorylation, and transcriptional activity. Mol Cell Biol. 2003;23:950–60. doi: 10.1128/MCB.23.3.950-960.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song JJ, Lee YJ. Role of the ASK1-SEK1-JNK1-HIPK1 signal in Daxx trafficking and ASK1 oligomerization. J Biol Chem. 2003;278:47245–52. doi: 10.1074/jbc.M213201200. [DOI] [PubMed] [Google Scholar]
- 18.Ohiro Y, Usheva A, Kobayashi S, et al. Inhibition of stress-inducible kinase pathways by tumorigenic mutant p53. Mol Cell Biol. 2003;23:322–34. doi: 10.1128/MCB.23.1.322-334.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21:701–10. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Wang P, Qiu W, Dudgeon C, et al. PUMA is directly activated by NF-kappaB and contributes to TNF-alpha-induced apoptosis. Cell Death Differ. 2009 doi: 10.1038/cdd.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–61. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 22.Wicovsky A, Muller N, Daryab N, et al. Sustained JNK activation in response to tumor necrosis factor is mediated by caspases in a cell type-specific manner. J Biol Chem. 2007;282:2174–83. doi: 10.1074/jbc.M606167200. [DOI] [PubMed] [Google Scholar]
- 23.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27:253–61. doi: 10.1007/s10555-008-9123-1. [DOI] [PubMed] [Google Scholar]
- 24.Takekawa M, Tatebayashi K, Saito H. Conserved docking site is essential for activation of mammalian MAP kinase kinases by specific MAP kinase kinase kinases. Mol Cell. 2005;18:295–306. doi: 10.1016/j.molcel.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Bunkoczi G, Salah E, Filippakopoulos P, et al. Structural and functional characterization of the human protein kinase ASK1. Structure. 2007;15:1215–26. doi: 10.1016/j.str.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laine A, Ronai Z. Ubiquitin chains in the ladder of MAPK signaling. Sci STKE. 2005;2005:re5. doi: 10.1126/stke.2812005re5. [DOI] [PubMed] [Google Scholar]
- 27.Ryo A, Hirai A, Nishi M, et al. A Suppressive Role of the Prolyl Isomerase Pin1 in Cellular Apoptosis Mediated by the Death-associated Protein Daxx. J Biol Chem. 2007;282:36671–81. doi: 10.1074/jbc.M704145200. [DOI] [PubMed] [Google Scholar]
- 28.Hwang J, Kalejta RF. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology. 2007;367:334–8. doi: 10.1016/j.virol.2007.05.037. [DOI] [PubMed] [Google Scholar]
- 29.Ang XL, Wade Harper J. SCF-mediated protein degradation and cell cycle control. Oncogene. 2005;24:2860–70. doi: 10.1038/sj.onc.1208614. [DOI] [PubMed] [Google Scholar]
- 30.Willems AR, Goh T, Taylor L, Chernushevich I, Shevchenko A, Tyers M. SCF ubiquitin protein ligases and phosphorylation-dependent proteolysis. Philos Trans R Soc Lond B Biol Sci. 1999;354:1533–50. doi: 10.1098/rstb.1999.0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitagawa D, Kajiho H, Negishi T, et al. Release of RASSF1C from the nucleus by Daxx degradation links DNA damage and SAPK/JNK activation. Embo J. 2006;25:3286–97. doi: 10.1038/sj.emboj.7601212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, Conze DB, Hanover JA, Ashwell JD. Tumor necrosis factor receptor 2 signaling induces selective c-IAP1-dependent ASK1 ubiquitination and terminates mitogen-activated protein kinase signaling. J Biol Chem. 2007;282:7777–82. doi: 10.1074/jbc.M609146200. [DOI] [PubMed] [Google Scholar]
- 33.Terzian T, Suh YA, Iwakuma T, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–44. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dittmer D, Pati S, Zambetti G, et al. Gain of function mutations in p53. Nat Genet. 1993;4:42–6. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 35.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945–51. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.