

Abstract
A comprehensive number of epidemiological and animal studies suggest that prenatal and early life events are important determinants for disorders later in life. Among them, prenatal stress (i.e. stress experienced by the pregnant mother with impact on the fetal ontogeny) has clear programming effects on the cardiovascular system. A fetus developing under adverse conditions becomes an adult who is susceptible to disease, which may include hypertension, insulin resistance, altered blood lipid levels and cardiovascular disease. Recent evidence demonstrates that maternal programming can occur in the absence of other adverse environmental factors. Obesity, which is becoming a problem of large proportions in Western countries, is a possible cause of programming. With over 30% of the US population currently obese, many mothers currently suffer from obesity during their child-bearing years (in fact, these conditions are often aggravated during pregnancy). One of the targets of programming is the cardiovascular system and reported consequences include hypertension, endothelial dysfunction and vascular abnormalities. The overall goal of our studies was to investigate the susceptibility of the heart to ischemia/reperfusion in an animal model of maternal obesity. Our data demonstrate that normal (non-mutant) offspring from obese Agouti mouse dams had an increased susceptibility to ischemia/reperfusion injury. These data may provide insights into the long-term cardiovascular consequences of programming.
Key terms: Fetal programming, heart disease, ischemia, reperfusion, animal model
Introduction
Originally proposed by Barker et al (Barker et al., 1989), the “fetal origins” hypothesis holds that a fetus developing under adverse conditions becomes an adult who is susceptible to disease, which includes hypertension, insulin resistance, altered blood lipid levels and cardiovascular disease. Originally, “programming” was described to result from undernourishment of mothers during pregnancy (Barker et al., 1989). We now know that it can also result from other insults during fetal development, which establishes a permanent physiological response that affects the individual for the rest of their lives (Fowden et al., 2006). Of particular concern are two conditions that affect a very large number of individuals in the US. These are obesity and diabetes. Maternal body mass index (BMI) is an independent predictor of birth weight and many studies have now shown that infants of obese women are prone to increased fat mass in childhood and young adulthood (Parsons et al., 2001; Salsberry & Reagan, 2005; Sacks et al., 2006; Boney et al., 2005; Sewell et al., 2006; Shields et al., 2006; Koupil & Toivanen, 2008). With over 30% of the US population currently obese, many mothers currently suffer from these conditions during their child-bearing years (in fact, these conditions are often aggravated during pregnancy). According to the “fetal origins” hypothesis, their children stand the chance of growing up to be adults in poor health, which, in addition to problems of morbidity and mortality, is a huge financial burden on our country. It is therefore imperative to understand the factors leading to, as well as the consequences of, fetal programming resulting from maternal obesity.
There are several reports showing that the heart can be persistently affected by adverse influences in-utero. Cardiac function in animal models of developmental programming has been infrequently addressed, although most studies suggest permanent influences on myocardial function. These include demonstration of ventricular hypertrophy arising from prenatal undernutrition (Battista et al., 2005). Prenatal exposure to under nutrition also leads to offspring tachycardia with reduced β1-adrenergic receptor (β-AR) expression and sympathetic function (Fernandez-Twinn et al., 2006), changes in cardiomyocytes number (Corstius et al., 2005), alterations in heart size (Xu et al., 2006; Williams et al., 2005; Xiao et al., 2000; Ostadalova et al., 2002) and interstitial fibrosis (Cheema et al., 2005). Maternal nutrient restriction in a sheep model is associated with expression in the fetal left ventricle of genes associated with cardiac hypertrophy (Han et al., 2004). Prenatal chronic hypoxia in the rat enhances susceptibility of the adult rat heart to ischemia/reperfusion injury (Li et al., 2003b) and differentially regulates the expression of β-AR subtypes (Roigas et al., 1996). These offspring have a greater than normal proportion of binucleate cardiomyocytes suggesting premature maturation and terminal differentiation of cardiomyocytes (Bae et al., 2003). Maternal anemia in the pregnant ewe altered coronary conductance which persists to adulthood (Broberg et al., 2003). Thus, prenatal programming can set the level of vulnerability for heart disease before birth. Chronic maternal hypoxia has also been demonstrated to lead to adaptive alterations in the heart, which increases the susceptibility of the heart to ischemic insults (Ostadalova et al., 2002; Xu et al., 2006; Li et al., 2003a). Our overall goal of this study was to examine effects of maternal obesity on the pathophysiological consequences of ischemia/reperfusion in the cardiovascular system. We used a genetic mouse model of maternal obesity. Normal (non-obese) offspring were raised on a normal diet for over a year, before assessing left ventricular function by echocardiography and subjecting the mice to an in vivo ischemia/reperfusion protocol.
Materials and Methods
Mouse model of maternal obesity
In mice, the lethal yellow (Ay) mutation is one of five dominant agouti mutations that result in ectopic agouti expression and is an excellent animal model for human obesity. The Ay mouse has been demonstrated as a valid model of fetal programming resulting from maternal obesity since normal offspring from Ay dams have impaired glucose tolerance and impaired pancreatic β-cell function (Han et al., 2005). We consequently used the Ay mouse model to characterize the cardiovascular consequences of maternal obesity-induced fetal programming. Agouti mice become obese around 6–8 weeks of age, at which time their blood glucose levels are normal (Han et al., 2005), which provides for a model of maternal obesity in the absence of diabetes. The breeding strategies and colony management are depicted in Figure 1. We bred heterozygote AY females (in a C57BL6 background; Jackson Laboratories: strain: B6.Cg-Ay/J; stock number: 000021) to non-mutant C57BL6J males. Non-mutant (black coat color; Ay-negative) offspring were used in these studies. Heterozygote offspring were not used. The control group consisted of non-mutant C57BL6J animals. Offspring were gender-matched and litter sizes were kept similar between groups. Forty-eight hours after delivery litters were reduced to 4 with equal sex ratios where possible (litters less than 4 were not used). The animals came from a total number of 10 litters. All animals had free access to normal chow and water. All animals were used at 46–48 weeks of age and received humane care in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society of Medical Research and the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, Revised 1996). The experimental protocol was reviewed and approved by the Institutional Animal Care and Use Committee of NYU School of Medicine and Emory University School of Medicine.
Figure 1.
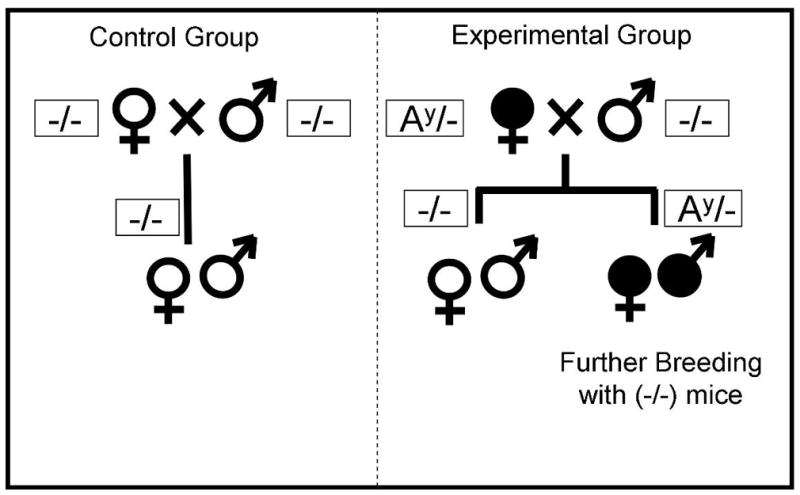
Breeding and colony management strategy to study normal offspring from obese mouse dams. In the experimental group, obese female heterozygous Agouti (Ay/−; filled symbols) mice were bred with non-mutant males. Non-mutant offspring were raised under normal conditions and used for experiments. The control animals came from normal breeding pairs (same strain mice). Litters were kept at similar sizes.
Echocardiographic Assessment of left ventricular structure and function
Mice were anesthetized with isoflourane in 100% O2 with continuous monitoring of ECG, core body temperature and respiration rate. Transthoracic high-resolution, two-dimensional ECG-based kilohertz visualization, B mode images were acquired at the rate of 1,000 frames/sec over 7 min. These images were used to calculate left ventricular (LV) dimensions as well as ejection fraction (EF). After the baseline images were acquired, the mice were subjected to LCA occlusion followed by reperfusion as described below. At 24 hr of reperfusion, post I/R echocardiographic images were obtained. Off-line measurements and calculations were made using Visual Sonics V2.2.3 software for the assessment of LV structure and function.
Myocardial ischemia and reperfusion protocol
All surgical procedures were performed using aseptic techniques as previously described in detail (Elrod et al., 2006). Briefly, mice were anesthetized via i.p. injection of sodium pentobarbital (50 mg/kg) and ketamine (60 mg/kg). Mice were intubated and ventilated with 100% oxygen by a rodent ventilator, Minivent (HugoSachs, Model 845) at a rate of 110 strokes/minute and 230 μL tidal volume. A thoracotomy was performed, the left coronary artery (LCA) was visually identified and ligated with a 7-0 silk suture rendering the left ventricle ischemic. Mice were subjected to 45 min of LCA ischemia followed by 24 hr of reperfusion.
Assessment of myocardial infarct size
After 24 of reperfusion, infarct size was determined as previously described (Elrod et al., 2006). Briefly, Evan’s blue dye was injected into the carotid artery catheter to delineate the ischemic zone from the nonischemic zone. The heart was rapidly excised and cross-sectioned into 1-mm thick sections, which were then incubated in 1.0% 2,3,5 triphenyltetrazolum chloride (TTC) for 5 minutes to demarcate the viable and nonviable myocardium within the risk zone. Images of each side of each section were acquired and weighed. The area of myocardial infarction (INF), area-at-risk (AAR), and area of nonischemic LV were assessed blinded using computer-assisted planimetry (NIH Image 1.57).
Statistical Analysis
All the data in this study are expressed as mean ± standard error (SEM). Differences in data between the groups were compared using Prism 4 (GraphPad Software, Inc) or SigmaStat v3.5. Statistical test employed included Student’s t-test, one-way analysis of variance (ANOVA) or Kruskal-Wallis One Way Analysis of Variance on Ranks as appropriate. Pairwise multiple comparisons were performed using the Tukey test or the Holm-Sidak method. A p value of less than 0.05 was considered significant.
Results
Long-term phenotypes of the Ay offspring
We assessed the non-mutant offspring of Ay dams in terms of their body weights and mortality. There were no differences in overall mortality between groups (data not shown). There were no differences in body weight at weaning (~3 weeks) or at the termination of the experiment (~46–48 weeks of age). The males in the experimental group weighed on average ~10% less than control mice at this time point, but this difference was not statistically significant (Table 1). Non-fasting blood glucose levels in blood samples from the mice just prior to the surgical procedure were 203 mg/dl in control (both genders) vs 200 mg/dl in the experimental group (the reason for the higher levels relative to that typically seen with younger mice is probably related to the old age of these mice).
Table 1.
Body weights at weaning and at the experimental age for control and experimental animals.
| At Weaning | Experimental Age | ||||
|---|---|---|---|---|---|
| Group | Gender | Age (weeks) | Weight (g) | Age (weeks) | Weight (g) |
| Control | Male (n=7) | 3.0+0.01 | 9.4+0.22 | 48.9+0.47 | 32.9+0.83 |
| Female (n=7) | 3.0+0.01 | 9.1+0.18 | 46.7+2.22 | 25.5+1.05* | |
| Experimental | Male (n=8) | 2.9+0.03 | 8.7+0.22 | 47.2+1.53 | 30.4+0.72 |
| Female (n=4) | 2.8+0.05 | 9.2+0.40 | 48.9+0.47 | 25.2+1.17* | |
p<0.05 when comparing to either male group.
Maternal programming is associated with an increased myocardial I/R injury
Control and experimental mice were subjected to 45 min of left coronary artery (LCA) ischemia and 24 hr of reperfusion. A total of 42 animals were subjected to this procedure (21 animals in each of the control and experimental groups). Overall, the survival rate from this procedure was ~62% (Table 2). The survival rate in the experimental female group was 50%, but this value is not significantly different from the others (Fisher’s Exact test).
Table 2.
Mortality following ischemia/reperfusion in the control and experimental animals.
| Group | Gender | Enrolled | Survived |
|---|---|---|---|
| Control | Male | 10 | 7 (70%) |
| Female | 11 | 7 (64%) | |
| Experimental | Male | 13 | 8 (62%) |
| Female | 8 | 4 (50%) | |
| Total | 42 | 26 (65%) | |
Quantitatively, control mice displayed a mean infarct per area-at-risk (INF/AAR) of 64.5 ± 2.7 % (Figure 2). There was no difference in the area-at-risk relative to the LV (AAR/LV), between control and experimental animals (56.1 ± 1.2% in control vs. 54.8 ± 2.3% in the experimental group, n= 14 and 12 respectively). Overall, animals in the experimental group had a significantly larger INF/AAR, but not when expressed as Inf/LV (Figure 2A).
Figure 2.
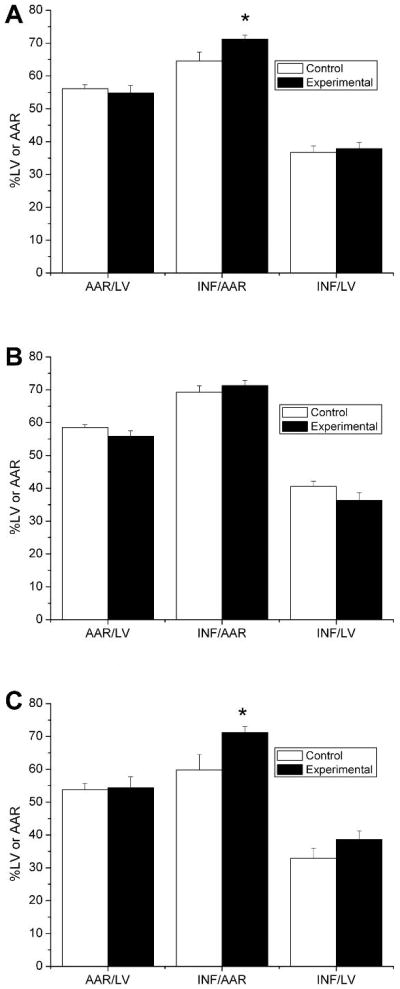
Male offspring from obese Agouti mouse dams have a larger myocardial infarct size following ischemia/reperfusion. Shown are the area-at-risk as a function of the left ventricular mass (AAR/LV) and the infarct size expressed as a percentage of the area-at-risk (INF/AAR) or as a percentage of the left ventricular mass (INF/LV). Data are shown for all animals combined (A), females only (B) or males only (C). The control group represents offspring from dams with normal body weights and the experimental group represents offspring from dams with increased body weights (agouti dams). The experimental numbers are given in Table 2.
Restricting the analysis to female animals only revealed no differences in INF/AAR between control and experimental animals (Figure 2B). The infarct size (expressed as a function of the area-at-risk) was 69.2 ± 2.0% in control females and 71.3 ± 1.6% in experimental females (n=7 and n=4 respectively). A corresponding analysis for male animals revealed that INF/AAR in experimental males was significantly larger when compared to control males (59.8 ± 4.7% in control males vs. 71.2 ± 1.8% in experimental males; p<0.05).
Left ventricular function and structure
Prior to the determination of infarct size (see above), left ventricular function was determined by echocardiography before and after ischemia/reperfusion. The left ventricular end-systolic diameter was significantly increased after ischemia/reperfusion with little change in the left ventricular end-diastolic diameter (Figure 3). The calculated ejection fraction was depressed following ischemia/reperfusion (Figure 3). There were no significant differences in these parameters between the control and experimental groups (regardless of gender). Other parameters that were measured included the LV systolic area, LV diastolic area, LV systolic volume and the LV diastolic volume (Table 3). The LV systolic area and volumes were significantly increased after ischemia/reperfusion. A between-group comparison shows that the LV diastolic area and volume were significantly larger in the experimental group following ischemia/reperfusion (Table 3). This difference was significant for males, but not females.
Figure 3.
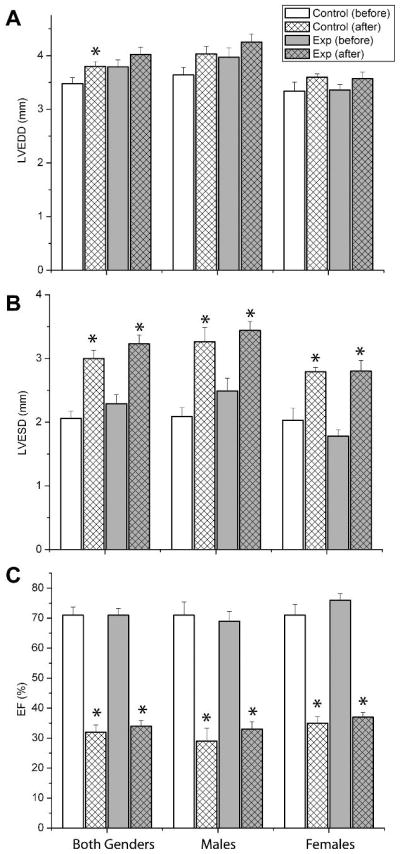
No differences in cardiac function with maternal programming. Left ventricular ejection fraction was determined by echocardiography before (BASE) or following transient ischemia and 24h hour reperfusion (POST). Data are shown for all animals combined (A), females only (B) or males only (C).
Table 3.
Echocardiography left ventricular parameters measured before and after ischemia/reperfusion for control and experimental animals
| Systolic Area (mm2) | Diastolic Area (mm2) | Systolic Volume (μL) | Diastolic Volume (μL) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gender | Group | Before | After | Before | After | Before | After | Before | After |
| Both | Control | 6.71±0.55 | 11.80±0.75* | 14.07±0.62 | 14.99±0.83 | 9.59±1.24 | 24.80±2.45* | 32.76±2.28 | 36.36±3.02 |
| Experimental | 8.26±0.85 | 14.12±1.01* | 16.60±1.07 | 18.00±1.07† | 13.66±2.31 | 32.27±3.27* | 42.60±4.28 | 48.52±4.21† | |
| Males | Control | 7.13±0.83 | 13.04±1.16* | 15.09±0.85 | 16.30±1.23 | 10.51±1.77 | 29.82±3.80* | 36.54±3.08 | 41.90±4.63 |
| Experimental | 9.10±1.36 | 15.60±1.03* | 17.62±1.88 | 19.72±0.93† | 15.99±3.81 | 37.49±3.28* | 47.18±7.37 | 55.53±3.89† | |
| Females | Control | 6.34±0.76 | 10.74±0.84* | 13.18±0.79 | 13.87±1.02 | 8.80±1.80 | 20.49±2.31* | 29.51±2.95 | 31.62±3.25 |
| Experimental | 5.55±0.80 | 11.18±1.36* | 12.82±0.82 | 14.55±1.62 | 6.80±1.50 | 21.81±3.50* | 27.12±2.41 | 34.50±5.07 | |
Discussion
Epidemiological studies performed over the past decade have demonstrated that the risk of coronary heart disease, stroke mortality and hypertension is strongly related to the environment encountered during an individual’s life while still a fetus. In particular, maternal undernutrition during pregnancy has been highlighted as a causal factor in the long-term programming of disease risk. The concept is now firmly established that a compromised intra-uterine environment is a major cause for this “fetal programming” of poor health in later life (Fowden et al., 2006). Although much further studies are needed, developmental programming and childhood disease have been cited as possible causes for the recent finding of the unexpected and significantly poorer health status of older non-Hispanic whites in the United States compared to those in England (Banks et al., 2006).
Fetal programming resulting from maternal obesity
Although developmental programming was initially described resulting from maternal malnutrition, it is now clear that other environmental factors occurring during fetal development also have major consequences for health later in life. For example, offspring from overweight mothers are susceptible to long-term health problems (Armitage et al., 2005b). This is a particularly important problem given that obesity is reaching epidemic proportions. At present, 65% of the US population is overweight (BMI > 25) and 31% of the adult population is obese (BMI > 30) (Ogden et al., 2004; Hedley et al., 2004; Ogden et al., 2002). Many overweight and obese women unknowingly enter pregnancy and continue overeating during the critical first trimester, when much of the fetal programming is thought to occur. Thus, the stage is set for adult-onset health problems to emerge in the next generation – even if the current weight problems can be curbed.
The cardiovascular system as a target of fetal programming
David Barker and colleagues performed population studies to examine the reasons for regional differences in the incidence of mortality from stroke and cardiovascular disease in England and Wales. They noted that ischemic heart disease and mortality due to stroke and cardiovascular disease was clearly associated with the poor health of mothers, which they named as a determinant of risk in the offspring (Barker et al., 1989; Barker & Osmond, 1986). Further studies also demonstrated an association between low infant weight and high rates of cardiovascular mortality (Kermack et al., 2001). The adverse cardiovascular consequences of fetal programming has now been firmly established (Remacle et al., 2004; Langley-Evans, 2001; McMillen & Robinson, 2005).
Several cardiovascular consequences may occur. One of these is hypertension. Of the 71.3 million American adults with one or more form of cardiovascular disease, hypertension is the most prevalent (91% of these have a high blood pressure) (Thom et al., 2006). Epidemiological and animal studies alike have provided ample evidence for hypertension as a consequence of adverse fetal programming. In general, there is an inverse relationship between blood pressure in the adult and their birth weights (Huxley et al., 2002; Huxley et al., 2000). There is also evidence that vascular and endothelial functions are affected by maternal programming (Leeson et al., 2001; Ozaki et al., 2001; Brawley et al., 2003). The latter is particularly interesting since endothelial dysfunction is a strong predictor of the development of coronary artery disease (Bugiardini et al., 2004).
The present study was designed to examine the consequences of ischemia/reperfusion in programming resulting from maternal obesity. Coronary artery disease remains the number 1 killer in America and caused 1 out of every 5 deaths in the United States in 2003 (Thom et al., 2006). About every 26 seconds an American suffers from a coronary event and about every minute, someone will die from it. About 40% of the people who experience a coronary event will die within a year as a result (Thom et al., 2006). Although fetal programming has not been linked directly to coronary heart disease in experimental studies, it should be noted that the fetal programming hypothesis first came into being when epidemiological studies linked mortality due to cardiovascular disease and stroke to poor health and physique in the mothers of the victims (Barker & Osmond, 1986; Barker et al., 1989; Barker, 2002). Yet, very little is known about fetal programming of the heart and or how the cardiac response to pathophysiological states is altered (such as with stress or during ischemia/reperfusion). It appears that fetal programming of the rat heart is associated with adaptive responses, including an increase in cell size (cellular hypertrophy), cell number (hyperplasia) and apoptosis (Langley-Evans et al., 1996; Bae et al., 2003). The consequences of these changes are not immediately apparent, but they may make the heart more susceptible to pathophysiological injury. Indeed, there are some indications that the severity of ischemia may be increased by fetal programming. For example, in a rat model of maternal hypoxia the susceptibility of the offspring heart to ischemia/reperfusion injury is impaired (Li et al., 2003a; Li et al., 2004; Xu et al., 2006). Our data are the first to directly demonstrate that maternal obesity is associated with impaired cardiac outcome after ischemia/reperfusion. Specifically, we found that male, but not female, offspring of obese mice have a significantly increased infarct size following an in-vivo ischemia/reperfusion protocol. These changes were not associated with clear alterations in cardiac function (ejection fraction as assessed by echocardiography). There were, however, differences in the LV diastolic area and volume. At the present, the underlying mechanisms for the worse prognostic outcome is unknown and future experimental studies are needed to examine the consequences of fetal programming on the susceptibility of the heart and coronary vasculature to pathophysiological insults. It is interesting to note, however, that alterations in heart sizes have been noted in prior studies. For example, in a model of diet-induced maternal obesity in mice, offspring are hypertensive and have evidence of metabolic syndrome. Interestingly, there was also an increase in heart mass (Samuelsson et al., 2008). The changes in cardiac weight and dimensions have also been previously reported for other models of fetal programming. Maternal hypoxia, for example, has been shown to be associated with in increase in heart weight in offspring (fetuses, neonates and adults) (Williams et al., 2005; Xu et al., 2006; Xiao et al., 2000; Ostadalova et al., 2002). Finally, a recent study demonstrated that 6 month old infants of women who gained the greatest weight in pregnancy have heavier hearts than offspring of women who gained appropriate weight (Geelhoed et al., 2008).
Possible gender-bias with maternal programming
Animal studies indicate that the phenotype observed with maternal programming depends not only on the timing and intensity of the manipulation, but also on the offspring gender. In some studies, there appears to be a preference for female offspring to be affected. For example, obesity-induced programming may lead to gender differences in renin-angiotensin axis and insulin release, with females being more susceptible than males (Armitage et al., 2005a; Han et al., 2005). If similar gender differences exist in the effects of programming on the cardiovascular system, then this may have important implications in understanding the emerging realization that cardiovascular disease is not restricted to males and is also the number one killer for women - in 2003, more female lives were claimed by CVD than were claimed by the next five leading causes of death combined (cancer, COPD, Alzheimer’s, diabetes and accidents) (Thom et al., 2006). Other studies suggest that males are also susceptible to the sequelae of maternal programming. For example, perinatal protein restriction during pregnancy in rats preferentially affects males in programming for adult hypertension or insulin resistance, whereas female rats are relatively resistant (Woods et al., 2005; Sugden & Holness, 2002), which is an effect that could even be carried to a second generation (Pinheiro et al., 2008). While experimental data regarding gender-specific effects on the response to ischemia/reperfusion are not known, human population studies suggest that cardiac mortality in males is affected (Banks et al., 2006). Our data demonstrate a clear increase in infarct size following ischemia/reperfusion in males, but not in female offspring from obese Agouti dams. Similarly, we observed an increased LV diastolic area and volume in males, but not in females. Caution should be used in over-interpreting these gender data; however, since the precise effects of programming on the cardiovascular system may depend on the species and/or model being used.
Possible Mechanisms for Increased Susceptibility to I/R injury in offspring from Obese dams
In the present study we did not define the cellular and molecular mechanisms responsible for the increase in susceptibility to myocardial ischemia-reperfusion (I/R) injury in the offspring from obese dams. This was largely the result of the very limited number of offspring that were available for these studies. It is possible to speculate on the mechanisms that aggravate the severity of myocardial cell death and myocardial contractile dysfunction. Primary culprits in the pathogenesis of myocardial I/R injury include the generation of reactive oxygen species (ROS), impaired endothelial function and loss of coronary nitric oxide (NO) bioavailability, myocardial inflammation, mitochondrial dysfunction, and apoptosis. The effects of adult obesity on the severity of myocardial I/R has been previously investigated in various animal models and there are strong data demonstrating that obesity directly enhances the severity of MI/R injury (Gundewar et al., 2007; Bouhidel et al., 2008; du Toit et al., 2008; Thakker et al., 2006). Obese, non-diabetic animals subjected to myocardial I/R exhibit a significant increase in myocardial infarct size and increased cardiac dysfunction (Gundewar et al., 2007; Bouhidel et al., 2008; du Toit et al., 2008; Thakker et al., 2006). Similar results are observed in obese animals with increased blood glucose levels (Greer et al., 2006). These studies also suggest that the cardioprotective effects of various therapeutic agents are largely attenuated in obese animals (Gundewar et al., 2007; Bouhidel et al., 2008; du Toit et al., 2008). Previous studies suggest that increased oxidative stress, enhanced inflammatory response, and a loss of cardioprotective signaling pathways such as the RISK pathway promote myocardial cell injury in obesity (Gundewar et al., 2007; Bouhidel et al., 2008; du Toit et al., 2008; Thakker et al., 2006). It is likely that obesity related factors such as adipokines are exchanged between the maternal and fetal circulations and “imprint” a number of latent signals that promote oxidative and inflammatory stress in the offspring of obese dams that enhance subsequent cardiac injury following I/R. Our future studies of the offspring of obese animals will be targeted at the elucidation of the pathological signaling involved in the exaggerated response to MI/R injury.
Limitations of the study
The Agouti (Ay) mouse has been previously used as a valid model of fetal programming resulting from maternal obesity. Normal (non-obese) offspring from Ay dams have impaired glucose tolerance and impaired pancreatic β-cell function (Han et al., 2005). However, this is not a commonly used model of programming resulting from maternal obesity and our data should be validated with the use of other experimental models. A further issue is that our experimental numbers were fairly low. Although we initiated the study with 42 animals (21 per experimental group), the post-procedural mortality was quite high, most likely due to the older age of the animals when compared to other studies. Nevertheless, despite the relatively small remaining group sizes, these data show a clear increase in infarct size in male offspring from obese dams.
Significance
Our data provide novel and important insights into the effects of maternal obesity on long-term cardiovascular health of the offspring. Further experiments of this nature are essential if we wish to understand how obesity, a major Western burden, may affect cardiovascular health of millions of individuals, generations from now. Resulting data may aid in the prediction of long-term cardiovascular outcomes of fetal programming from maternal obesity in the human population. This would allow the medical community to alter medical management of these patients in hopes of preventing poor cardiac outcomes. Additionally, such data may help set the stage to prepare the general population (through advocacy agencies) about the very real dangers that maternal obesity (and possibly also diabetes) may hold for their children.
Acknowledgments
The authors are grateful to Joan Hoffman, Opal Prevatt, Peter C. Rajacic, Laura L. Carrihill, Mark Duranski, Ji Sang and Saurabh Jha for assistance in maintaining the mouse colony, assistance with experiments, and some of the data collection.
Funding Sources
This study was supported by the National Institutes of Health (HL-060849-08 to D.J.L. HL085820-01 to W.A.C and F32 DK 077380-01 to J.W.C.), the American Diabetes Association (7-04-RA-59 to D.J.L.) and by the 7th Masonic District (to W.A.C.).
Footnotes
Disclosures
None of the authors have any disclosures.
References
- Armitage JA, Lakasing L, Taylor PD, Balachandran AA, Jensen RI, Dekou V, Ashton N, Nyengaard JR, Poston L. Developmental programming of aortic and renal structure in offspring of rats fed fat-rich diets in pregnancy. J Physiol. 2005a;565:171–184. doi: 10.1113/jphysiol.2005.084947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage JA, Taylor PD, Poston L. Experimental models of developmental programming: consequences of exposure to an energy rich diet during development. J Physiol. 2005b;565:3–8. doi: 10.1113/jphysiol.2004.079756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Xiao Y, Li G, Casiano CA, Zhang L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. Am J Physiol Heart Circ Physiol. 2003;285:H983–H990. doi: 10.1152/ajpheart.00005.2003. [DOI] [PubMed] [Google Scholar]
- Banks J, Marmot M, Oldfield Z, Smith JP. Disease and disadvantage in the United States and in England. JAMA. 2006;295:2037–2045. doi: 10.1001/jama.295.17.2037. [DOI] [PubMed] [Google Scholar]
- Barker DJ. Fetal programming of coronary heart disease. Trends Endocrinol Metab. 2002;13:364–368. doi: 10.1016/s1043-2760(02)00689-6. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–567. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battista MC, Calvo E, Chorvatova A, Comte B, Corbeil J, Brochu M. Intra-uterine growth restriction and the programming of left ventricular remodelling in female rats. J Physiol. 2005;565:197–205. doi: 10.1113/jphysiol.2004.078139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290–e296. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- Bouhidel O, Pons S, Souktani R, Zini R, Berdeaux A, Ghaleh B. Myocardial ischemic postconditioning against ischemia-reperfusion is impaired in ob/ob mice. Am J Physiol Heart Circ Physiol. 2008;295:H1580–H1586. doi: 10.1152/ajpheart.00379.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawley L, Itoh S, Torrens C, Barker A, Bertram C, Poston L, Hanson M. Dietary protein restriction in pregnancy induces hypertension and vascular defects in rat male offspring. Pediatr Res. 2003;54:83–90. doi: 10.1203/01.PDR.0000065731.00639.02. [DOI] [PubMed] [Google Scholar]
- Broberg CS, Giraud GD, Schultz JM, Thornburg KL, Hohimer AR, Davis LE. Fetal anemia leads to augmented contractile response to hypoxic stress in adulthood. Am J Physiol Regul Integr Comp Physiol. 2003;285:R649–R655. doi: 10.1152/ajpregu.00656.2002. [DOI] [PubMed] [Google Scholar]
- Bugiardini R, Manfrini O, Pizzi C, Fontana F, Morgagni G. Endothelial function predicts future development of coronary artery disease: a study of women with chest pain and normal coronary angiograms. Circulation. 2004;109:2518–2523. doi: 10.1161/01.CIR.0000128208.22378.E3. [DOI] [PubMed] [Google Scholar]
- Cheema KK, Dent MR, Saini HK, Aroutiounova N, Tappia PS. Prenatal exposure to maternal undernutrition induces adult cardiac dysfunction. Br J Nutr. 2005;93:471–477. doi: 10.1079/bjn20041392. [DOI] [PubMed] [Google Scholar]
- Corstius HB, Zimanyi MA, Maka N, Herath T, Thomas W, van der LA, Wreford NG, Black MJ. Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatr Res. 2005;57:796–800. doi: 10.1203/01.PDR.0000157726.65492.CD. [DOI] [PubMed] [Google Scholar]
- du Toit EF, Smith W, Muller C, Strijdom H, Stouthammer B, Woodiwiss AJ, Norton GR, Lochner A. Myocardial susceptibility to ischemic-reperfusion injury in a prediabetic model of dietary-induced obesity. Am J Physiol Heart Circ Physiol. 2008;294:H2336–H2343. doi: 10.1152/ajpheart.00481.2007. [DOI] [PubMed] [Google Scholar]
- Elrod JW, Greer JJ, Bryan NS, Langston W, Szot JF, Gebregzlabher H, Janssens S, Feelisch M, Lefer DJ. Cardiomyocyte-specific overexpression of NO synthase-3 protects against myocardial ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;26:1517–1523. doi: 10.1161/01.ATV.0000224324.52466.e6. [DOI] [PubMed] [Google Scholar]
- Fernandez-Twinn DS, Ekizoglou S, Wayman A, Petry CJ, Ozanne SE. Maternal low protein diet programs cardiac beta-adrenergic response and signalling in 3 month old male offspring. Am J Physiol Regul Integr Comp Physiol. 2006;291:R429–R436. doi: 10.1152/ajpregu.00608.2005. [DOI] [PubMed] [Google Scholar]
- Fowden AL, Giussani DA, Forhead AJ. Intrauterine programming of physiological systems: causes and consequences. Physiology (Bethesda ) 2006;21:29–37. doi: 10.1152/physiol.00050.2005. [DOI] [PubMed] [Google Scholar]
- Geelhoed JJ, Osch-Gevers VAN, Verburg BO, Steegers EA, Hofman A, Helbing W, Witteman JC, Jaddoe VW. Maternal anthropometrics in pregnancy are associated with left ventricular mass in infancy. The generation R study. Pediatr Res. 2008;63:62–66. doi: 10.1203/PDR.0b013e31815b4449. [DOI] [PubMed] [Google Scholar]
- Greer JJ, Ware DP, Lefer DJ. Myocardial infarction and heart failure in the db/db diabetic mouse. Am J Physiol Heart Circ Physiol. 2006;290:H146–H153. doi: 10.1152/ajpheart.00583.2005. [DOI] [PubMed] [Google Scholar]
- Gundewar S, Calvert JW, Elrod JW, Lefer DJ. Cytoprotective effects of N,N,N-Trimethyl sphingosine (TMS) during Ischemia-Reperfusion Injury are lost in the setting of obesity and diabetes. Am J Physiol Heart Circ Physiol. 2007 doi: 10.1152/ajpheart.00392.2007. [DOI] [PubMed] [Google Scholar]
- Han HC, Austin KJ, Nathanielsz PW, Ford SP, Nijland MJ, Hansen TR. Maternal nutrient restriction alters gene expression in the ovine fetal heart. J Physiol. 2004;558:111–121. doi: 10.1113/jphysiol.2004.061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Xu J, Epstein PN, Liu YQ. Long-term effect of maternal obesity on pancreatic beta cells of offspring: reduced beta cell adaptation to high glucose and high-fat diet challenges in adult female mouse offspring. Diabetologia. 2005;48:1810–1818. doi: 10.1007/s00125-005-1854-8. [DOI] [PubMed] [Google Scholar]
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. JAMA. 2004;291:2847–2850. doi: 10.1001/jama.291.23.2847. [DOI] [PubMed] [Google Scholar]
- Huxley R, Neil A, Collins R. Unravelling the fetal origins hypothesis: is there really an inverse association between birthweight and subsequent blood pressure? Lancet. 2002;360:659–665. doi: 10.1016/S0140-6736(02)09834-3. [DOI] [PubMed] [Google Scholar]
- Huxley RR, Shiell AW, Law CM. The role of size at birth and postnatal catch-up growth in determining systolic blood pressure: a systematic review of the literature. J Hypertens. 2000;18:815–831. doi: 10.1097/00004872-200018070-00002. [DOI] [PubMed] [Google Scholar]
- Kermack WO, McKendrick AG, McKinlay PL. Death-rates in Great Britain and Sweden. Some general regularities and their significance. Int J Epidemiol. 2001;30:678–683. doi: 10.1093/ije/30.4.678. [DOI] [PubMed] [Google Scholar]
- Koupil I, Toivanen P. Social and early-life determinants of overweight and obesity in 18-year-old Swedish men. Int J Obes (Lond) 2008;32:73–81. doi: 10.1038/sj.ijo.0803681. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC. Fetal programming of cardiovascular function through exposure to maternal undernutrition. Proc Nutr Soc. 2001;60:505–513. doi: 10.1079/pns2001111. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Gardner DS, Jackson AA. Association of disproportionate growth of fetal rats in late gestation with raised systolic blood pressure in later life. J Reprod Fertil. 1996;106:307–312. doi: 10.1530/jrf.0.1060307. [DOI] [PubMed] [Google Scholar]
- Leeson CP, Kattenhorn M, Morley R, Lucas A, Deanfield JE. Impact of low birth weight and cardiovascular risk factors on endothelial function in early adult life. Circulation. 2001;103:1264–1268. doi: 10.1161/01.cir.103.9.1264. [DOI] [PubMed] [Google Scholar]
- Li G, Bae S, Zhang L. Effect of prenatal hypoxia on heat stress-mediated cardioprotection in adult rat heart. Am J Physiol Heart Circ Physiol. 2004;286:H1712–H1719. doi: 10.1152/ajpheart.00898.2003. [DOI] [PubMed] [Google Scholar]
- Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J Soc Gynecol Investig. 2003a;10:265–274. doi: 10.1016/s1071-5576(03)00074-1. [DOI] [PubMed] [Google Scholar]
- Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ, Chen AF. Endothelin-1 increases vascular superoxide via endothelin(A)-NADPH oxidase pathway in low-renin hypertension. Circulation. 2003b;107:1053–1058. doi: 10.1161/01.cir.0000051459.74466.46. [DOI] [PubMed] [Google Scholar]
- McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Flegal KM, Carroll MD, Johnson CL. Prevalence and trends in overweight among US children and adolescents, 1999–2000. JAMA. 2002;288:1728–1732. doi: 10.1001/jama.288.14.1728. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Fryar CD, Carroll MD, Flegal KM. Mean body weight, height, and body mass index, United States 1960–2002. Adv Data. 2004:1–17. [PubMed] [Google Scholar]
- Ostadalova I, Ostadal B, Jarkovska D, Kolar F. Ischemic preconditioning in chronically hypoxic neonatal rat heart. Pediatr Res. 2002;52:561–567. doi: 10.1203/00006450-200210000-00016. [DOI] [PubMed] [Google Scholar]
- Ozaki T, Nishina H, Hanson MA, Poston L. Dietary restriction in pregnant rats causes gender-related hypertension and vascular dysfunction in offspring. J Physiol. 2001;530:141–152. doi: 10.1111/j.1469-7793.2001.0141m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons TJ, Power C, Manor O. Fetal and early life growth and body mass index from birth to early adulthood in 1958 British cohort: longitudinal study. BMJ. 2001;323:1331–1335. doi: 10.1136/bmj.323.7325.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro AR, Salvucci ID, Aguila MB, Mandarim-de-Lacerda CA. Protein restriction during gestation and/or lactation causes adverse transgenerational effects on biometry and glucose metabolism in F1 and F2 progenies of rats. Clin Sci (Lond) 2008;114:381–392. doi: 10.1042/CS20070302. [DOI] [PubMed] [Google Scholar]
- Remacle C, Bieswal F, Reusens B. Programming of obesity and cardiovascular disease. Int J Obes Relat Metab Disord. 2004;28(Suppl 3):S46–53. S46–S53. doi: 10.1038/sj.ijo.0802800. [DOI] [PubMed] [Google Scholar]
- Roigas J, Roigas C, Heydeck D, Papies B. Prenatal hypoxia alters the postnatal development of beta-adrenoceptors in the rat myocardium. Biol Neonate. 1996;69:383–388. doi: 10.1159/000244335. [DOI] [PubMed] [Google Scholar]
- Sacks DA, Liu AI, Wolde-Tsadik G, Amini SB, Huston-Presley L, Catalano PM. What proportion of birth weight is attributable to maternal glucose among infants of diabetic women? Am J Obstet Gynecol. 2006;194:501–507. doi: 10.1016/j.ajog.2005.07.042. [DOI] [PubMed] [Google Scholar]
- Salsberry PJ, Reagan PB. Dynamics of early childhood overweight. Pediatrics. 2005;116:1329–1338. doi: 10.1542/peds.2004-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, Piersma AH, Ozanne SE, Twinn DF, Remacle C, Rowlerson A, Poston L, Taylor PD. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51:383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- Sewell MF, Huston-Presley L, Super DM, Catalano P. Increased neonatal fat mass, not lean body mass, is associated with maternal obesity. Am J Obstet Gynecol. 2006;195:1100–1103. doi: 10.1016/j.ajog.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Shields BM, Knight BA, Powell RJ, Hattersley AT, Wright DE. Assessing newborn body composition using principal components analysis: differences in the determinants of fat and skeletal size. BMC Pediatr. 2006;6:24, 24. doi: 10.1186/1471-2431-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Holness MJ. Gender-specific programming of insulin secretion and action. J Endocrinol. 2002;175:757–767. doi: 10.1677/joe.0.1750757. [DOI] [PubMed] [Google Scholar]
- Thakker GD, Frangogiannis NG, Bujak M, Zymek P, Gaubatz JW, Reddy AK, Taffet G, Michael LH, Entman ML, Ballantyne CM. Effects of diet-induced obesity on inflammation and remodeling after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H2504–H2514. doi: 10.1152/ajpheart.00322.2006. [DOI] [PubMed] [Google Scholar]
- Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng ZJ, Flegal K, O’donnell C, Kittner S, Lloyd-Jones D, Goff DC, Jr, Hong Y, Adams R, Friday G, Furie K, Gorelick P, Kissela B, Marler J, Meigs J, Roger V, Sidney S, Sorlie P, Steinberger J, Wasserthiel-Smoller S, Wilson M, Wolf P. Heart Disease and Stroke Statistics--2006 Update. A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–e151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- Williams SJ, Campbell ME, McMillen IC, Davidge ST. Differential effects of maternal hypoxia or nutrient restriction on carotid and femoral vascular function in neonatal rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R360–R367. doi: 10.1152/ajpregu.00178.2004. [DOI] [PubMed] [Google Scholar]
- Woods LL, Ingelfinger JR, Rasch R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1131–R1136. doi: 10.1152/ajpregu.00037.2003. [DOI] [PubMed] [Google Scholar]
- Xiao D, Ducsay CA, Zhang L. Chronic hypoxia and developmental regulation of cytochrome c expression in rats. J Soc Gynecol Investig. 2000;7:279–283. [PubMed] [Google Scholar]
- Xu Y, Williams SJ, O’Brien D, Davidge ST. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB J. 2006;20:1251–1253. doi: 10.1096/fj.05-4917fje. [DOI] [PubMed] [Google Scholar]