

Abstract
The α subunit of heterotrimeric G13 protein is required for the embryonic angiogenesis (Offermanns et al., Science 275:533–536, 1997). However, the molecular mechanism of Gα13-dependent angiogenesis is not understood. Here, we show that myocyte-specific enhancer factor-2 (MEF2) mediates Gα13-dependent angiogenesis. Our data showed that constitutively activated Gα13Q226L stimulated MEF2-dependent gene transcription. In addition, downregulation of endogenous Gα13 inhibited thrombin-stimulated MEF2-dependent gene transcription in endothelial cells. Both Ca2+/calmodulin-dependent kinase IV (CaMKIV) and histone deacetylase 5 (HDAC5) were involved in Gα13-mediated MEF2-dependent gene transcription. Gα13Q226L also increased Ca2+/calmodulin-independent CaMKIV activity, while dominant negative mutant of CaMKIV inhibited MEF2-dependent gene transcription induced by Gα13Q226L. Furthermore, Gα13Q226L was able to derepress HDAC5-mediated repression of gene transcription and induce the translocation of HDAC5 from nucleus to cytoplasm. Finally, downregulation of endogenous Gα13 and MEF2 proteins in endothelial cells reduced cell proliferation and capillary tube formation. Decrease of endothelial cell proliferation that was caused by the Gα13 downregulation was partially restored by the constitutively active MEF2-VP16. Our studies suggest that MEF2 proteins are an important component in Gα13-mediated angiogenesis.
Keywords: Heterotrimeric G13 protein, MEF2-dependent transcription, Endothelium, CaMKII, HDAC5
Introduction
Heterotrimeric guanine nucleotide-binding proteins (G-proteins) transmit the signals from cell membrane receptors to the signaling molecules, such as enzymes and ion channels. The G12 family consists of ubiquitously expressed Gα12 and Gα13 subunits [47], which regulate a variety of cellular responses, including transformation of fibroblasts [51, 54], activation of Jun N-terminal kinase and serum response element (SRE) [12, 15, 38], and actin stress fiber formation [10]. Although Gα12 and Gα13 proteins share a high amino acid sequence homology (67%), their functional properties are not completely overlapped. For example, Gα13 knockout mice die during early embryonic development due to vascular system defect [36], whereas mice with a disrupted Gα12 gene are viable and fertile [19]. Endothelial cell-specific Gα13 knockout mice show phenotype similar to that of the global Gα13 knockout mice [42]. A recent report also indicates that Gα13 but not Gα12 is responsible for platelet-derived growth factor (PDGF)-stimulated cell migration [43].
Myocyte enhancer factor-2 (MEF2) transcription factors [18] are the members of family of MADS (MCM1, agamous, deficiens, serum response factor)-box transcription factors [8]. MEF2 factors participate in diverse gene regulatory programs, including muscle and neural differentiation, cardiac morphogenesis, blood vessel formation, and growth factor responsiveness (reviewed in Ref. [8]). The importance of MEF2 proteins in vascular formation was determined by knockout mice studies [7, 26]. Deletion of MEF2C gene resulted in embryonic lethality by 9.5 day due to impaired vascular formation and cardiac defect. It was reported that MEF2C is important for endothelial cell survival since expression of constitutively active MEF2C partially rescued endothelial apoptosis caused by BMK1 deficiency [21]. In addition, mutations of MEF2A were suggested as a cause of coronary artery disease [52]. Importantly, vascular endothelial growth factor is a key regulator of physiological and pathologic angiogenesis and was shown to induce MEF2 activity in endothelial cells [29]. G-protein-coupled receptors, such as M1 muscarinic receptor and thrombin receptor, proteinase-activated receptor 1 (PAR-1), have been shown to regulate c-Jun expression through activation MEF2 transcription factors [16].
The Ca2+/calmodulin-dependent protein kinase (CaMK) is a potent activator of MEF2 activity. It was shown that CaMK phosphorylates the transcriptional repressor histone deacetylases (HDACs), HDAC4, HDAC5, and HDAC7, leading to disruption of the interaction between MEF2 and HDACs, which results in the translocation of HDACs from nucleus to cytoplasm and releases the repression [31, 32]. Importantly, global inhibition of HDAC activity inhibits angiogenesis and tumor growth both in vitro and in vivo [34]. Interestingly, it was reported that Gbetagamma binds histone deacetylase 5 (HDAC5) and inhibits its transcriptional co-repression activity [46].
As Gα13 is required for embryonic angiogenesis, we tested the hypothesis that MEF2 and HDACs are involved in the signaling cascade regulated by Gα13. Here, we showed that Gα13 stimulates MEF2-dependent gene transcription in vascular endothelial cells. We also characterized the molecular pathway that allows Gα13 to stimulate MEF2-dependent gene transcription in endothelial cells. Finally, we determined that MEF2 proteins mediate Gα13-dependent angiogenic response.
Materials and methods
Materials
Constitutively activated Gα12Q229L and Gα13Q226L with internal EE tag were purchased from Guthrie Research Institute (Sayre, PA). An internal EE epitope tag was introduced into the human G-protein alpha 12 subunit (wild type; GNA1200000) via the Quickchange mutagenesis kit (Stratagene). Residues NYFPSK (193–198) were mutated to EYMPTE. An internal EE epitope tag was introduced into the human G-protein alpha 13 subunit (wild type; GNA130000) via the Quickchange mutagenesis kit (Stratagene). Residues DYIPSQ (188–193) were mutated to EYMPTE. Dominant negative CaMKII (K42M) and CaMKIV (dCTK75E) were obtained from Dr. M. Rosner (University of Chicago, IL) and Dr. J. Xie (University of Manitoba, Canada), respectively. pGL2-MEF2-luc, HA-HDAC4, and HA-HDAC5 were generated as described [25] and were provided by Dr. S. Khochbin (INSERM, France). MEF2A in pCMV-Sport6 was purchased from Open Biosystems. MEF2C was from Dr. L. Kedes (University of Southern California, CA) and MEF2-VP16 was from Dr. E. Olson (University of Texas at Dallas, TX). Activated mutants of MEK5 and MEK6, MEK5(DD) and MKK6b(E), were from Dr. M. Cobbs (University of Texas at Dallas, TX) and Dr. J. Han (The Scripps Research Institute, CA), respectively.
Monoclonal antibodies against Hsp90 and HA epitope and polyclonal antibodies against phospho-CaMKIV, Gα12, Gα13, and MEF2 were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Monoclonal antibody against EE epitope was from Covance (Berkeley, CA). MEF2C and CaMKIV polyclonal antibodies were from Cell Signaling (Beverly, MA). Goat CaMKIV antibody and CaMKIV substrate were purchased from Santa Cruz Biotechnology. Thrombin was purchased from Enzyme Research Laboratories (South Bend, IN) and okadaic acid was purchased from Sigma (St Louis, MO). Cell-permeable myristoylated protein kinase A (PKA) inhibitor (PKI) [14–22] amide and H-89 were from Calbiochem (San Diego, CA). [–32P]ATP (3000 Ci/mmol) was purchased from Amersham Pharmacia Biotechnology (Piscataway, NJ).
Cell culture and transfection
NIH 3T3 fibroblasts were grown in DMEM medium supplemented with 10% heat-inactivated calf serum, 100 U/ml streptomycin, and 100 U/ml penicillin. LipofectAmine 2000 reagent (Invitrogen, Carlsbad, CA) was used for transfection according to the manufacturer’s instruction. The human umbilical vein endothelial cells (HUVECs) obtained from Cambrex (Walkersville, MD) were cultured for up to six passages in EBM-2 medium (Cambrex) supplemented with 10% (v/v) fetal bovine serum (FBS, Invitrogen) and EGM-2 SingleQuots (Cambrex). CytoPure™-huv (Q. Biogene, Morgan Irvine, CA) and Targefect-HUVEC (Targeting systems, Santee, CA) were used for transfection in HUVECs. When both cDNA plasmid and siRNA were used, HUVECs were transfected with cDNA plasmid for 3 h, recovered for 2 h, and subsequently transfected with siRNA overnight. Transfection efficiency of cDNA constructs was ~90% for NIH 3T3 cells and ~35% for HUVECs. Transfection efficiency of siRNA was ~95–99% for both cell types.
Downregulation of Gα13, Gα12, and MEF2 proteins
Downregulation of the endogenous Gα13 and Gα12 was achieved using siRNA. Gα13-specific siRNA was made by Qiagen (Valencia, CA) to target the sequence AAGGAGATCGACAAATGCCTG (94–114) of human Gα13. Gα12-specific (CCGGATCGGCCAGCTGAATTA) and MEF2A-specific (CACGCATGAGATATTCAGAAA) siRNA duplexes were purchased Qiagen. The non-silencing control siRNA was from Qiagen. The siRNA transfection was performed using siRNA transfection reagent and medium from Santa Cruz Biotechnology. The effects of siRNAs were confirmed by real-time PCR and Western blot at 24 or 48 h after transfection, respectively.
Real-time PCR
Total RNA was prepared using RNeasy RNA extraction kit with DNaseI treatment following the manufacturer’s instructions (Qiagen). Using random primers and Superscript III transcriptase (Invitrogen), 1 μg total RNA was converted into cDNA. Specific primers for Gα12, Gα13, and four members of MEF2 proteins were purchased from Qiagen. The housekeeping gene glyceraldehydes-3-phosphate dehydrogenase (GAPDH) was used as a reference gene for quantification. GAPDH primers were synthesized by Integrated DNA Technologies. PCR was performed with 50 ng cDNA in a 25 μl reaction volume containing a SYBR Green Master Mix (Applied Biosystems). Amplification was carried out utilizing an ABI PRISM 7000 sequence detection system (Applied Biosystems). Cycling conditions were 50°C for 2 min, 95°C for 10 min followed by a 40-cycle amplification at 95°C for 15 s, and 57°C for 1 min. Experiments were repeated two times and samples were analyzed in triplicate. Results of the real-time PCR data were represented as Ct values, where Ct is defined as the threshold cycle of PCR at which amplified product was first detected. To compare the different RNA samples, we used the comparative Ct method and compared the RNA expression in samples to that of the control in each experiment.
Reporter gene assay
MEF2-dependent gene transcription was determined using MEF2-luc [25] reporter system and SRE-mediated gene expression was determined by SRE.L reporter system (Stratagene). NIH 3T3 cells were grown on 24-well plates at 90% confluence. Cells were transfected with the following plasmids (per well): 50 ng pGL2-MEF2-luc or pSRE.L reporter, 50 ng pCMV–β-galactosidase, and 50–200 ng other plasmids as indicated in figure legends. Cells were serum-starved overnight, washed with PBS buffer, lysed, and assayed following the manufacturer’s instruction for luciferase and galactosidase activities using Promega assay kit (Promega, Madison, WI). HUVECs were grown at 90% confluence on 12-well plates and were transfected with the following plasmids (per well): 0.7 μg pGL2-MEF2-luc, 0.1 μg Renilla (control plasmid for transfection efficiency), and 0.3 μg of other plasmids as indicated in figure legends. Cells were serum-starved overnight. In some cases, cells were incubated with 20 nM thrombin for 6 h. Promega Dual-luciferase Reporter Assay System was used to determine the luciferase activity. Luciferase activity was normalized to the activity of Renilla to correct the difference caused by different transfection efficiency.
Cell proliferation assay
After transfection with indicated siRNA for 48 h in serum-free transfection medium, HUVECs (5 × 103 cells/well) in 96-well plates were incubated with EBM-2 medium with or without 1% One Solution FBS for 24 h. Then, cell proliferation was determined with CellTiter 96 AQueous Cell Proliferation Assay from Promega (Madison, WI).
Endothelial tube formation assay
After transfection with indicated siRNAs for 24 h in the serum-free transfection medium, confluent HUVECs in 60-mm plates were harvested with trypsin, washed with EBM-2 medium twice, and counted. Growth factor-reduced Matrigel gels (BD labware, Bedford, MA) were formed at 37°C for 1 h. Transfected HUVECs were seeded in 30 × 103 cells/well in 48-well plate. After 20 h, the tube formation were assessed (4× magnification) with an inverted phase contrast Nikon microscope and the image captured with digital camera. For quantitative measure of tube formation, the branch number in each experiment was counted.
Western blotting
Cells were washed with ice-cold PBS, lysed in lysis buffer (20 mM HEPES, pH 7.4, 50 mM NaCl, 1% Triton X-100, and Sigma cocktail of protease and phosphatase inhibitors), and briefly sonicated on ice. Lysates were cleared by centrifugation. Lysates were subjected to SDS-polyacrylamide gel electrophoresis (SDS–PAGE). Proteins were transferred to PVDF membrane and were analyzed by immunoblotting with appropriate antibodies. Densitometry of protein bands was performed on scanned images using NIH Image 1.63 software. Data were normalized as described in Fig. 1.
Fig. 1.

Gα13Q226L stimulates MEF2-dependent gene transcription. a The expression levels of EE-tagged Gα13Q226L and Gα12Q229L in NIH 3T3 cells. NIH 3T3 cells were grown on 24-well plates at 90% confluence and transfected with 50 ng/well Gα12Q229L and Gα13Q226L. Proteins were visualized by Western blotting with antibodies as indicated. Actin staining is used as a loading control. Experiment shown is representative of three similar experiments. b, c MEF2-dependent and SRE-dependent gene transcription induced by Gα12Q229L and Gα13Q226L in NIH 3T3 cells. NIH 3T3 cells were grown on 24-well plates at 90% confluence. Cells were transfected with the following plasmids (per well): 50 ng pGL2-MEF2-luc or pSRE.L reporter, 50 ng pCMV–β-galactosidase, and 50 ng EE-tagged Gα13Q226L and Gα12Q229L as indicated. Transfection of 50 ng pcDNA3.1 was used as a vector control. Luciferase activity was normalized against β-galactosidase activity and was expressed as fold stimulation of the basal level. Data points represent the means ± SE from at least three independent experiments performed in triplicates
Phosphorylation of CaMKIV and Ca2+/calmodulin-independent CaMKIV kinase assay
The phospho-CaMKIV and total CaMKIV were detected using appropriate antibodies. To analyze Ca2+/CaM-independent CaMKIV kinase activity, in vitro kinase assay was performed. Briefly, NIH 3T3 cells (100 mm dishes) were lysed in the lysis buffer (20 mM HEPES, pH 7.4, 2 mM EGTA, 1 mM DTT, 1% Triton X-100, 10% glycerol, Sigma cocktails of protease and phosphatase inhibitors). Clarified cell lysates were incubated with the antibody against CaMKIV for 1 h at 4°C followed by the overnight incubation with protein A/G agarose beads at 4°C. Precipitated complexes were washed and Ca2+/calmodulin-independent CaMKIV activity was assayed in the 100 μl volume of the kinase buffer (25 mM HEPES, pH 7.5, 10 mM MgCl2, 0.5 mM DTT, 200 μM ATP, 1 μCi – 32P-ATP [3000–5000 cpm/pmol], 150 μg substrate peptide [KSDGGVKKRKSSSS], 2 mM EGTA) for 10 min at 30°C. Reaction was stopped by brief centrifugation and aliquots were spotted on Whatmann P81 P-cellulose paper, washed with 75 mM phosphoric acid buffer. Free [γ–32P]ATP was eluted by four washes in 150 mM phosphoric acid, and radioactivity incorporated into peptide substrate was measured by scintillation counting. The average radioactivity of two blank samples (range = 150–350 cpm) containing no immune complex was subtracted from the result of each sample.
PP2A activity assay
Immunoprecipitation and activity measurement of protein phosphatase 2A (PP2A) were conducted by using a kit from Upstate according to the manufacturer’s instruction.
Immunofluorescence microscopy
HUVECs were seeded on gelatinized coverslips and transfected with 0.4 μg HA-tagged HDAC5, with or without 0.2 μg EE-tagged Gα13Q226L or Gα12Q229L, respectively. After 20 h transfection, cells were fixed with 3.7% paraformaldehyde in PBS for 20 min, washed, and permeabilized with 0.1% Triton X-100 for 5 min, and blocked with 1% BSA in 0.2% fish skin gelatin HBSS (blocking solution) for 1 h. Coverslips were incubated with 1 μg/ml polyclonal EE antibody for 1 h at room temperature. After five washes with PBS containing 0.1% Tween-20, cells were incubated with anti-rabbit secondary antibody conjugated to immunofluorescent dye Alexa 594 (red fluorescence) in blocking solution for 1 h at room temperature. Thereafter, cells were incubated with FITC-conjugated (green fluorescence) monoclonal HA antibody. Coverslips were mounted using Vectashield mounting medium with DAPI (Vector Laboratories, Inc., Burlingame, CA). Analysis of fluorescent staining was performed using a Zeiss LSM 510 confocal microscopy.
Statistical analysis
For statistical analysis, ANOVA test was used to compare data between two groups. Values are expressed as mean ± SD of triplicate from a representative of two to four experiments, otherwise indicated in figure legends. P < 0.05 was considered statistically significant.
Results
MEF2-dependent gene transcription is stimulated by Gα13Q226L
We tested whether Gα12 and Gα13 can regulate MEF2-dependent gene transcription. In order to achieve equal expression of G-proteins, we used EE-tagged constitutively active mutants of Gα12 and Gα13, EE-tagged Gα13Q226L and Gα12Q229L. Western blot showed that at 50 ng cDNAs, the expression levels of Gα13Q226L and Gα12Q229L were similar (Fig. 1a). Expression level of EE-tagged Gα subunits was ~50% of expression level of endogenous Gα subunits (data not shown). MEF2-dependent gene transcription was measured using reporter assay with a plasmid encoding binding site of MEF2 fused with luciferase [25]. NIH 3T3 cells were transfected with MEF2-driven luciferase reporter. To correct variations in transfection efficiency, an expression vector coding for β-galactosidase was co-transfected with the above constructs and the expressed β-galactosidase activity was used for normalization of MEF2 luciferase data. Twenty-four hours after transfection, cells were serum starved for additional 16 h. Then, cells were collected and MEF2 luciferase reporter activity was measured. To evaluate functional activity of Gα13Q226L and Gα12Q229L, SRE-dependent gene transcription was measured.
Data showed that Gα13Q226L stimulated both MEF2-dependent and SRE-dependent gene transcription by sixfold (Fig. 1b, c). By contrast, Gα12Q229L stimulated only SRE-dependent gene transcription (Fig. 1b, c).
Involvement of Gα13 in thrombin-stimulated MEF2-dependent gene transcription
In endothelial cells, thrombin receptor, PAR-1, is coupled to multiple G-proteins, including Gi, Gq, G12, and G13 [17, 41, 50]. We examined whether Gα13 mediates thrombin-induced MEF2-dependent gene transcription in endothelial cells using siRNA targeted to Gα13 and Gα12. HUVECs were transfected with 20 pg control, Gα12, or Gα13 siRNA. Expression of mRNA was examined using real-time PCR. The housekeeping gene GAPDH was used as a reference gene for quantification (Fig. 2a). Twenty-four hours after transfection, Gα13 mRNA was decreased by ~80% and Gα12 mRNA was decreased by ~85% (Fig. 2a). In control experiments, we determined that Gα12 and Gα13 siRNAs did not induce apoptosis in HUVECs (data not shown). Western blotting showed that Gα13 expression was reduced by 70%, whereas expression of Gα12 and Hsp90 was not affected (Fig. 2b, c). The siRNA targeted to Gα12 reduced the expression of endogenous Gα12 by 80% but did not affect the expression of Gα13 and Hsp90 (Fig. 2b, c).
Fig. 2.
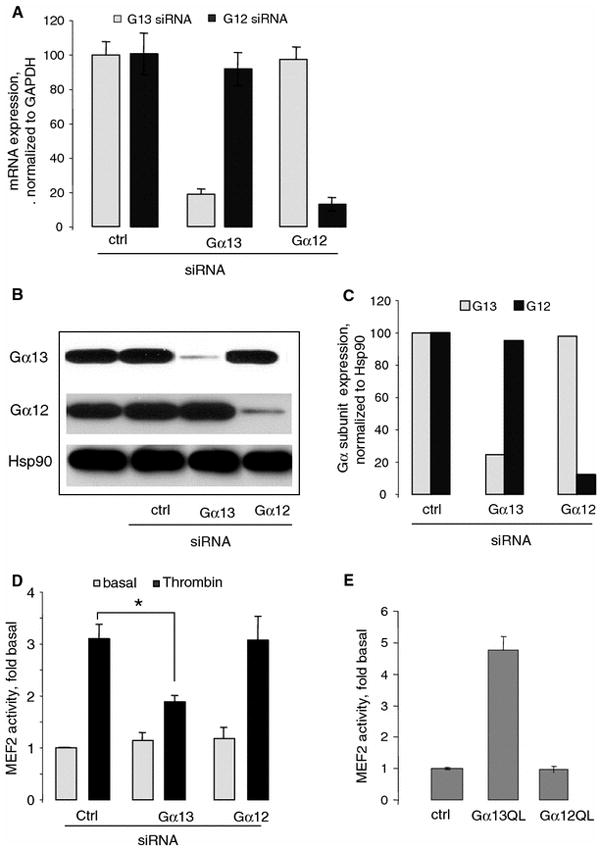
Involvement of Gα13 in thrombin-stimulated MEF2-dependent gene transcription. a Specific downregulation of the endogenous Gα13 and Gα12 mRNAs in HUVECs 24 h after transfection with indicated siRNAs. HUVECs grown on six-well plates to confluency were transfected with 50 pg indicated siRNAs. Twenty-four hours after transfection, indicated mRNAs were quantified using real-time PCR. Expression of Gα12 or Gα13 treated with control siRNA was normalized to GAPDH expression (100%). The data are shown as mean ± SD of triplicate after normalization with control GAPDH. b HUVECs grown on six-well plates to confluency were transfected with 50 pg indicated siRNAs for 24 hours. Reduction of the expressed Gα13 and Gα12 proteins after 48 h siRNA transfection by Western blot analysis using appropriate antibodies. c Western blotting data were normalized to Hsp90 content in respective samples. Data shown are the means of two replicates. d Reduction of endogenous Gα13 but not Gα12 inhibits the thrombin-stimulated MEF2-dependent gene transcription in HUVECs. HUVECs grown on six-well plates were transfected with 50 ng pGL2-MEF2-luc, 50 ng pCMV–β-galactosidase, and 50 pg indicated siRNA. Twenty-four hours after transfection, HUVECs were stimulated with thrombin for 6 h and MEF2 activity was measured. Luciferase activity was normalized against β-galactosidase activity and was expressed as fold stimulation from basal level. Data are shown as mean ± SD from three experiments. *P < 0.05. e Gα13Q226L stimulates MEF2-dependent gene transcription in HUVECs. HUVECs grown on six-well plates were transfected with 100 ng Gα12Q229L or Gα13Q226L. MEF2 activity was measured 36 h after transfection. Data points represent the means ± SE from at least three independent experiments performed in triplicates. *P < 0.05
We tested how downregulation of Gα12 and Gα13 would affect thrombin-induced MEF activity. HUVECs grown on six-well plates were transfected with 50 ng pGL2-MEF2-luc, 50 ng pCMV–β-galactosidase, and 50 pg indicated siRNA. Twenty-four hours after transfection, HUVECs were stimulated with thrombin for 6 h and MEF2 activity was measured. Reduction of endogenous Gα13 by siRNA inhibited the thrombin-stimulated MEF2-dependent gene transcription by 50% (Fig. 2d). By contrast, the control and Gα12 siRNAs did not affect thrombin-induced MEF2-dependent gene transcription (Fig. 2d). These results suggest that the endogenous Gα13 but not Gα12 is required for thrombin-induced MEF2-dependent gene transcription in HUVECs.
To corroborate the role of Gα13, we examined whether Gα12Q229L and Gα13Q226L can stimulate MEF2-dependent gene transcription in HUVECs. Gα13Q226L induced a fivefold increase in MEF2-dependent gene transcription. By contrast, Gα12Q229L did not affect the transcription (Fig. 2e).
Gα13 and MEF2 proteins are required for HUVECs proliferation and capillary tube formation Angiogenesis is a tightly regulated process that involves the degradation of extracellular matrix, disruption of cell–cell contacts, migration and proliferation, and capillary sprout formation of endothelial cells [48]. To examine the role of Gα13 and MEF2 proteins, we have used two in vitro assays: Matrigel capillary tube formation and endothelial cell proliferation. HUVECs were transfected with 20 pg control or MEF2A siRNAs. Twenty-four hours after transfection, expression of MEF2A, MEF2B, MEF2C, and MEF2D mRNAs was analyzed using RT-PCR. The housekeeping gene GAPDH was used as a reference gene for quantification (Fig. 3a). Real-time PCR showed siRNA MEF2A primary targeted to MEF2A and did not affect other members of MEF2 protein (Fig. 3a). Western blotting using the antibody that detect all the members of MEF2 proteins showed that the expression of endogenous MEF2 proteins was decreased by 75%; the expression of Hsp90 was not affected (Fig. 3b).
Fig. 3.
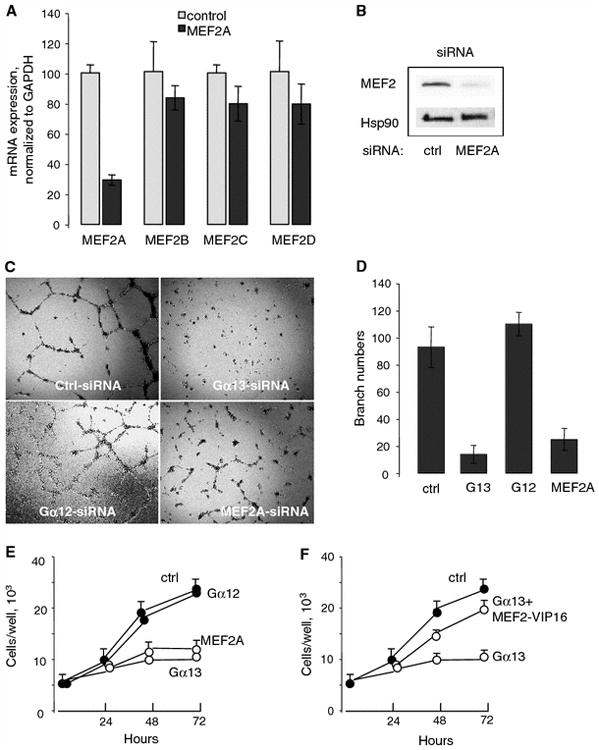
Essential roles of Gα13 and MEF2 proteins in endothelial cell tube formation and proliferation. a The selectivity of siRNA for MEF2A against other members of MEF2 protein. HUVECs grown on six-well plate were transfected with 50 pg siRNA as indicated; 24 h after transfection, mRNA levels of each MEF2 were analyzed with real-time PCR. The data are shown in mean ± SD of triplicate after normalization with control GAPDH. b HUVECs were transfected with 50 pg MEF2A siRNA and, 48 h after transfection, MEF2 proteins were analyzed using MEF2 antibody. The antibody was recommended by the manufacturer for detection MEF2A and to lesser extent, MEF2C and MEF2D. Two additional experiments showed similar results. c A representative data (d) quantity data show that Gα13 and MEF2 proteins are required for tube formation. HUVECs grown on six-well plate were transfected with 50 pg siRNAs for control, Gα13, Gα12, and MEF2A for 24 h. The cells were then harvested, counted, and seeded in 3 × 104 cells/well in 48-well plate with formed growth factor-reduced Matrigel gels. The tube formation were assessed (4× magnification) with an inverted phase contrast Nikon microscope and the image captured with digital camera after 20 h. Quantity of branch number in tube formation assay is shown in mean ± SD from three experiments. e Gα13 and MEF2A proteins are required in HUVECs proliferation. HUVECs grown on six-well plate were transfected with 50 pg siRNA for 24 hours as indicated. Thereafter, cells were seeded in 24-well plate (5 × 104 cells/well) and stimulated with 1% FBS for 24, 48, and 72 h and counted. Data are shown in mean ± SD from a representative of three experiments performed in triplicate. f Constitutively active MEF2-VP16 partially rescues the cell proliferation in HUVECs transfected with Gα13 siRNA. HUVECs grown on six-well plate were transfected with 50 pg Gα13 or control siRNA and 50 ng MEF2-VP16 for 48 hours. Proliferation was determined as described earlier. Data are shown in from a representative of experiments in triplicate. *P < 0.05
To test capillary formation, we used Matrigel assay. HUVECs were transfected with indicated siRNAs for 24 h in the serum-free transfection medium; confluent HUVECs in 60-mm plates were harvested with trypsin, washed with EBM-2 medium twice, and counted. Growth factor-reduced Matrigel gels (BD labware, Bedford, MA) were formed at 37°C for 1 h. Transfected HUVECs were seeded in 304 cells/well in 48-well plate. After 20 h, the tube formation were assessed (4× magnification) with an inverted phase contrast Nikon microscope and the image captured with digital camera. For quantitative measure of tube formation, the number of branch points is counted in ten random fields. Data showed that downregulation of both Gα13 and MEF2A disrupted formation of capillary tubes whereas control and Gα12 siRNAs did not affect capillary tube formation (Fig. 3c, d).
Heterotrimeric G13 protein is known to activate cell proliferation [1]. Importantly, involvement of MEF2 proteins in proliferation is not understood. We analyzed the effect of downregulation of Gα13 or MEF2A on HUVECs proliferation. Downregulation of Gα13 or MEF2A significantly inhibited HUVECs proliferation (Fig. 3e). To test whether MEF2 proteins contribute to Gα13-mediated endothelial cell proliferation, we examine whether constitutively activated MEF2-VP16 could restore HUVECs proliferation that was inhibited by Gα13 downregulation. Data showed that MEF2-VP16 partially restored HUVECs proliferation that was inhibited by Gα13 downregulation (Fig. 3f).
CaMKIV mediates Gα13Q226L-stimulated MEF2-dependent gene transcription
We have recently reported that Gα13Q226L can increase the phosphorylation of CaMKII [28]. We have also shown that Gα13-induced activation of SRE-dependent gene transcription is partially mediated by CaMKII and CaMKIV [28], suggesting that CaM kinases are involved in the Gα13 signaling. CaM kinases were shown to be potent activators of MEF2 activity [31, 32]. Therefore, we tested whether CaMKII or CaMKIV is involved in Gα13-activated MEF2-dependent gene transcription.
As shown on Fig. 4a, MEF2-dependent gene transcription stimulated by Gα13Q226L was inhibited by the dominant negative CaMKIV by 50%, whereas dominant negative CaMKII had no effect. Functional activity of dominant negative CaMKII was confirmed by its ability to inhibit Gα13Q226L-mediated SRE-dependent gene transcription (Fig. 4b). These data suggested that CaMKIV but not CaMKII is involved in Gα13-mediated MEF2-dependent gene transcription.
Fig. 4.

Involvement of CaMKIV in Gα13Q226L-stimulated MEF2-dependent gene transcription. a, b Dominant negative CaMKIV inhibits Gα13Q226L-induced MEF2-dependent gene transcription. HUVECs grown on six-well plates were transfected with 100 ng Gα13Q226L, CaMKII (K42M), or CaMKIV (dCTK75E) as indicated. Forty-eight hours after transfection, a MEF2- and b SRE-dependent gene transcription was measured. Data points represent the means ± SE from at least three independent experiments performed in triplicates. *P < 0.05. c Gα13Q226L increases the phosphorylation of CaMKIV. HUVECs grown on six-well plates were transfected with 100 ng with indicated plasmids. The total cell lysates were subjected to SDS–PAGE. The phosphorylated and total CaMKIV were detected with the appropriate antibodies. Representative blot is shown. d Gα13Q226L increases the Ca2+/calmodulin-independent CaMKIV activity. Using the antibody for CaMKIV, the endogenous CaMKIV was immunoprecipitated from HUVECs transfected with 100 ng vector, Gα13Q226L, and Gα12Q229L, and then the Ca2+/CaM-independent CaMKIV activity was measured in the presence of 2 mM EGTA without Ca2+ and calmodulin. Data points represent the means ± SE from at least three independent experiments performed in triplicates. *P < 0.05
Next, we examined the effect of Gα13Q226L on the phosphorylation of CaMKIV. We tested whether Gα13Q226L affected the phosphorylation of CaMKIV at the position of Thr196 using phospho-specific antibody. Western blot showed that Gα13Q226L but not Gα12Q229L induces an increase in the phosphorylation of CaMKIV (Fig. 4c).
It has been reported that autonomous activity of CaMKIV, which is Ca2+/calmodulin independent, is important in the regulation of gene transcription [11]. To determine whether Gα13Q226L can activate Ca2+/calmodulin-independent activity of CaMKIV, we transfected HUVECs with Gα13Q226L and endogenous CaMKIV was immunoprecipitated from cell lysates as described in “Materials and methods” section. Ca2+/calmodulin-independent CaMKIV activity was measured in the presence of 2 mM EGTA without Ca2+ and calmodulin. As shown in Fig. 4d, Gα13Q226L but not Gα12Q229L induces an increase in autonomous activity of CaMKIV, further suggesting the involvement of CaMKIV in Gα13-mediated MEF2-dependent gene transcription.
HDAC5 repression of MEF2-dependent gene transcription is released by Gα13Q226L
One of the functions of class II HDACs is to repress the MEF2-dependent gene transcription by their direct binding to MEF2 proteins [6, 25]. Phosphorylation of HDAC5 by CaMKIV results in the derepression of the MEF2-dependent gene transcription [31, 32]. We examined whether Gα13Q226L can release the repression of MEF2-dependent gene transcription induced by HDAC5.
HUVECs transfected with MEF2A or MEF2C showed a 2.5-fold increase in MEF2-dependent gene transcription (Fig. 5a, b). In agreement with previous studies [25], HDAC5 completely repressed MEF2A- and MEF2C-induced MEF2-dependent gene transcription (Fig. 5a, b). Gα13Q226L released the HDAC5-mediated repression of MEF2-dependent gene transcription (Fig. 5a, b). By contrast, Gα12Q229L showed no effect. Similarly, only Gα13Q226L released the HDAC4-mediated repression of MEF2-dependent gene transcription (data not shown).
Fig. 5.

Gα13Q226L releases the repression of MEF2-dependent gene transcription-induced HDAC5. a, b HUVECs grown on six-well plates were transfected with 200 ng empty vector, 50 ng Gα13Q226L, 200 ng MEF2A, 200 ng MEF2C, and 25 ng HDAC5 as indicated. MEF2 activity was measured 48 h after transfection. Overexpression of MEF2 proteins was ~100% over basal level, overexpression of HDAC5 was ~50% over basal level. Data points represent the means ± SE from at least three independent experiments performed in triplicates. *P < 0.05. c, d p115RhoGEF and RhoA pathway is not required for releasing HDAC5 repression. HUVECs were transfected with 50 ng p115RhoGEF, 50 ng RhoA (G14V), 200 ng MEF2A, 200 ng MEF2C, and 25 ng HDAC5 as indicated. MEF2 activity was measured 48 h after transfection. Data points represent the means ± SE from at least three independent experiments performed in triplicates. *P < 0.05
Gα13 is known to activate Rho signaling pathway via guanine nucleotide exchange factor, p115RhoGEF [20, 24]. We tested whether Rho signaling pathway is involved in the derepression of HDAC5 in MEF2-dependent gene transcription. Neither p115RhoGEF nor constitutively activated RhoA, Rho14V, was able to induce derepression of MEF2-dependent gene transcription (Fig. 5c, d). Functional activity of p115RhoGEF and Rho14V was confirmed by their ability to activate SRE-dependent gene transcription (data not shown). Therefore, our data suggested that Gα13Q226L-mediated derepression of MEF2-dependent gene transcription is RhoA independent.
Gα13Q226L induced the translocation of HDAC5 from nucleus to cytoplasm
Phosphorylation of the transcriptional repressors HDAC4 and HDAC5 by CaMKIV leads to their translocation from nucleus to cytoplasm that results in the release of their repression of the MEF2-dependent gene transcription [31, 32]. Because Gα13Q226L activated CaMKIV, we examined whether Gα13Q226L can induce translocation of HDAC5 from nucleus to cytoplasm, thereby releasing the repression of MEF2-dependent gene transcription. HUVECs were transfected with HA-tagged HDAC5 and EE-tagged Gα13Q226L or Gα12Q229L and stained with appropriate antibodies. Nuclei were visualized with DAPI staining. As expected, in cells transfected only with HA-HDAC5, HDAC5 was expressed in the nucleus (Fig. 6). Gα13Q226L induced translocation of HDAC5 to the cytoplasm (Fig. 6). By contrast, Gα12Q229L did not affect HDAC5 localization (Fig. 6).
Fig. 6.

Gα13Q226L induces the translocation of HDAC5 from nucleus to cytoplasm. HUVECs grown on 24-mm coverslips were transfected with 100 ng EE-tagged Gα13Q226L or Gα12Q229L and HA-tagged HDAC5. The expressions of Gα13Q226L and Gα12G229L were examined with EE antibody and anti-mouse secondary antibody conjugated to immunofluorescent dye Alexa 594 (red fluorescence). HDAC5 was detected with FITC-conjugated HA antibody. The nuclei were identified with DAPI. Two additional experiments showed similar results
PKA but not PP2A is involved in Gα13Q226L-induced MEF2-dependent gene transcription Gα12 family was shown to interact with PP2A and stimulate its activity [55]. Because PP2A is stably associated with CaMKIV and inactivates it by dephosphorylation [2], it was tempting to study the role of PP2A in Gα13-induced MEF2-dependent gene transcription. Data showed that endogenous PP2A activity was not affected in HUVECs transfected with Gα13Q226L or Gα12Q229L (Fig. 7a). Similarly, neither Gα13Q226L nor Gα12Q229L affected the expression of endogenous PP2A (data not shown). Finally, PP2A inhibitor, okadaic acid, did not affect Gα13Q226L-induced MEF2-dependent gene transcription (Fig. 7b). These results suggested that PP2A is not involved in Gα13-induced MEF2-dependent gene transcription.
Fig. 7.

PKA but not PP2A is involved in Gα13Q226L-induced MEF2-dependent gene transcription. a HUVECs grown in six-well plate were transfected with 100 ng empty vector, Gα13Q226L, and Gα12Q229L for 48 hours. The endogenous PP2A was precipitated with anti PP2A antibody and its activity was measured. Data are shown in mean ± SD from four experiments. b Effects of the potent PP2A inhibitor okadaic acid on MEF2-dependent gene transcription in HUVECs transfected with 100 ng vector, Gα13Q226L, and Gα12Q229L. The cells were incubated with okadaic acid at indicated concentrations for 18 h after transfection. Data points represent the means ± SE from at least three independent experiments performed in triplicates. *P < 0.05. c Effects of the potent PKI and H-89 on MEF2-dependent gene transcription in HUVECs transfected with 100 ng Gα13Q226L. The cells were incubated with PKI and H-89 at indicated concentrations for 6 h after transfection. Data points represent the means ± SE from at least three independent experiments performed in triplicates. *P < 0.05. d Gα13Q226L-induced phosphorylation of CaMKIV is inhibited by PKI and H-89. HUVECs transfected with 100 ng Gα13Q226L were treated with 30 μM PKI and 1 μM H–89 for 1 h before assay. The total cell lysates were subjected to SDS–PAGE. The phosphorylation form and total CaMKIV were detected with the appropriate antibodies. Two additional experiments showed similar results. e Inhibition of Gα13QL induced HUVEC proliferation by dominant negative form of CaMKIV and PKA inhibitor H-89. HUVECs grown on six-well plate were transfected with 100 ng Gα13Q226L and 100 ng dead CaMKIV or incubated with 1 μM H-89 for 24 h in the absence of FBS, and counted. Data are shown in from a representative of three experiments in triplicate
Studies from our laboratory have shown that Gα13 can stimulate PKA [35, 39]. Therefore, we tested the effects of PKA inhibitors, PKI and H-89, on Gα13Q226L-stimulated MEF2-dependent gene transcription. Both PKI and H-89 partially inhibited MEF2-dependent gene transcription (Fig. 7c). Since PKA may affect the ability of MEF2 proteins binding to DNA [53], we examined whether PKA inhibitors are able to inhibit the phosphorylation of CaMKIV. Indeed, both PKI and H-89 inhibited phosphorylation of CaMKIV induced by Gα13Q226L (Fig. 7d). These results suggest that Gα13-PKA pathway may be involved in regulation of CaMKIV activity and subsequent MEF2-dependent gene transcription.
Activated Gα13 protein is known to activate proliferation of fibroblasts [1]. We tested whether Gα13Q226L can affect HUVECs proliferation. Data showed increased proliferation of HUVECs transfected with activated Gα13Q226L (Fig. 7e). Importantly, this increase was inhibited by dominant negative CaMKIV or PKA inhibitor H-89 (Fig. 7e), suggesting that both CaMKIV and PKA are involved in Gα13-mediated endothelial cell proliferation.
Discussion
In this study, we determined that alpha subunit of heterotrimeric G13 protein regulates angiogenic response via MEF2-dependent gene transcription. We showed that molecular mechanism of Gα13-dependent activation of MEF2 includes Gα13–CaMKIV axis resulting in the activation of MEF2 via phosphorylation of transcription repressor HDAC. Finally, we determined that Gα13-dependent activation of MEF2 is RhoA independent.
MEF2 is required for Gα13-dependent angiogenesis
Gene knockout mice studies have shown the different physiological roles of Gα12 and Gα13 proteins during vascular development [19, 36]. Gα13 deficiency resulted in embryonic lethality at 9.5 day due to impaired vascular formation, whereas Gα12 deficiency had no obvious phenotype. Importantly, endothelial-specific deletion of Gα13 also resulted in embryonic lethality, indicating the essential role of Gα13 in endothelial cells during embryonic development [42]. Molecular mechanisms underlying mechanism of Gα13-mediated vascular development remain unknown.
We have shown the essential role of Gα13 in endothelial cell proliferation because downregulation of Gα13 inhibited proliferation of HUVECs. Furthermore, downregulation of Gα13 lead to inhibition of capillary tube formation in Matrigel assay.
Role of MEF2 proteins in vascular formation was determined by knockout mice studies [7, 26]. Deletion of MEF2C gene resulted in embryonic lethality by 9.5 day due to impaired vascular formation and cardiac defect [21]. It was reported recently that vascular endothelial growth factor (VEGF), which is a key regulator of physiological and pathologic angiogenesis, induces MEF2C and MEF2-dependent activity in endothelial cells [29].
Our data have shown that Gα13 induces MEF2-dependent gene transcription. Importantly, we showed that Gα13 is required for thrombin-stimulated MEF2-dependent gene transcription.
Rho family of proteins signals downstream of VEGF receptors (for review [9]). Rho proteins are implicated in several stages of angiogenesis, such as endothelial barrier function, endothelial migration and proliferation, and capillary survival [9]. Rho proteins are shown to be major effector proteins of Gα13 [24]. Importantly, our data suggested that Gα13-dependent, MEF2-dependent gene transcription is RhoA independent.
Role of PKA in angiogenic processes is poorly defined. Thus, it was reported that COX-2 inhibitors suppress endothelial cell migration and angiogenesis by preventing cAMP/PKA-dependent activation of the small GTPases Rac and Cdc42 [14]. We have identified a novel signaling pathway mediated by Gα13 [35, 39]. We determined that Gα13 can stimulate PKA in cAMP-independent manner [35, 39]. Here, we showed that Gα13Q226L-stimulated MEF2-dependent gene transcription is mediated via PKA, defining a novel signaling pathway mediated by Gα13-dependent activation of PKA.
Gα13–CaMKIV axis results in the activation of MEF2 via phosphorylation of transcription repressor HDAC
Ca2+/calmodulin-independent CaMK activity, also called autoactivity of CaMKIV, is important in the regulation of gene transcription [11]. CaMK is able to stimulate the MEF2-dependent gene transcription [33]. CaMK phosphorylates the gene transcription repressor class II HDAC. Phosphorylation of HDAC leads to dissociation of HDAC from MEF2 proteins and translocation of HDAC from nucleus to cytoplasm [33]. We found that Gα13 can activate MEF2-dependent gene transcription via activation of CaMKIV. Thus, Gα13Q226L increased phosphorylation of CaMKIV and activated Ca2+/calmodulin-independent activity of the kinase. Inhibition of Gα13-mediated MEF2-dependent gene transcription by a dominant negative CaMKIV further supported the notion that CaMKIV is involved in Gα13-mediated MEF2-dependent gene transcription. CaMKII has been shown to inactivate HDAC4 and HDAC5 [4, 27]. Since Gα13 can activate CaMKII [28], it is possible that CaMKII is also involved in Gα13-mediated MEF2-dependent gene transcription.
In HUVECs, Gα13Q226L and thrombin (data not shown) induced the translocation of HDAC5 from nucleus to cytoplasm. Furthermore, the repression of HDAC in MEF2-dependent gene transcription was released by Gα13Q226L. It was recently reported that Gβγ subunits is able to inhibit transcriptional co-repression activity of HDAC5 [46]. Our data suggest that a novel member of heterotrimeric G-proteins family, Gα13, is involved in regulation of class II HDACs.
Gα13 activates CaMKIV via PKA but is RhoA independent
Gα12 and Gα13 regulate distinct intracellular pathways often due to the interaction with distinct signaling proteins [3, 22, 35, 40, 44, 49, 55]. Thus, Gα13 can induce phosphorylation of vasodilator-stimulated phosphoprotein in endothelial cells [39] and activation of the chloride conductance in neuronal cells [37]. By contrast, Gα12 can stimulate the serum-induced release of arachidonic acid [13]. Recent studies have shown that Gα12 interacts with Hsp90 [49], PP2A [55], and αSNAP [3], whereas Gα13 interacts with AKAP110 [35], Hax-1 [40], RGS16 [22], and PYK2 [44]. Our data showed that Gα12 had no role in endothelial cell proliferation, capillary tube formation, and stimulation of MEF2-dependent gene transcription.
RhoA pathway is strongly activated by both Gα12 and Gα13. Our data showed that p115RhoGEF and activated form of RhoA could not release the repression of HDAC5, suggesting that RhoA pathways is not involved in Gα13-induced activation of CaMKIV.
We showed previously that Gα13 stimulates PKA [35, 39]. Although cAMP inhibits MEF2D-mediated gene transcription in hippocampal neurons [5], it was reported that PKA phosphorylates MEF2 proteins thereby increasing their DNA binding [53]. We found that PKA inhibitors, PKI and H-89, inhibited Gα13Q226L-stimulated MEF2-dependent gene transcription. In addition, we showed that PKI and H-89 inhibited phosphorylation of CaMKIV induced by Gα13Q226L. It should be pointed out that PKA induced phosphorylation of CaMK kinase, kinase upstream of CaMKIV leads to inhibition of CaMK pathway [30], suggesting that PKA may play dual roles in CaMKIV-mediated signaling. In addition, PKA-dependent Ca2+ influxes through NMDA receptors and Ca2+ channel were reported [45]. On the other hand, PKA may also affect the CaMKII activity by activating PP1 inhibitor peptides [23].
In summary, we demonstrated that Gα13 can activate Ca2+/calmodulin-independent activity of CaMKIV via PKA pathway. Activation of CaMKIV induces translocation of the transcription repressor HDAC5 and releases the repression of HDAC5 on MEF2-dependent gene transcription leading to vascular remodeling (Fig. 8).
Fig. 8.
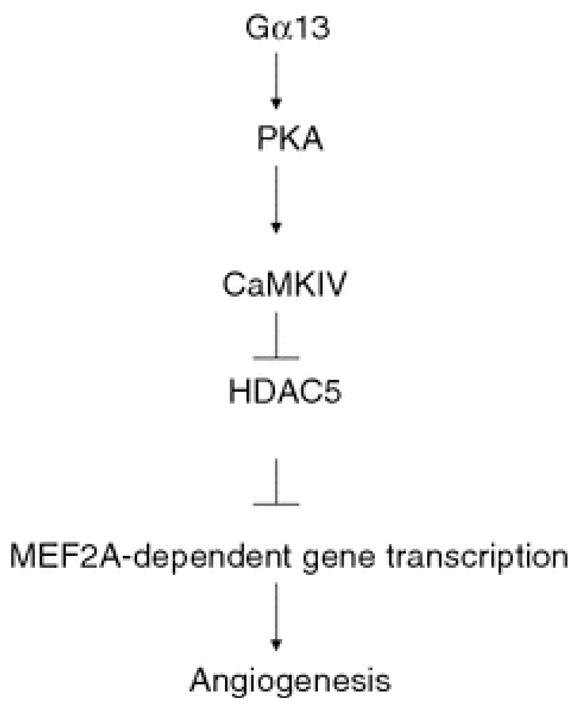
The proposed signaling pathway mediated by Gα13 in MEF2-dependent gene transcription. Activation of Gα13 results in the activation of PKA, which in turn activates CaMKIV. This results in the inhibition of HDCA5 followed by the derepression of MEF2-dependent gene transcription. This event contributes to the Gα13-dependent angiogenic response
Acknowledgments
We thank Drs. M. Cobbs, J. Han, L. Kedes, S. Khochbin, E. Olson, M. Rosner, and J. Xie for providing us with their constructs. This work was supported by National Institutes of Health Grants GM56159 and HL06078 and by a grant from the American Heart Association (to TVY).
References
- 1.Adarichev VA, Vaiskunaite R, Niu J, Balyasnikova IV, Voyno-Yasenetskaya TA. Ga13-mediated transformation and apoptosis is permissively dependent on basal ERK activity. Am J Physiol Cell Physiol. 2003;285:C922–C934. doi: 10.1152/ajpcell.00115.2003. [DOI] [PubMed] [Google Scholar]
- 2.Anderson KA, Noeldner PK, Reece K, Wadzinski BE, Means AR. Regulation and function of the calcium/calmodulin-dependent protein kinase IV/protein serine/threonine phosphatase 2A signaling complex. J Biol Chem. 2004;279:31708–31716. doi: 10.1074/jbc.M404523200. [DOI] [PubMed] [Google Scholar]
- 3.Andreeva AV, Kutuzov MA, Vaiskunaite R, Profirovic J, Meigs TE, Predescu S, Malik AB, Voyno-Yasenetskaya T. G alpha12 interaction with alphaSNAP induces VE-cadherin localization at endothelial junctions and regulates barrier function. J Biol Chem. 2005;280:30376–30383. doi: 10.1074/jbc.M502844200. [DOI] [PubMed] [Google Scholar]
- 4.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belfield JL, Whittaker C, Cader MZ, Chawla S. Differential effects of Ca2+ and cAMP on transcription mediated by MEF2D and cAMP-response element-binding protein in hippocampal neurons. J Biol Chem. 2006;281:27724–27732. doi: 10.1074/jbc.M601485200. [DOI] [PubMed] [Google Scholar]
- 6.Bertos NR, Wang AH, Yang XJ. Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol. 2001;79:243–252. doi: 10.1139/bcb-79-3-243. [DOI] [PubMed] [Google Scholar]
- 7.Bi W, Drake CJ, Schwarz JJ. The transcription factor MEF2C-null mouse exhibits complex vascular malformations and reduced cardiac expression of angiopoietin 1 and VEGF. Dev Biol. 1999;211:255–267. doi: 10.1006/dbio.1999.9307. [DOI] [PubMed] [Google Scholar]
- 8.Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- 9.Bryan BA, D’Amore PA. What tangeled webs they weave: Rho-GTPase control of angiogenesis. Cell Mol Life Sci. 2007;64:2053–2065. doi: 10.1007/s00018-007-7008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buhl AM, Johnson NL, Dhanasekaran N, Johnson GL. G alpha 12 and G alpha 13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J Biol Chem. 1995;270:24631–24634. doi: 10.1074/jbc.270.42.24631. [DOI] [PubMed] [Google Scholar]
- 11.Chow FA, Anderson KA, Noeldner PK, Means AR. The autonomous activity of calcium/calmodulin-dependent protein kinase IV is required for its role in transcription. J Biol Chem. 2005;280:20530–20538. doi: 10.1074/jbc.M500067200. [DOI] [PubMed] [Google Scholar]
- 12.Collins LR, Minden A, Karin M, Brown JH. Galpha12 stimulates c-Jun NH2-terminal kinase through the small G proteins Ras and Rac. J Biol Chem. 1996;271:17349–17353. doi: 10.1074/jbc.271.29.17349. [DOI] [PubMed] [Google Scholar]
- 13.Dermott JM, Reddy MR, Onesime D, Reddy EP, Dhanasekaran N. Oncogenic mutant of Galpha12 stimulates cell proliferation through cycloxygenase-2 signaling pathway. Oncogene. 1999;18:7185–7189. doi: 10.1038/sj.onc.1203345. [DOI] [PubMed] [Google Scholar]
- 14.Dormond O, Rüegg C. Regulation of endothelial cell integrin function and angiogenesis by COX-2, cAMP and protein kinase A. Thromb Haemost. 2003;90:577–585. doi: 10.1160/TH03-03-0196. [DOI] [PubMed] [Google Scholar]
- 15.Fromm C, Coso OA, Montaner S, Xu N, Gutkind JS. The small GTP-binding protein Rho links G protein-coupled receptors and Galpha12 to the serum response element and to cellular transformation. Proc Natl Acad Sci USA. 1997;94:10098–10103. doi: 10.1073/pnas.94.19.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukuhara S, Marinissen MJ, Chiariello M, Gutkind JS. Signaling from G protein-coupled receptors to ERK5/big MAPK 1 involves Galpha q and Galpha 12/13 families of heterotrimeric G proteins. Evidence for the existence of a novel Ras and Rho-independent pathway. J Biol Chem. 2000;275:21730–21736. doi: 10.1074/jbc.M002410200. [DOI] [PubMed] [Google Scholar]
- 17.Gilchrist A, Vanhauwe JF, Li A, Thomas TO, Voyno-Yasenetskaya T, Hamm HE. G alpha minigenes expressing C-terminal peptides serve as specific inhibitors of thrombin-mediated endothelial activation. J Biol Chem. 2001;276:25672–25679. doi: 10.1074/jbc.M100914200. [DOI] [PubMed] [Google Scholar]
- 18.Gossett LA, Kelvin DJ, Sternberg EA, Olson EN. A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol Cell Biol. 1989;9:5022–5033. doi: 10.1128/mcb.9.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu JL, Muller S, Mancino V, Offermanns S, Simon MI. Interaction of G alpha(12) with G alpha(13) and G alpha(q) signaling pathways. Proc Natl Acad Sci USA. 2002;99:9352–9357. doi: 10.1073/pnas.102291599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson EN, Seasholtz TM, Waheed AA, Kreutz B, Suzuki N, Kozasa T, Jones TL, Brown JH, Druey KM. RGS16 inhibits signalling through the G alpha 13-Rho axis. Nat Cell Biol. 2003;5:1095–1103. doi: 10.1038/ncb1065. [DOI] [PubMed] [Google Scholar]
- 23.Kawaguchi SY, Hirano T. Signaling cascade regulating long-term potentiation of GABA(A) receptor responsiveness in cerebellar Purkinje neurons. J Neurosci. 2002;22:3969–3976. doi: 10.1523/JNEUROSCI.22-10-03969.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- 25.Lemercier C, Verdel A, Galloo B, Curtet S, Brocard MP, Khochbin S. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J Biol Chem. 2000;275:15594–15599. doi: 10.1074/jbc.M908437199. [DOI] [PubMed] [Google Scholar]
- 26.Lin Q, Lu J, Yanagisawa H, Webb R, Lyons GE, Richardson JA, Olson EN. Requirement of the MADS-box transcription factor MEF2C for vascular development. Development. 1998;125:4565–4574. doi: 10.1242/dev.125.22.4565. [DOI] [PubMed] [Google Scholar]
- 27.Linseman DA, Cornejo BJ, Le SS, Meintzer MK, Laessig TA, Bouchard RJ, Heidenreich KA. A myocyte enhancer factor 2D (MEF2D) kinase activated during neuronal apoptosis is a novel target inhibited by lithium. J Neurochem. 2003;85:1488–1499. doi: 10.1046/j.1471-4159.2003.09799.x. [DOI] [PubMed] [Google Scholar]
- 28.Liu G, Voyno-Yasenetskaya TA. Radixin stimulates Rac1 and Ca2 +/calmodulin-dependent kinase, CaMKII: Crosstalk with Galpha 13 signaling. J Biol Chem. 2005;280 (47):39042–39049. doi: 10.1074/jbc.M504341200. [DOI] [PubMed] [Google Scholar]
- 29.Maiti D, Xu Z, Duh EJ. Vascular endothelial growth factor induces MEF2C and MEF2-dependent activity in endothelial cells. Invest Ophthalmol Vis Sci. 2008;49:3640–3648. doi: 10.1167/iovs.08-1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsushita M, Nairn AC. Inhibition of the Ca2+/calmodulin-dependent protein kinase I cascade by cAMP-dependent protein kinase. J Biol Chem. 1999;274:10086–10093. doi: 10.1074/jbc.274.15.10086. [DOI] [PubMed] [Google Scholar]
- 31.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci USA. 2000;97:14400–14405. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–47. doi: 10.1016/S0968-0004 (01)02031-X. [DOI] [PubMed] [Google Scholar]
- 34.Mottet D, Bellahcène A, Pirotte S, Waltregny D, Deroanne C, Lamour V, Lidereau R, Castronovo V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101:1237–1246. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- 35.Niu J, Vaiskunaite R, Suzuki N, Kozasa T, Carr DW, Dulin N, Voyno-Yasenetskaya TA. Interaction of heterotrimeric G13 protein with an A-kinase-anchoring protein 110 (AKAP110) mediates cAMP-independent PKA activation. Curr Biol. 2001;11:1686–1690. doi: 10.1016/S0960-9822(01)00530-9. [DOI] [PubMed] [Google Scholar]
- 36.Offermanns S, Mancino V, Revel JP, Simon MI. Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science. 1997;275:533–536. doi: 10.1126/science.275.5299.533. [DOI] [PubMed] [Google Scholar]
- 37.Postma FR, Jalink K, Hengeveld T, Offermanns S, Moolenaar WH. Galpha(13) mediates activation of a depolarizing chloride current that accompanies RhoA activation in both neuronal and nonneuronal cells. Curr Biol. 2001;11:121–124. doi: 10.1016/S0960-9822(01) 00030-6. [DOI] [PubMed] [Google Scholar]
- 38.Prasad MV, Dermott JM, Heasley LE, Johnson GL, Dhanasekaran N. Activation of Jun kinase/stress-activated protein kinase by GTPase-deficient mutants of G alpha 12 and G alpha 13. J Biol Chem. 1995;270:18655–18659. doi: 10.1074/jbc.270.31.18655. [DOI] [PubMed] [Google Scholar]
- 39.Profirovic J, Gorovoy M, Niu J, Pavlovic S, Voyno-Yasenetskaya T. A novel mechanism of G protein-dependent phosphorylation of vasodilator-stimulated phosphoprotein. J Biol Chem. 2005;280:32866–32876. doi: 10.1074/jbc.M501361200. [DOI] [PubMed] [Google Scholar]
- 40.Radhika V, Onesime D, Ha JH, Dhanasekaran N. Galpha13 stimulates cell migration through cortactin-interacting protein Hax-1. J Biol Chem. 2004;279:49406–49413. doi: 10.1074/jbc.M408836200. [DOI] [PubMed] [Google Scholar]
- 41.Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB. Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ Res. 2002;91:398–405. doi: 10.1161/01.RES.0000033520.95242.A2. [DOI] [PubMed] [Google Scholar]
- 42.Ruppel KM, Willison D, Kataoka H, Wang A, Zheng YW, Cornelissen I, Yin L, Xu SM, Coughlin SR. Essential role for Galpha13 in endothelial cells during embryonic development. Proc Natl Acad Sci USA. 2005;102:8281–8286. doi: 10.1073/pnas.0503326102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shan D, Chen L, Wang D, Tan YC, Gu JL, Huang XY. The G protein G alpha(13) is required for growth factor-induced cell migration. Dev Cell. 2006;10:707–718. doi: 10.1016/j.devcel.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 44.Shi CS, Sinnarajah S, Cho H, Kozasa T, Kehrl JH. G13alpha-mediated PYK2 activation. PYK2 is a mediator of G13alpha-induced serum response element-dependent transcription. J Biol Chem. 2000;275:24470–24476. doi: 10.1074/jbc.M908449199. [DOI] [PubMed] [Google Scholar]
- 45.Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- 46.Spiegelberg BD, Hamm HE. G betagamma binds histone deacetylase 5 (HDAC5) and inhibits its transcriptional co-repression activity. J Biol Chem. 2005;280:41769–41776. doi: 10.1074/jbc.M504066200. [DOI] [PubMed] [Google Scholar]
- 47.Strathmann MP, Simon MI. G alpha 12 and G alpha 13 subunits define a fourth class of G protein alpha subunits. Proc Natl Acad Sci USA. 1991;88:5582–5586. doi: 10.1073/pnas.88.13.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res. 2006;71:226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 49.Vaiskunaite R, Kozasa T, Voyno-Yasenetskaya TA. Interaction between the G alpha subunit of heterotrimeric G(12) protein and Hsp90 is required for G alpha(12) signaling. J Biol Chem. 2001;276:46088–46093. doi: 10.1074/jbc.M108711200. [DOI] [PubMed] [Google Scholar]
- 50.Vanhauwe JF, Thomas TO, Minshall RD, Tiruppathi C, Li A, Gilchrist A, Yoon EJ, Malik AB, Hamm HE. Thrombin receptors activate G(o) proteins in endothelial cells to regulate intracellular calcium and cell shape changes. J Biol Chem. 2002;277:34143–34149. doi: 10.1074/jbc.M204477200. [DOI] [PubMed] [Google Scholar]
- 51.Voyno-Yasenetskaya TA, Pace AM, Bourne HR. Mutant alpha subunits of G12 and G13 proteins induce neoplastic transformation of Rat-1 fibroblasts. Oncogene. 1994;9:2559–2565. [PubMed] [Google Scholar]
- 52.Wang L, Fan C, Topol SE, Wang Q. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science. 2003;302:1578–1581. doi: 10.1126/science.1088477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Tang X, Li M, Marshall J, Mao Z. Regulation of neuroprotective activity of myocyte-enhancer factor 2 by cAMP-protein kinase A signaling pathway in neuronal survival. J Biol Chem. 2005;280:16705–16713. doi: 10.1074/jbc.M501819200. [DOI] [PubMed] [Google Scholar]
- 54.Xu N, Voyno-Yasenetskaya T, Gutkind JS. Potent transforming activity of the G13 alpha subunit defines a novel family of oncogenes. Biochem Biophys Res Commun. 1994;201:603–609. doi: 10.1006/bbrc.1994.1744. [DOI] [PubMed] [Google Scholar]
- 55.Zhu D, Kosik KS, Meigs TE, Yanamadala V, Denker BM. Galpha12 directly interacts with PP2A: evidence for Galpha12-stimulated PP2A phosphatase activity and dephosphorylation of microtubule-associated protein, tau. J Biol Chem. 2004;279:54983–54986. doi: 10.1074/jbc.C400508200. [DOI] [PubMed] [Google Scholar]