

The challenges of developing medical adhesives for the wet environment of open surgery are analogous to the adhesion problems solved by marine organisms living at the watery interface of land and ocean. These organisms routinely bond dissimilar materials together under seawater with little if any surface preparation. One such organism is the sandcastle worm (Phragmatopoma californica). Our goal is to copy this marine worm’s mechanisms of underwater bonding to create synthetic water-borne underwater medical adhesives, and in turn, to use the synthetic adhesives to test mechanistic hypotheses about the natural adhesive. Biomimetic underwater adhesives were formulated with polyelectrolytic analogues of the natural glue proteins. The copolymers condensed into complex coacervates—dense partially water-immiscible cohesive fluids poised between soluble polymers and insoluble polymeric salts. The boundary between fluid coacervate phases and solid or gelled states was dependent on divalent cation species as well as the pH and temperature, which demonstrated that these environmental factors can trigger the adhesive setting reaction (Fig. 1). The results provide, respectively, empirical support for the natural pH-triggered set hypothesis and practical triggers for controlled setting of mimetic medical adhesives.
Figure 1.
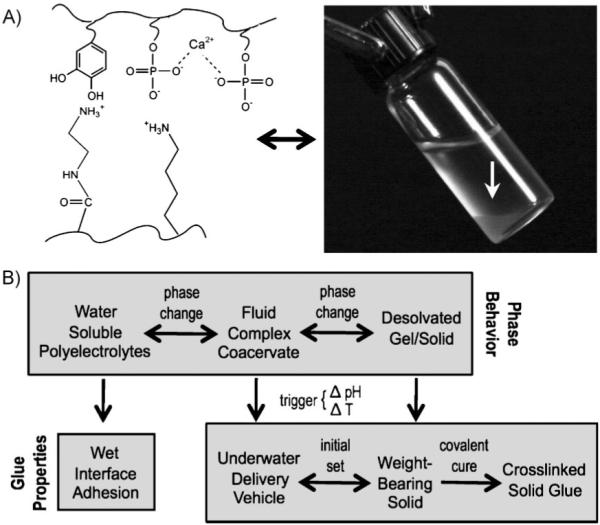
Complex coacervates as adhesives. A) The adhesive comprises dopamide containing poly-phosphate, poly-aminated gelatin, and divalent cations. Under the right conditions, the polyelectrolyte solution condenses into a complex coacervate phase (white arrow in photo). B) The top row represents the phase behavior of the polyelectrolytes. The bottom row connects the features of the phase behavior to solving the several problems of creating an underwater glue. The change from fluid complex coacervate to insoluble solid—the intial setting reation—is triggered by a change in the pH, temperature, or both. Irreversible covalent hardening occurs through oxidative coupling between catechol and primary amine sidechains.
The sandcastle worm lives in protective tubular shells, which it assembles underwater by gluing together sand and seashell hash with a proteinaceous adhesive.[1-3] To build underwater sandcastles, the worm had to solve three major problems: first, its adhesive must form strong chemical bonds with wet surfaces and to do so it must displace interfacial water;[4,5] second, its water-borne fluid adhesive must not dissolve into the ocean when it is secreted underwater; and third, setting of the adhesive must be accurately timed. If it sets too fast, the glue would plug up the worm’s adhesive ducts, an obvious problem, while setting too slowly would be inefficient. The sandcastle glue is composed of oppositely charged proteins, plus calcium and magnesium ions. A high proportion of charged sidechains (phosphates and amines) and dihydroxyphenyl-alanine (dopa) residues with catechol sidechains help the adhesive breech the interfacial water barrier to solve the first problem. Dopa residues have been implicated as promoters of strong adhesion to wet metal oxide surfaces.[2,6,7] To solve the second problem, the oppositely charged proteins may associate electrostatically into a dense cohesive fluid—a complex coacervate[8,9]—that is partially immiscible in water, yet spreads over and wets submerged surfaces.[3] Glue protein analogues synthesized as water-soluble acrylates containing phosphate, amine, and catechol sidechains in similar proportions as the natural adhesive proteins condensed into complex coacervates when mixed under the right conditions.[10] Timing the hardening reaction, the third problem, is likely coupled to the pH differential between secretory granules (pH ≈ 5) and seawater (pH = 8.2).[3,11] Secretion into seawater triggers a quick set within 30 s followed by slower covalent curing through redox active catechol sidechains.[12]
In addition to wet field deliverability, controlled setting, and sufficient bond strength, a practical adhesive to be used for internal fixation of mineralized tissue should degrade over time to allow natural healing. An adhesive complex coacervate expected to be biodegradable was created using a low molecular weight (MW, 3–5 kDa) non-gelling collagen hydrolysate as the polycation. As received, the collagen hydrolysate did not form complex coacervates with the phosphodopa copolymer (poly(MAEP)85-co-dopamide15 where MAEP = monoacryloxyethyl phosphate) at physiological pH. Amination of carboxylic acid sidechains with ethylenediamine increased the amine concentration to ≈16 mol% and shifted the pI from 5.5 to 10.4, values analogous to the glue protein, Pc1, which contains 14.6 mol% lysine and has a pI of 9.75.[11] In this regard, the amine-modified collagen hydrolysate is a reasonable analogue of the polycationic glue proteins. When mixed, the aminated collagen and phosphodopa copolymer formed dense coacervates at 25 °C (Fig. 1A, Supporting Information) over a broad range of compositions. At pH 5.0, concentrated coacervates formed at amine-to-phosphate sidechain ratios of 0.5–1.0 and Ca2+-to-phosphate ratios up to 0.8 (Fig. 2A). None of the compositions precipitated. At pH 7.4, the coacervation space was more confined; at Ca2+ ratios higher than 0.2, the copolymers precipitated as hard solids, reflecting the decreased solubility of the mixed polyelectrolytes and Ca2+ with increasing pH (Fig. 2B).
Figure 2.
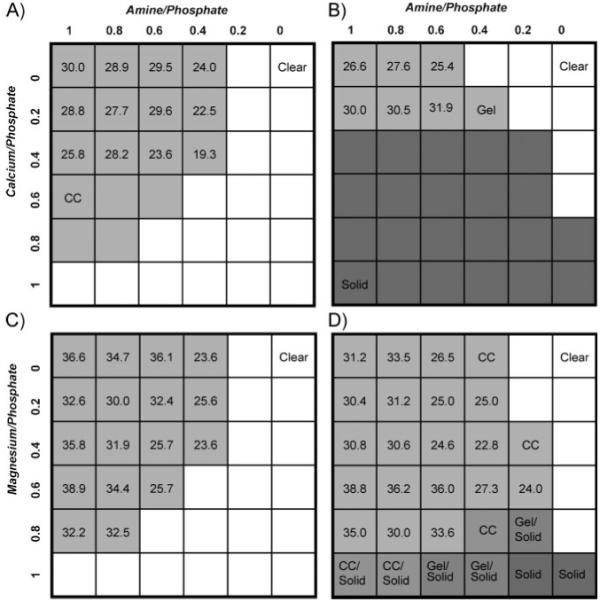
Phase diagrams of polyphosphate–gelatin–divalent cation mixtures. The top axis represents the molar ratio of amine sidechain to phosphate sidechain. The left axis represents the molar ratio of divalent cation to phosphate sidechain. A) Ca2+ compositions, pH 5.0. B) Ca2+ compositions, pH 7.4. C) Mg2+ compositions, pH 5.0. D) Mg2+ compositions, pH 7.4. The total concentration of copolymers in each mixture was 5.0 wt%. Soluble compositions are represented by white squares, compositions that condensed into complex coacervates are represented by light grey squares, compositions that formed gels or hard solid precipitates are represented by darker grey squares. The numbers in the light grey squares are the concentrations (wt/vol %) of the complex coacervate phase. Grey squares without numbers contained complex coacervates, but with volumes too low to allow accurate measurement of the concentration. The Mg2+ solid phases were softer and more gel-like than the Ca2+ solids.
Investigation of the separate effect of Mg2+ on coacervation of the polyelectrolytes revealed significant differences when compared with Ca2+. At pH 5.0, the coacervated region was larger. At ratios up to a 1:1 ratio of Mg2+ to phosphate, none of the compositions precipitated (Fig. 2C). With Mg2+ the copolymers condensed into more concentrated coacervates, in some cases >380 mg mL−1, an almost eightfold increase from the initial copolymer concentration (50 mg mL−1). However, the volume and total mass of polymers in the coacervate phase were lower than the Ca2+ coacervates. Further experiments are required to determine if this is due to partitioning of a subset of the copolymers selected by size or charge density into the Mg2+ coacervates. At pH 7.4 the coacervation range is broader, and at high Mg2+ ratios compositions with mixed phases of fluid and solid occur due to decreased solubility with increased pH (Fig. 2D). The expanded coacervation space at higher pH again illustrates that the dense fluid coacervates are stably balanced intermediates between soluble polyelectrolytes and insoluble solids. The physical nature of the solidified state at high Mg2+ ratios is non-fluid, but softer and more gel-like than the hard Ca2+ precipitates, reflecting perhaps an intermediate state of desolvation relative to fluid coacervates and solids. The distinct physical nature and solubility profile of the Mg2+ complexes are likely consequences of the smaller radius, higher charge density, and smaller coordination number of Mg2+ ions compared to Ca2+ ions.[13] Mg2+ tends to coordinate single bulky ligands, like phosphate, because multiple ligands will not fit around the small ion. As a consequence much of its solvation sphere is retained. The larger Ca2+ ion, on the other hand, can accommodate several bulky ligands resulting in displacement of its solvation sphere and cross-link formation between ligands. Coacervates prepared with mixed Mg2+ and Ca2+ occupied space in between the coacervated regions of the individual cations.[10]
The phase diagrams illustrate empirically how the pH differential between secretory granules and seawater could trigger a phase change that drives the rapid, but well-timed initial setting reaction of the natural adhesive. The condensed fluid complex coacervate phase is thermodynamically balanced between stable colloidal complexes and gelled or precipitated polymeric salts.[10] The composition of the natural adhesive may be adapted to fall just inside the coacervation boundary within the secretory pathway, but to be outside of the coacervated region at the elevated pH of seawater. In other words, they are composed to undergo a pH-dependent phase change upon secretion. For example, row 3 compositions (Fig. 2a,b), with ratios of 0.4 Ca2+ and greater than 0.3 amine are coacervated at pH 5.0, but solid at pH 7.4 and higher. The adhesive could issue from secretory pores in the building organ as a cohesive low-viscosity fluid coacervate that does not disperse into the ocean, yet it has low interfacial tension, readily spreads on wet mineral substrates, fills cracks and gaps, and allows the worm a few seconds to position a sandgrain before hardening. Taken together, the Mg2+ and Ca2+ phase diagrams also establish that mixing divalent cations provides another dimension for nature to adapt the phase behavior of the worm’s bioadhesive to the physical conditions of the ocean, or for human technologists to adjust a mimetic adhesive to the exact specifications of open surgery. Although the complex coacervation model has been a useful guide for designing mimetic adhesives, it is an incomplete description of the natural process because it does not include details of how the natural glue proteins are packaged into dense secretory granules,[14] or where and when complex coacervation occurs during secretion. The coacervated phase may be a transient state formed briefly when glue components mix just after exiting the secretory pores. A more complete and quantitative description of the natural setting mechanism will be published elsewhere.
In principle, adhesive complex coacervates for tissue repair could be designed to be delivered at pH 5.0 and to solidify as they equilibrate to physiological pH to mimic the natural trigger mechanism. The phase behavior experiments were done at pH 7.4 rather than pH 8.2 to make this point. However, a more efficient trigger mechanism, not available in the practically isothermal world of the worm, may be to exploit the difference between ambient and internal body temperature of warm blooded animals. At 0 °C the coacervated region in Figure 2b is shifted approximately one row lower, while at 37 °C it is shifted one row higher (not shown). The temperature-dependent phase transition of several compositions at pH 7.4 with increasing Ca2+-to-phosphate ratios and a fixed amine ratio of 0.6 were investigated in more detail by dynamic oscillatory rheology (Fig. 3b). At low temperature, the viscous shear moduli (G”) were greater than the elastic moduli (G’) consistent with the fluid character of the complex coacervates.[15] With increasing temperature G’ rose sigmoidally in a Ca2+-ratio-dependent manner. The crossover points at which G’ = G” (Fig. 3a), taken as the transition temperature where the compositions begin to change from viscous fluids to load-bearing elastic solids,[16] were 36, 21, 12, and 9 °C for Ca2+ ratios of 0.15, 0.20, 0.25, and 0.30, respectively. The Mg2+-containing coacervates demonstrated qualitatively similar behavior: there was no crossover of G’ and G” at Mg2+-to-phosphate ratios up to 0.8 at pH 7.4; at higher ratios the crossover temperature again decreased with increasing Mg2+ ratios. The elastic moduli at 37 °C were much lower with Mg2+ than with Ca2+ (Fig. 3b), consistent with the more hydrated gel-like quality of the solidified Mg2+ coacervates.
Figure 3.
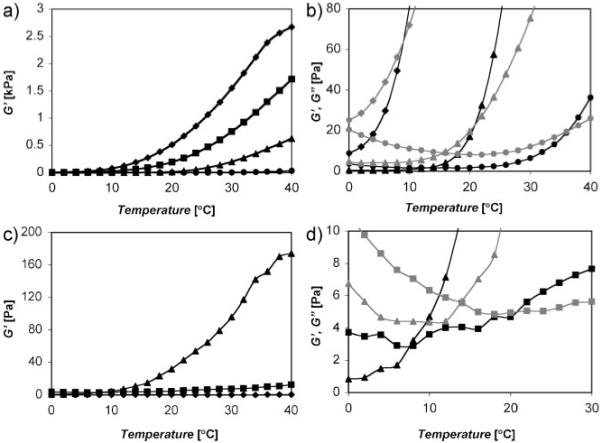
Transition temperatures determined by dynamic oscillatory rheology. a) Ca2+/gelatin/polyphosphate rheology. The elastic modulus (G’, black symbol) increased sigmoidally as the temperature was raised from 0 to 40 °C at Ca2+ ratios greater than 0.15. b) Expanded scale to demonstrate the crossover temperature of the elastic (G’) and viscous (G”, grey symbol) moduli. The solidification or gellation temperature decreased with increasing Ca2+ ratio. The 0.25 Ca2+ ratio was excluded for clarity. (Symbols: ◆ 0.3/0.6, ■ 0.25/0.6, ▲ 0.2/0.6, ●0.15/0.6 Ca2+ ratios). c) Mg2+/gelatin/polyphosphate rheology. (Symbols: ◆ 0.8/01.0, ■ 0.9/1.0, ▲ 1.0/01.0 Mg2+ ratios). d) Expanded scale to demonstrate the crossover temperature of the elastic (G’) and viscous (G”, grey symbol) moduli. The comparative measurements were made with constant strain of 0.1% and frequency of 1.0 Hz.
Bonds formed with Ca2+ ratios ranging from 0 to 0.3 with an amine-to-phosphate ratio fixed at 0.6 were evaluated in a standardized lap shear mechanical test using wet polished aluminum adherends. The bonded adherends were cured and tested while fully submerged in a temperature-controlled water bath. At 37 °C, well above the transition temperatures of the compositions, the lap shear strength increased with increasing Ca2+ up to a ratio of 0.3 (Fig. 4a, black bars). The 0.2 and 0.25 Ca2+/0.6 amine compositions were also tested slightly below their respective transition temperatures at 10 and 20 °C. In both cases, the bond strengths above the transition temperature were greater than below the transition temperature (Fig. 4a, white bars). Under the conditions of the test set-up, there is likely to be little covalent oxidative crosslinking between the dopamide sidechains of the polyphosphate and the amines of gelatin: the rate of dopa oxidation is much slower at pH 7.4 than 8.2; diffusion of dissolved O2 into the narrow bond gap (62 μm) was restricted, and there was no evident browning of the adhesive indicative of dopa oxidation. Therefore the increase in bond strength above the transition temperature was predominantly due to the state change of the adhesive. Similar tests with the 1.0 Mg2+ ratio demonstrated a more dramatic increase, a more than sixfold increase in bond strength above the transition temperature than below (Fig. 4b). As a practical matter, the results demonstrated that temperature differentials can be exploited as a convenient means to trigger the initial set of the synthetic adhesive and that the temperature trigger can be adjusted within a physiologically relevant range by small changes in the divalent cation ratio.
Figure 4.
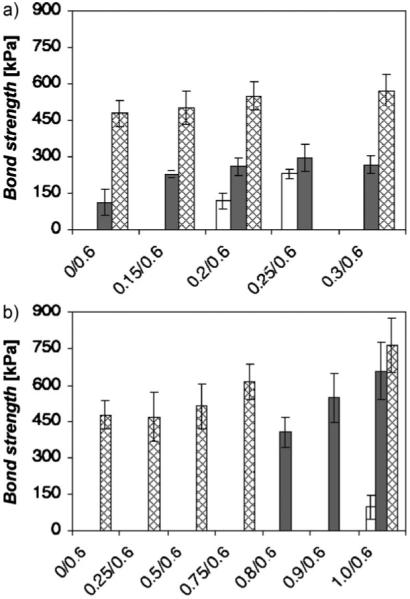
Shear strength as a function of divalent cation ratio and temperature. a) The ratio of Ca2+ to phosphate was varied at a constant amine ratio. b) The Mg2+ ratio was varied at a constant amine ratio. Standardized mechanical tests were done with clean aluminum adherends fully submerged in a temperature-controlled water bath (pH 7.4). Dark bars represent shear tests done at 37 °C without oxidative crosslinking. White bars indicate shear tests done below the transition temperature without oxidative crosslinking. Cross-hatched bars represent shear tests done at 37 °C after oxidative crosslinking with NaIO4 at a molar ratio of 1:2 relative to dopamide sidechains. The crosslinked specimens were cured (24 h) and tested while fully submerged in a temperature-controlled water bath. The bars represent the average +/− standard deviations (s.d.; n = 9 for all compositions and conditions).
The quick set of the natural P. californica adhesive is followed by a slower covalent curing process which likely adds considerably to bond strength. To investigate the contribution of covalent crosslinking to bond strength of the synthetic adhesive, oxidative coupling between the polyphosphate dopamide sidechains and the gelatin amines was initiated by adding 0.5 equivalents NaIO4 relative to the dopamide sidechains during the bonding procedure. The bonds were cured and tested at 37 °C while fully submerged in water adjusted to pH 7.4. The bonds strengths increased with increasing divalent cation ratio for both Ca2+ and Mg2+ (Fig. 4, hatched bars). Maximum bond strengths with Mg2+, 765 kPa, were ≈40% higher than the bond strength of Ca2+. This is also more than twice the estimated bond strength of the natural P. californica adhesive.[17]
In conclusion, complex coacervates are dense, partially water-immiscible fluids precariously balanced between colloidal complexes and insoluble polymeric salts. The fluid coacervate phase can be injected underwater and adheres to wet surfaces. These properties make complex coacervates an ideal foundation for underwater adhesives, perhaps discovered eons ago by marine organisms, but only recently exploited for creating injectable medical adhesives for wet tissue. Small environmental changes during delivery of the fluid coacervate can trigger an initial setting reaction (Fig. 1). Covalent crosslinking through catechol sidechains permanently hardens the adhesive. Experiments to determine the rate of biodegradation of the gelatin-based adhesive complex coacervates are in progress. Finally, the coacervate-based adhesives are formed in water from prepolymerized components, so repaired tissues are not exposed to toxic solvents, reactants, reaction by-products, or heat generated by exothermic in situ polymerization. It may be possible to simultaneously fix tissue and deliver bioactive agents to provide greater patient comfort, accelerate bone healing, and/or prevent infections.
Experimental
Materials
Low endotoxin, non-gelling, gelatin (Mw 3–5 kDa) was a gift from Gelita Inc. (Sioux City, IA). 1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) and ethylenediamine dihydrochloride were purchased from Thermo Scientific Inc. MAEP and 2,2′-azobisisobutyronitrile (AIBN) were purchased from Polysciences, Inc. Sodium periodate (NaIO4), Sephadex LH-20, and dopamine hydrochloride were obtained from Sigma–Aldrich.
Polyphosphodopamide Synthesis
Poly(MAEP85-dopamide15) was synthesized as described previously [10] by free radical polymerization of MAEP and dopamine methacrylamide (DMA) using AIBN as initiator. The copolymer was recovered by size-exclusion chromatography (SEC) in MeOH on a Sephadex LH-20 column (Sigma–Aldrich). MeOH was removed; the copolymer resuspended in water, lyophilized, and stored at −80 °C. The mol% of dopamide sidechains in the copolymers were determined by UV/vis spectroscopy: the catechol form of dopamide has an absorption peak at 279 nm (λ279 = 2600 M−1 cm−1). Copolymer MWs were determined by size exclusion chromatography on an AKTA FPLC using a Superose 6 HR 10/30 column (GE Healthcare) with an Optilab RI detector and a miniDawn TREOS light scattering detector (Wyatt Technology Corp.). Samples were run in 150 mM NaCl, 50 mM phosphate, pH 7.2, and 0.4 mL min−1 flow rate. The mass average Mw of polyphosphodopamide was 57 kDa with a polydispersion index (PDI) of 1.55.
Gelatin Modification
Gelatin (100 mg mL−1) was mixed with ethylenediamine dihydrochloride (1.2:1 molar ratio to the gelatin carboxyl groups). The pH was adjusted to 5.2 with 6 M HCl. EDC at a 1:1 molar ratio to the gelatin carboxyl groups was added to the reaction mixture while stirring. The reaction proceeded for 2 h at room temperature. The amine-modified gelatin was dialyzed against DI water for 3 days, then lyophilized. The primary amine sidechain concentration was determined by ninhydrin assay using glycine as a standard. Zeta potential measurements of gelatin (1 mg mL−1 in water) were determined by electrophoresis using a Malvern Zetasizer Nano-ZS ZEN 3600 (Malvern Instruments Ltd.).
Gelatin Coacervate Formation
A 50 mg mL−1 aqueous solution of amine-modified gelatin (pH 5.0) was added dropwise while stirring to a 50 mg mL−1 aqueous solution (pH 5.0) of poly(MAEP85-dopamide15) containing various ratios of divalent cation (Ca2+ or Mg2+) until reaching the target amine/phosphate ratio. The pH of the mixture was raised to 7.4 with 0.1 m NaOH. The coacervate phase was allowed to settle for 24 h. The upper polymer-depleted equilibrium phase was removed from above the dense polymer-rich coacervate phase. The volumes of both phases were measured. The coacervate phase was lyophilized then weighed. The coacervate concentration was determined from the measured volume and dry mass of the solids. The numbers in the grey squares in Figure 2 are the coacervate concentration in wt/vol %.
Dynamic Rheology
The elastic (G’) and storage (G”) moduli were measured with a cone and plate configuration (20 mm diameter, 4 °C cone) on a stress-controlled rheometer (TA Instruments, AR 500). To compare coacervate compositions, the measurements were made with a constant frequency of 1 Hz and dynamic strain of 0.1% as the temperature was ramped from 0 to 40 °C at a rate of 0.5 °C min−1.
Adhesive Bond Strength
An earlier adhesive complex coacervate formulation was shown to bond wet human cortical bone specimens [10]. Milling adherends out of bone is costly, time consuming, and less reproducible than metal substrates. Therefore, for rapid parametric studies of adhesive formulations and bonding conditions, aluminum adherends were used for mechanical testing of adhesive bonds. A water saw was used to cut hundreds of aluminum strips, 0.6 cm × 5.0 cm, from 0.050 inch (1 inch = 2.54 cm) thick sheets of 5052 aluminum. The strips were polished with 600 grit super fine sandpaper and then cleaned following the procedure of ASTM D2651. In brief, the adherends were sonicated twice in MeOH, air-dried, dipped into a solution of sulfuric acid and nochromix for 15 min, then rinsed thoroughly with DI water and stored in DI water until bonded. The adherends were bonded within 12 h of cleaning. For each adhesive test condition, 9 wet aluminum test specimens were bonded. For covalently crosslinked bonds, NaIO4 at a 1:2 molar ratio to dopamide sidechains was evenly applied to the bond area of two aluminum adherends. The test coacervate solution (6 μL) was applied to wet adherends with a pipette, which were then pressed together with an overlap of about 20 mm, clamped with stainless steel clips, and immediately submerged in water adjusted to pH 7.4 with NaOH. The bonded specimens cured fully submerged in water for ≈24 h at the specified temperature. Shear strengths were measured while the aluminum adherends where fully submerged in a temperature-controlled water bath mounted on an Instron 3342 materials testing system with a 100 N load cell. The loading rate was 1.2 mm min−1. The instrument was controlled and the data acquired using Bluehill Lite software (Instron, Inc.).
Supplementary Material
Acknowledgements
This work was supported by a grant from the NIH (R01 EB006463).
Footnotes
The authors have no conflicts of interest.
References
- [1].Jensen RA, Morse DE. J. Comp. Physiol. B. 1988;158:317. [Google Scholar]
- [2].Waite JH, Jensen RA, Morse DE. Biochemistry. 1992;31:5733. doi: 10.1021/bi00140a007. [DOI] [PubMed] [Google Scholar]
- [3].Stewart RJ, Weaver JC, Morse DE, Waite JH. J. Exp. Biol. 2004;207:4727. doi: 10.1242/jeb.01330. [DOI] [PubMed] [Google Scholar]
- [4].Baier RE, Shafrin EG, Zisman WA. Science. 1968;162:1360. doi: 10.1126/science.162.3860.1360. [DOI] [PubMed] [Google Scholar]
- [5].Waite JH, Andersen NH, Jewhurst S, Sun C. J. Adhes. 2005;81:297. [Google Scholar]
- [6].Deming TJ. Curr. Opin. Chem. Biol. 1999;3:100. doi: 10.1016/s1367-5931(99)80018-0. [DOI] [PubMed] [Google Scholar]
- [7].Lee H, Scherer NF, Messersmith PB. Proc. Natl. Acad. Sci. USA. 2006;103:12999. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bungenberg de Jong HG. In: Colloid Science. Kruyt HR, editor. II. Elsevier; Amsterdam: 1949. p. 431. [Google Scholar]
- [9].Xia JL, Dubin PL. In: Macromolecular Complexes in Chemistry and Biology. Dubin P, Bock J, Davis R, Schulz D, Thies C, editors. Springer; Heidelberg: 1994. p. 247. [Google Scholar]
- [10].Shao H, Bachus KN, Stewart RJ. Macromol. Biosci. 2009;9:464. doi: 10.1002/mabi.200800252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhao H, Sun C, Stewart RJ, Waite JH. J. Biol. Chem. 2005;280:42938. doi: 10.1074/jbc.M508457200. [DOI] [PubMed] [Google Scholar]
- [12].Stevens MJ, Steren RE, Hlady V, Stewart RJ. Langmuir. 2007;23:5045. doi: 10.1021/la063765e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cowan JA. The Biologial Chemistry of Magnesium. VCH; New York: 1995. [Google Scholar]
- [14].Vovelle J. Arch. Zool. Exp. Gen. 1965;106:1. [Google Scholar]
- [15].Goodwin JW, Hughes RW. Rheology for Chemists: An Introduction. Royal Society of Chemistry; Cambridge, UK: 2000. [Google Scholar]
- [16].Gil ES, Spontak RJ, Hudson SM. Macromol. Biosci. 2005;5:702. doi: 10.1002/mabi.200500076. [DOI] [PubMed] [Google Scholar]
- [17].Sun C, Fantner GE, Adams J, Hansma PK, Waite JH. J. Exp. Biol. 2007;210:1481. doi: 10.1242/jeb.02759. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.