

Abstract
mRNA transfection is a useful approach for temporal cell reprogramming with minimal risk of transgene-mediated mutagenesis. We applied this to redirect lymphocyte cytotoxicity toward malignant cells. Using the chimeric immune receptor (CIR) constructs anti-CD19 CIR and 8H9 CIR, we achieved uniform expression of CIRs on virtually the entire population of lymphocytes. We reprogrammed CD3+ CD8+, CD3+ CD4+, and natural killer (NK ) cells toward autologous and allogeneic targets such as B cells, Daudi lymphoma, primary melanoma, breast ductal carcinoma, breast adenocarcinoma, and rhabdomyosarcoma. The reprogramming procedure is fast. Although most of the experiments were performed on lymphocytes obtained after 7-day activation, only 1-day activation of T cells with anti-CD3, anti-CD28 antibodies, and interleukin-2 is sufficient to develop both lymphocyte cytotoxicity and competence for mRNA transfer. The entire procedure, which includes lymphocyte activation and reprogramming, can be completed in 2 days. The efficiency of mRNA-modified human T cells was tested in a murine xenograft model. Human CD3+CD8+ lymphocytes expressing anti-CD19 CIR mRNA inhibited Daudi lymphoma growth in NOD/SCID mice. These results demonstrate that a mixed population of cytotoxic lymphocytes, including T cells together with NK cells, can be quickly and simultaneously reprogrammed by mRNA against autologous malignancies. With relatively minor modifications the described method of lymphocyte reprogramming can be scaled up for cancer therapy.
Introduction
Adoptive transfer of activated lymphocytes can be highly effective in some patients with melanoma and renal carcinoma (Dudley and Rosenberg, 2007; June, 2007). However, generation of cytotoxic CD8+ T cells (CTLs) in vivo requires that tumor-associated antigens be presented in an immunogenic MHC context, a condition not usually observed for a variety of malignancies. Moreover, the activity of tumor-specific T cells, even if they were present, is often hampered by immune evasion, such as inhibition of MHC expression in tumor cells (Leen et al., 2007; Gross and Walden, 2008).
An alternative approach is the use of genetically modified, ex vivo-propagated CTLs expressing chimeric immune receptors (CIRs). A CIR transgene contains three basic modules: an extracellular single-stranded antibody, which recognizes tumor antigen; a transmembrane anchor, placing the protein in the cellular membrane; and a signal domain, which triggers a specific cellular cytotoxic response (Eshhar et al., 2001; Ren-Heidenreich et al., 2002; Biagi et al., 2007).
Lymphocytes reprogrammed with CIR transgenes can possess cytotoxicity toward a variety of tumor types in a non-MHC-restricted manner and therefore can be applied to patients of all MHC haplotypes. Also, they can target not only tumor-associated peptides but any antibody-recognizable antigens present on tumor cells, for instance, carbohydrate or glycolipid moieties (Kershaw et al., 2005). Consequently, this approach would not be compromised by tumor loss of MHC alleles.
The methods generally used for lymphocyte reprogramming include viral transduction and plasmid DNA transfection (Cheung et al., 2003; Cooper et al., 2006). However, these methods carry the risk of irreversible genomic alterations resulting from exogenous DNA integration (Hacein-Bey-Abina et al., 2003). Also, these methods employ prolonged ex vivo lymphocyte propagation, which can result in undesirable functional modifications of the cells. DNA transfer is a permanent modification of the cells. This can create a permanent burden of lymphocytes reacting against antigens that may be present not only on cancer cells but also on normal cells.
One way to address these problems is by the addition of a suicide agent such as the herpes simplex virus thymidine kinase gene (HSV-tk ) (Bonini et al., 1997). However, expression of HSV-tk may generate an undesirable immune response (Berger et al., 2006). Another way is by the use of nonintegrated (episomal) viral vectors (Griffiths et al., 2006). Neither method could be considered as highly reliable.
We have been working on an alternative procedure in which lymphocyte reprogramming is achieved by transfer of CIR mRNA. Unlike DNA or viral particles, mRNA would have no permanent effect on the host cells because of its inability to replicate or integrate into the genome. We developed a fast and effective approach for in vitro synthesis of mRNA that can be transferred into human lymphocytes. RNA electroporation produces a uniform level of expression in lymphocytes and permits simultaneous coexpression of several mRNA transgenes (Rabinovich et al., 2006). In this paper, we report our investigation of various types of lymphocytes in the targeting of hematological and solid tumor cells. We have demonstrated CIR-specific killing of tumor cells by mRNA-transfected lymphocytes in vitro and also in vivo, in a murine xenograft model. The method we describe represents a relatively safe and efficient approach for the preparation of T and natural killer (NK) cells reprogrammed against autologous malignancies.
Materials and Methods
Cells
Primary human cells were obtained from healthy donors, after informed consent was given according to a protocol approved by the institutional review board. Activated B cells were generated by cultivating peripheral blood mononuclear cells (PBMCs) in the presence of CD40 ligand as described (Schultze et al., 1995). Freshly isolated (or thawed) PBMCs were washed and plated on a preformed layer of previously irradiated (96 Gy) 3T3-CD40 ligand (CD40L) cells (a kind gift from R.W. Childs, National Institutes of Health, Bethesda, MD) in Iscove's modified Dulbecco's medium (IMDM; GIBCO/Invitrogen, Carlsbad, CA), with 10% human serum (Gemini Bio-Products, Woodland, CA) and interleukin (IL)-4 (200 U/ml) and in the presence of 550 nM cyclosporine A (Sigma-Aldrich, St. Louis, MO). The cultured B cells were transferred to freshly prepared plates with irradiated 3T3-CD40L cells every 3–4 days. Cultures were kept up to 21 days. The percentage of CD19+ cells was 85–95% after 10 days of cultivation.
Populations of CD3+ T cells were separated from PBMCs with Xcyte Dynabeads (Xcyte Therapies [Seattle, WA]; product currently marketed by Invitrogen as Dynabeads ClinExVivo CD3/CD28), containing supermagnetic beads covalently linked with anti-CD3 and anti-CD28 monoclonal antibodies (anti-CD3/CD28 beads). PBMCs were resuspended at 10 × 106/ml in Dulbecco's phosphate-buffered saline (DPBS; GIBCO/Invitrogen) with 0.5% human albumin, and then anti-CD3/CD28 beads (final dilution, 1:20) were added. The mixture was incubated for 30 min in a refrigerator with rotation. The CD3+ fraction was isolated with a magnetic particle concentrator (Dynal MPC; Invitrogen). In the standard procedure, unless otherwise indicated, lymphocytes were activated for 7 days by incubation in IMDM with 5% human serum (Gemini Bio-Products) in the presence of anti-CD3/CD28 beads (final dilution, 1:20) and IL-2 (100 IU/ml; PeproTech, Rocky Hill, NJ). The beads were removed from the culture before electroporation. CTLs and CD4+ T cells were purified with a CD8+ T cell isolation kit II or CD4+ T cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's recommendations. The selected cells were >95% CD3+CD8+ or CD3+CD4+ as determined by flow cytometric analysis.
NK cells were isolated from PBMCs by positive immunomagnetic selection with CD56 MicroBeads (Miltenyi Biotec) and were expanded in IMDM, supplemented with 10% human serum, in the presence of IL-2 (1000 U/ml) for 3 weeks. Before 51Cr release assay the unwanted CD3+CD56+ cells were removed with anti-CD3/CD28 beads as described previously. The selected cells were >95 % CD56+ as determined by flow cytometric analysis.
A primary melanoma specimen was processed immediately after surgery, dissected free of surrounding normal tissue, and small chunks of tumor measuring about 2 mm each were cut with a sterile scalpel blade. The tumor fragments were then physically disaggregated with a BD Medimachine (BD Biosciences, San Jose, CA) under sterile conditions. The cell suspension was filtered with a BD Falcon filter (pore size, 50 nm; BD Biosciences), washed, resuspended in Opti-MEM (GIBCO/Invitrogen) with 10% fetal bovine serum (FBS), and plated in 25-cm2 flasks. After overnight incubation, nonadherent cells were removed and adherent melanoma cells were further cultivated. Cells were split after a confluent layer was formed, using trypsin–EDTA solution. T47D (breast ductal carcinoma), MCF7 (breast adenocarcinoma), HTB-82 (rhabdomyosarcoma), and A2058 (melanoma) cells were grown in Dulbecco's modified Eagle's medium (DMEM; GIBCO/Invitrogen) supplemented with 10% FBS. For dissociation of solid tumor cells we routinely used trypsin–EDTA solution (GIBCO/Invitrogen). However, in preparing targets for 51Cr release assay nonenzymatic cell dissociation solution (Sigma-Aldrich) was used, to preserve cell surface antigens.
Daudi lymphoma, NALM6 (pre-B cell acute leukemia), and K562 (myeloid leukemia) cells were grown in RPMI medium supplemented with 10% FBS (GIBCO/Invitrogen). Firefly luciferase-positive (ffLuc+) Daudi lymphoma cells, a kind gift from M.C. Jensen (Department of Pediatric Hematology-Oncology, City of Hope National Medical Center, Duarte, CA), had an integrated firefly luciferase gene. These cells were grown in medium supplemented with Zeocin (0.2 mg/ml) (Cooper et al., 2005).
DNA
Anti-CD19 CIR was a kind gift from D. Campana (Hematology-Oncology and Pathology, St. Jude Children's Research Hospital, University of Tennessee, Memphis, TN) (Imai et al., 2004). 8H9 CIR was constructed in the laboratory of N.-K. Cheung (Memorial Sloan-Kettering Cancer Center, New York, NY) (Cheung et al., 2003). pmaxGFP plasmid, carrying the gene encoding green fluorescent protein (GFP) under the regulation of the cytomegalovirus (CMV) promoter, was supplied by Amaxa (Gaithersburg, MD) as a part of an electroporation kit. The CD19 receptor gene was obtained from OriGene Technologies (Rockville, MD).
Polymerase chain reaction
DNA templates were made by polymerase chain reaction (PCR) according to the protocol we have described (Rabinovich et al., 2006). Gene amplification was performed with AccuPrime Pfx DNA polymerase (Invitrogen) according to the manufacturer's protocol. Twenty-five to 30 cycles of PCR were performed in standard 50-μl reaction using 0.1 μg of template DNA. The forward primer contained the T7 RNA promoter and an anchoring sequence in the proximal part of the gene expression cassette. The reverse primer, with anchoring sequence in the distal part of the gene expression cassette, contained a stretch of 100 dT residues. Three-step PCR to delete the 4-1BB signaling part of the anti-CD19 CIR was performed by a standard procedure (Elion et al., 2007).
RNA synthesis
mRNA synthesis with T7 RNA polymerase has been described (Rabinovich et al., 2006). This was performed with an mMESSAGE mMACHINE T7 Ultra kit (Ambion, Austin, TX), using the procedure recommended by the manufacturer. One hundred to 200 ng of DNA made by PCR with no further purification was used for the standard 20-μl transcription reaction. The product was treated with Escherichia coli poly(A) polymerase (from the same kit) in the presence of 1 mM ATP according to the Ambion polyadenylation protocol. The yield of mRNA was 20 to 60 μg of mRNA per reaction. The final product was treated with DNase I (Ambion) and purified with an Ambion MEGAclear kit or by LiCl precipitation. RNA quality was verified by agarose gel electrophoresis, and RNA was stored at −80°C.
Transfection
Electroporation was performed with an Amaxa Nucleofector II (Amaxa, Gaithersburg, MD) in accordance with manufacturer recommendations. T lymphocytes and NK cells were electroporated with “human T cell kit solution” and various regimens of electroporation. K562 cells were electroporated with “V solution” and program T16. A2058 cells were electroporated with “R solution” and program R01. All cells were electroporated with DNA (20 mg/ml) or mRNA (30 mg/ml) per sample. Cells were used at a concentration of 10–250 million/ml. In this interval of values the efficiency of transfection does not depend on cell density (Rabinovich et al., 2006). The efficiency of transfection was determined by flow cytometry 18 hr after transfection. Cell viability was determined by trypan blue exclusion.
Flow cytometry
Flow cytometric analysis of cell subpopulations was performed at the Yale Cancer Center Flow Cytometry Shared Resource (New Haven, CT), using a FACSCalibur (BD Biosciences). Fluorescence signals were collected on a logarithmic scale. At least 10,000 events were acquired for each sample. Data were analyzed with FlowJo software (TreeStar, Ashland, OR). Mouse anti-human CD4–FITC (anti-CD4 antibody, fluorescein isothiocyanate conjugated), CD8–PE (anti-CD8 antibody, phycoerythrin conjugated), CD19–PE (anti-CD19 antibody, phycoerythrin conjugated), and CD3–PerCP/Cy5.5 (anti-CD3 antibody, peridinin chlorophyll protein [PerCP]–Cy5.5 conjugated) were purchased from BD Biosciences. CIRs, which contain mouse single-stranded variable fractions (sFvs) as the recognition domain, were detected with biotin-conjugated goat anti-mouse IgG, F(ab′)2 fragment specific, purchased from Jackson ImmunoResearch Laboratories (West Grove, PA), and with streptavidin–PerCP (streptavidin, PerCP conjugated) purchased from BD Biosciences. 8H9 murine IgG1 monoclonal antibody directed at gp58 has been previously described (Modak et al., 2001).
Electrophoresis
DNA samples were run in 1% agarose in Tris–acetate buffer (Sigma-Aldrich) at 2 V/cm. RNA samples were run in 1% agarose in morpholinepropanesulfonic acid (MOPS)–formaldehyde buffer (Ambion) at 2 V/cm, using an RNA Millennium marker (Invitrogen) as size standard.
Cytotoxicity assay
The cytotoxic activity of electroporated CTLs and NK cells was evaluated by the standard 51Cr release assay. Target tumor cells were labeled with 0.25 mCi of Na251CrO4 (MP Biomedicals, Solon, OH) for 1 hr, extensively washed, and seeded at a density of 1 × 104 cells per well in V-bottom 96-well microplates. Transfected effector (E) cells were suspended in IMDM supplemented with 10% FBS and coincubated with target (T) cells at various E:T ratios. The plates were incubated at 37°C for 4 hr in 5% CO2. Aliquots of supernatants of each sample were harvested and radioactivity was measured with a γ counter (Beckman G 4000; Beckman Coulter, Fullerton, CA). Assays were carried out in duplicate or triplicate. Specific lysis was calculated as follows:
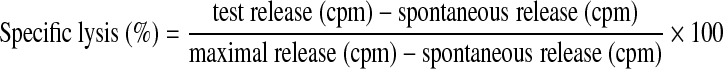 |
where cpm is counts per minute released by the targets. Spontaneous release represents 51Cr release from target cells in medium alone. Maximal release is the 51Cr release from target cells treated with 2% Triton X-100. In general, we performed the experiments two or three times. Each time we performed duplicate analysis. The average absolute difference between duplicates as well as standard deviation did not exceed 5%. In each measurement the difference in chromium release between “experiment” and “control” cells was statistically significant with p < 0.01. The Friedman test (Hollander and Wolfe, 1973) was used to determine the significance of differences in chromium release. It was performed with Matlab Statistics Toolbox 1997 (The MathWorks, Natick, MA).
Xenograft tumor model
The xenograft tumor model was previously described (Cooper et al., 2005). Murine experiments were performed under institutional IACUC-approved protocols. On day 0, 6-week-old female nonobese diabetic/severe combined immunodeficiency (NOD/LtSz-Prkdcscid/J) mice (Jackson Laboratory, Bar Harbor, ME) were injected intraperitoneally with ffLuc+ Daudi cells. On days 2 and 3 tumor engraftment was evaluated by biophotonic imaging. Mice with progressively growing tumors were segregated into three groups receiving additional intraperitoneal injections either of RPMI medium or 5 × 106 mock-electroporated CTLs or 5 × 106 CTLs electroporated with anti-CD19-CIR mRNA.
Biophotonic tumor imaging
Anesthetized mice were imaged with a Xenogen IVIS-50 imaging system (Caliper Life Sciences, Hopkinton, MA) beginning 15 min after intraperitoneal injection of 150 μl of a freshly thawed aqueous solution of d-luciferin (30 mg/ml) (Xenogen, Alameda, CA). Each animal was serially imaged in an anterior–posterior orientation at the same relative time point after d-luciferin injection. Photons emitted from ffLuc+ Daudi xenografts were quantified with the software program Living Image (Xenogen), and the bioluminescence signal was measured as total photon flux normalized for exposure time and surface area. For anatomic localization, a pseudocolor image representing light intensity (blue, least intense; red, most intense) was superimposed over a digital grayscale body surface reference image.
Statistical methods to analyze biophotonic data
To measure differences between mouse treatment groups, we considered evaluating tumor biophotonic signal over time. Geometric means of the signal were used for presentation on the basis of the assumption of a log-normal distribution for tumor sizes (Spratt, 1969).
The Student t test (Press et al., 1992), Mann–Whitney U test, and Friedman test (Hollander and Wolfe, 1973) are most commonly used to determine the significance of differences seen between control and treatment groups. We performed all these tests with Matlab Statistics Toolbox 1997 (The MathWorks) to determine the significance of differences seen between tumor growth curves.
Results
Factors determining CIR activity
Initial experiments were performed with an anti-CD19 CIR construct (Fig. 1A). This contains a leader sequence, an anti-CD19 single-chain variable region (scFv) antibody domain, a transmembrane domain, and two intracellular signal transduction domains derived from 4-1BB (CD137) and CD3-ζ (ζ) chains (Imai et al., 2004). We have shown previously that CTLs transfected with anti-CD19 CIR mRNA possessed cytotoxicity toward CD19+ B cells. Virtually all effector cells express CIR receptor when electroporated with CIR mRNA (Rabinovich et al., 2006). Here we studied the killing of lymphoma and leukemia cells. CTLs obtained after a standard 7 days of ex vivo activation with anti-CD3/CD28 beads and IL-2 and electroporated with anti-CD19 CIR mRNA lysed all CD19+ targets tested, specifically Daudi lymphoma and NALM6 pre-B cell acute leukemia cell lines, as well as autologous B cells, whereas mock-electroporated CTLs were not cytotoxic. The CD19-negative target cell line K562 (myeloid leukemia) was resistant to the killing (Fig. 1B).
FIG. 1.

Cytotoxicity of CTLs transfected with anti-CD19 CIR mRNA against CD19+ tumor cells. (A) Structure of anti-CD19 CIR, which contains a signal peptide, VL and VH domains of single-chain anti-CD19 antibody, a transmembrane (TM) domain, and signal transduction domains originated from 4-1BB and CD3 ζ proteins. (B) Cytotoxicity of mRNA-transfected CTLs against target cells loaded with 51Cr. The vertical axis represents normalized chromium release, and the horizontal axis indicates the effector-to-target (E:T) ratio. CTLs, after the standard 7 days of activation, were electroporated with CIR mRNA or without mRNA (mock ). K562 cells were used as a CD19-negative control. All other target cells expressed CD19 protein.
Next, we analyzed whether CTL reprogramming depends on their prior ex vivo activation. As the efficiencies of Amaxa electroporation programs were similar for both DNA and RNA (Amaxa, 2006; Rabinovich et al., 2006), we first determined CTL transfectability under various conditions by using a pmaxGFP plasmid DNA. CTLs, prepared by the standard 7 days of activation, were transfectable as expected: cells electroporated with programs T13 and T7 expressed GFP with corresponding viabilities of 40 and 55%; up to 40% of viable cells expressed detectable GFP. On the other hand, nonactivated (resting) CTLs were found to be virtually untransfectable in all sets of the programs recommended by Amaxa for these cells: T7, T13, T20, U1, U8, U10, U5, U14, and U9 (transfection efficiency, <2%). We then proceeded to mRNA transfection with program T7. CTLs were activated for 1 or 7 days, and then electroporated with anti-CD19 CIR mRNA. The transfection efficiency (Fig. 2A, panels 1 and 2) and the cytotoxicity against CD19+ target (Fig. 2B, panels 1 and 2) were similar for both groups. This suggests that prior activation of CTLs was essential for mRNA-mediated reprogramming and that even 1 day of incubation with anti-CD3/CD28 beads and IL-2 was sufficient for full activation.
FIG. 2.

CIR mRNA activity under various conditions. CTLs were obtained after short (1 day) or standard (7 days) ex vivo activation with anti-CD3/CD28 beads and IL-2, and were electroporated with CIR mRNA or without mRNA (mock). Surface CIR expression was visualized with a goat anti-mouse (Fab)2 polyclonal antibody conjugated with biotin and streptavidin PerCP (x axes). (A) Expression of anti-CD19 CIR in CTLs activated for 1 day (panel 1) or for 7 days (panel 2) (Elion et al., 2007); expression of anti-CD19 CIR δ(4–1BB) in CTLs activated for 7 days (panel 3). (B) Cytotoxicity of CTLs under the conditions indicated in (A). Targets, CD19+ autologous B cells, were loaded with 51Cr and analyzed at the indicated E:T ratios.
We also analyzed whether CTL killing ability depends on the presence of two signaling domains in CIR. It had been demonstrated that CIRs containing a “double” signaling structure, such as 4-1BB-CD3 ζ in comparison with a single CD3ζ domain, can increase in vivo lymphocyte viability and efficiency, probably emulating the effect of antigen-specific and antigen-independent costimulation (Moeller et al., 2004; Kowolik et al., 2006; Marin et al., 2007). It was not clear, however, whether two signal domains can synergistically enhance cytotoxicity. We deleted the 4-1BB signaling part of the anti-CD19 CIR by three-step PCR, and compared this construct with the original CIR. CTLs were transfected with mRNA made from original anti-CD19 CIR and its δ (4-1BB) variant. Both transfectants showed similar levels of CIR expression (Fig. 2A, panels 2 and 3), and CD19+ target killing (Fig. 2B, panels 2 and 3).
mRNA transfer in various types of lymphocytes
We investigated whether CD4+ T lymphocytes and NK cells from the same donor could be reprogrammed in the same way as CD8+ cells. Isolated NK cells showed high-level killing of K562 cells (data not shown). It has been reported that NK cells transduced with anti-CD19 CIR possessed CD19-specific killing activity (Imai et al., 2005). In pilot experiments using pmaxGFP DNA we tested various electroporation programs on CD4+ and CD8+ T cells, and NK cells (data not shown). The T7 Amaxa program, which was efficient for all samples, was used for mRNA transfection.
We prepared four groups of lymphocytes: CD8+ T cells, CD4+ T cells, their mix, and NK cells and electroporated them with anti-CD19 CIR mRNA. After transfection, all types of cells possessed specific killing activity against autologous B cells (Fig. 3).
FIG. 3.

Cytotoxicity of various types of lymphocytes against autologous targets. CD8+ T cells, CD4+ T cells, a 1:1 mix of both (CD8+ and CD4+ cells), and NK cells were transfected with anti-CD19 CIR mRNA or without mRNA (mock). Targets, autologous CD19+ B cells, were loaded with 51Cr and analyzed for cytotoxicity at the indicated E:T ratios.
Lymphocyte cytotoxicity against non-B cell malignancies
First, we tested the validity of our approach on CD19– tumors by converting the targets to a CD19+ phenotype. The CD19 gene was transcribed in vitro and CD19 mRNA was introduced by electroporation into K562 myeloid leukemia and A2058 melanoma cells. Within 18 hr, the whole populations of K562 and A2058 cells uniformly expressing CD19 (Fig. 4A) were loaded with 51Cr and used as targets for CTLs transfected with anti-CD19 CIR mRNA. Reprogrammed CTLs were able to kill both converted CD19+ targets (Fig. 4B).
FIG. 4.

Cytotoxicity of anti-CD19CIR+ CTLs against tumor cells transfected with CD19 mRNA. (A) Expression of CD19 protein in two target cell lines, K562 and A2058, 18 hr after their electroporation with CD19 mRNA (CD19+) or without mRNA (CD19–). Surface receptor expression was visualized with PE-conjugated anti-CD19 monoclonal antibody. (B) Cytotoxicity of CTLs transfected with anti-CD19 CIR mRNA against targets characterized in (A). Targets were loaded with 51Cr and analyzed for cytotoxicity at the indicated E:T ratios.
Next, we analyzed cell targeting by using another chimeric receptor, 8H9 CIR (Fig. 5A). This contains 8H9scFv antibody (Modak et al., 2001), which recognizes a glycoprotein gp58, presented on a broad spectrum of human solid tumors (Onda et al., 2004). CTLs transduced with 8H9 CIR DNA construct lysed a variety of malignancies, such as breast cancer, neuroblastoma, and osteosarcoma (Cheung et al., 2003). Here we tested whether solid tumor cells can be killed by CTLs transfected with 8H9 CIR mRNA. First, we determined the presence of gp58 antigen on the chosen targets. Primary melanoma cells and three established cell lines (T47D [breast ductal carcinoma], MCF7 [breast adenocarcinoma], and HTB-82 [rhabdomyosarcoma]) were stained with 8H9scFv antibody and found to be gp58 positive. K562 cells were shown to be gp58 negative (see Suppl. Fig. S1 at http://www.liebertonline.com/doi/pdfplus/10.1089/hum.2008.068). Autologous CTLs from the melanoma patient were loaded with 8H9 CIR mRNA and tested against various targets. Reprogrammed CTLs possessed cytotoxicity to all gp58+ tumors, but not to gp58-negative K562 cells (Fig. 5B).
FIG. 5.

Cytotoxicity of 8H9 CIR+ CTLs against some solid tumors. (A) Structure of 8H9 CIR, which contains signal peptide (Spratt), VH and VL domains of single-chain 8H9 antibody, a transmembrane domain (TM), and signal transduction domains originated from CD28 and CD3ζ proteins. (B) Cytotoxicity of CTLs isolated from a melanoma patient against autologous melanoma and other tumors. CTLs obtained after the standard 7 days of ex vivo activation were electroporated with 8H9 CIR mRNA or without mRNA (mock) and tested against various tumor cells. We have verified that 8H9 antigen, gp58, was expressed on all solid tumor targets used, but not on K562 cells (negative control). Targets were loaded with 51Cr and analyzed for cytotoxicity at the indicated E:T ratios.
Cytotoxic activity of anti-CD19 CIR+ CTLs in vivo
For these experiments we used a murine xenograft model (Cooper et al., 2005). To determine the kinetics of human lymphoma growth in mice, nine NOD/LtSz-Prkdcscid/J (NOD/SCID) mice were allocated to three groups. Each group was injected intraperitoneally with either 1 × 106, 3 × 106, or 9 × 106 ffLuc+ Daudi cells per mouse, and analyzed by biophotonic measurements. Exponentially growing tumors were established in all mice 3 days after injection. In the experiments with CTLs, we chose to make multiple injections of human CTLs at 3-day intervals, because anti-CD19 CIR protein remains on the lymphocyte surface for 3–4 days. In a pilot experiment we used six mice that had been injected intraperitoneally with 3 × 106 ffLuc+ Daudi cells per mouse. The mice were injected intraperitoneally with 5 × 106 CTLs twice, on days 3 and 6. Treatment with anti-CD19 CIR+ CTLs resulted in marked regression of tumors compared with exponential tumor growth in the control group (data not shown).
In the next experiment 24 mice were injected intraperitoneally with ffLuc+ Daudi cells (3 × 106 cells per mouse). Three days later the mice were analyzed by biophotonic measurements and were distributed into three groups (8 mice per group, with median ffLuc signal (∼6 × 108 photons/sec/cm2/sr) similar for each group. The mice were injected intraperitoneally with 5 × 106 CTLs three times, on days 3, 6, and 9. In comparison to the control group, given RPMI medium alone (group 1), and mock-transfected CTLs (group 2), there was a significant reduction of tumor ffLuc signal in the treatment group (group 3), which we had injected with CTLs loaded with anti-CD19 CIR mRNA.
The Student t test, Mann–Whitney U test, and Friedman test were used to determine the significance of differences seen between control and treatment groups. All three tests showed that tumor growth in both controls (groups 1 and 2) were indistinguishable (p = 0.6620 for the Student t test, p = 0.8810 for the Mann–Whitney U test, and p = 0.4795 for the Friedman test). In group 3, however, tumor growth curve was significantly different (p < 0.0001 for all three tests mentioned). Tumor inhibition in group 3 was still evident 6 days after the last CTL injection on days 12 and 15 (Fig. 6).
FIG. 6.

In vivo activity of anti-CD19 CIR+ CTLs. Twenty-four NOD/SCID mice injected with 3 × 106 luciferase (ffLuc)-expressing Daudi cells, and developed exponentially growing tumors by day 3 after injection were imaged and divided into three groups: treated with RPMI medium (1), mock transfected (2), or anti-CD19 CIR-transfected (3) CTLs. Groups received injections of 5 × 106 CTLs on days 3, 6, and 9. The mice were imaged on days 3, 6, 9, 12, and 15. (A) Pseudocolor image representing light intensity and anatomic localization of the ffLuc– Daudi cells in representative mice. (B) Longitudinal monitoring of the bioluminescent signals of ffLuc+ Daudi cells injected into 3 groups of 24 NOD/SCID mice. Data are presented as geometric means of total photon flux normalized for exposure time, surface area, and initial signal for each group of mice; error bars represent the geometric SD.
Discussion
Viral transduction or DNA transfection of lymphocytes to express CIRs is a workable method to redirect the specificity of immune system toward tumor antigens, which are not readily recognized by the endogenous lymphocyte receptors. However, a potential disadvantage of such methods is genomic integration of exogenous DNA. On the other hand, mRNA transfection is essentially transient, and the mRNA transgene can be delivered into lymphocytes as a minimal expression cassette without any additional vector-derived sequences. Under these conditions genomic integration of the transgene is improbable. Another disadvantage of DNA transfer is that it usually takes weeks or months to clone and accumulate an adequate amount of transduced or transfected lymphocytes with the desired level of antitumor activity (Brentjens et al., 2003; Cooper et al., 2006). Such extensive lymphocyte propagation transforms the majority of cells to the effector memory phenotype and may lead to insufficient antitumor activity in vivo (Dudley et al., 2002; Gattinoni et al., 2005; Cooper et al., 2006).
With our method, mRNA-mediated lymphocyte reprogramming can be completed in 2 days: 1 day for cell isolation and activation and another day for mRNA electroporation. The efficiency of mRNA transfer eliminates the need for cell cloning because virtually the whole population of electroporated cells uniformly expressed CIR proteins. Moreover, we demonstrated that both CD4+ and CD8+ T cells as well as NK cells obtained from the same donor can be reprogrammed against chosen targets. Consequently, mRNA transfer can be applied to the whole population of cytotoxic lymphoid cells with the potential synergistic benefit of their different homing potentials and cytotoxicity pathways.
The activation of CTLs for mRNA transfection was crucial. This could result from morphological or metabolic changes, such as increased cell radius/volume, translation enhancement, or membrane remodeling, but the actual role of these various factors requires further investigation.
T cell propagation requires two signals, one through the T cell receptor and a secondary signal through costimulatory molecules. The presence of two signaling domains in CIRs enhances in vivo persistence and antitumor efficacy of stably transfected lymphocytes (Moeller et al., 2004; Kowolik et al., 2006; Marin et al., 2007). In our study, removal of the 4-1BB signal domain did not change the parameters of CIR-mediated killing. This suggests that in short-term experiments with insignificant lymphocyte proliferation, the presence of CD3ζ as a sole signaling domain is sufficient for cell cytotoxicity.
The method of T cell redirection is not limited to hematological malignancies. We have shown that T cells modified with anti-CD19 CIR can kill CD19-negative tumor lines if targets were modified with CD19 mRNA. CTLs transfected with 8H9 CIR mRNA possessed cytotoxicity toward all tested solid tumors expressing gp58 antigen. Most importantly, CTLs isolated from a melanoma patient killed the patient's melanoma cells, indicating that this method can be used in autologous setting.
We tested the antitumor activity of redirected T cells in a mouse xenograft model. Daudi lymphoma cells grew aggressively in NOD/SCID mice and developed exponentially growing tumors within 3 days of injection. Treatment with CIR mRNA-transfected CTLs inhibited tumor growth, whereas mock-transfected T cells had no inhibitory effect. The tumors did resume growth in the treatment group after the end of CTL injection, presumably when CIR proteins on modified CTLs were degraded. The outcome of this experiment is comparable to the results obtained by others, in which CTLs expressing CIRs alone were not able to completely eliminate the tumor (Cooper et al., 2005). It is probably worth noting that an approach that does not cure the disease in an immune-deficient host may potentially lead to a better outcome in an immune-competent environment. However, in such an environment short-term lymphodepletion can also enhance the survival and proliferation of transferred cells. We plan to investigate whether an appropriate method of immunodepletion would facilitate the activity of lymphocytes loaded with chimeric receptor mRNA.
One potential drawback of the mRNA transfection method that may be contributing to tumor persistence is the transient nature of CIR expression. This may be overcome by repeated injection of effector cells as well as by a combination of various types of lymphocytes. Another possibility is to modify the mRNA to prolong CIR expression. We are currently studying those approaches.
Our results provide encouraging preclinical data for the development of a clinical-scale mRNA transfection method to be used in clinical trials. It would probably be most applicable in an autologous setting for minimal residual disease, following chemo- or immunotherapy.
Supplementary Material
Acknowledgments
The authors thank Dr. Dario Campana for the anti-CD19 CIR construct and highly valuable discussions of the obtained results, Dr. Michael C. Jensen for ffLuc+ Daudi lymphoma cell line, Dr. Faye Rogers for help with luciferase assay, and Ms. Janet Hernandez for help in preparing this paper. This work was supported by the National Institutes of Health (grants N01-HV-28186, 1P01-HL-63357, 1P50 CA121974, and CA-106450).
Author Disclosure Statement
No competing financial interest exists.
References
- Amaxa. Amaxa Biosystems. 2006. http://www.amaxa.com/no_cache/meta/faq/faq-details/article/25/ [Dec;2008 ]. http://www.amaxa.com/no_cache/meta/faq/faq-details/article/25/
- Berger C. Flowers M.E. Warren E.H. Riddell S.R. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagi E. Marin V. Giordano Attianese G.M. Dander E. D'amico G. Biondi A. Chimeric T-cell receptors: New challenges for targeted immunotherapy in hematologic malignancies. Haematologica. 2007;92:381–388. doi: 10.3324/haematol.10873. [DOI] [PubMed] [Google Scholar]
- Bonini C. Ferrari G. Verzeletti S. Servida P. Zappone E. Ruggieri L. Ponzoni M. Rossini S. Mavilio F. Traversari C. Bordignon C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia [see comment] Science. 1997;276:1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- Brentjens R.J. Latouche J.B. Santos E. Marti F. Gong M.C. Lyddane C. King P.D. Larson S. Weiss M. Riviere I. Sadelain M. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15 [see comment] Nat. Med. 2003;9:279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- Cheung N.K. Guo H.F. Modak S. Cheung I.Y. Anti-idiotypic antibody facilitates scFv chimeric immune receptor gene transduction and clonal expansion of human lymphocytes for tumor therapy. Hybrid Hybridomics. 2003;22:209–218. doi: 10.1089/153685903322328938. [DOI] [PubMed] [Google Scholar]
- Cooper L.J. Al-Kadhimi Z. Serrano L.M. Pfeiffer T. Olivares S. Castro A. Chang W.C. Gonzalez S. Smith D. Forman S.J. Jensen M.C. Enhanced antilymphoma efficacy of CD19-redirected influenza MP1-specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood. 2005;105:1622–1631. doi: 10.1182/blood-2004-03-1208. [DOI] [PubMed] [Google Scholar]
- Cooper L.J. Ausubel L. Gutierrez M. Stephan S. Shakeley R. Olivares S. Serrano L.M. Burton L. Jensen M.C. Forman S.J. Digiusto D.L. Manufacturing of gene-modified cytotoxic T lymphocytes for autologous cellular therapy for lymphoma [see comment] Cytotherapy. 2006;8:105–117. doi: 10.1080/14653240600620176. [DOI] [PubMed] [Google Scholar]
- Dudley M.E. Rosenberg S.A. Adoptive cell transfer therapy. Semin. Oncol. 2007;34:524–531. doi: 10.1053/j.seminoncol.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley M.E. Wunderlich J.R. Yang J.C. Hwu P. Schwartzentruber D.J. Topalian S.L. Sherry R.M. Marincola F.M. Leitman S.F. Seipp C.A. Rogers-Freezer L. Morton K.E. Nahvi A. Mavroukakis S.A. White D.E. Rosenberg S.A. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J. Immunother. 2002;25:243–251. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion E.A. Marina P. Yu L. Current Protocols in Molecular Biology. Wiley Interscience; New York: 2007. Constructing recombinant DNA molecules by PCR; pp. 3.17.11–13.17.12. [DOI] [PubMed] [Google Scholar]
- Eshhar Z. Waks T. Bendavid A. Schindler D.G. Functional expression of chimeric receptor genes in human T cells. J. Immunol. Methods. 2001;248:67–76. doi: 10.1016/s0022-1759(00)00343-4. [DOI] [PubMed] [Google Scholar]
- Gattinoni L. Klebanoff C.A. Palmer D.C. Wrzesinski C. Kerstann K. Yu Z. Finkelstein S.E. Theoret M.R. Rosenberg S.A. Restifo N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells [see comment] J. Clin. Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths R.A. Boyne J.R. Whitehouse A. Herpesvirus saimiri-based gene delivery vectors. Curr. Gene Ther. 2006;6:1–15. doi: 10.2174/156652306775515529. [DOI] [PubMed] [Google Scholar]
- Gross S. Walden P. Immunosuppressive mechanisms in human tumors: Why we still cannot cure cancer. Immunol. Lett. 2008;116:7–11. doi: 10.1016/j.imlet.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S. Von Kalle C. Schmidt M. Le Deist F. Wulffraat N. McIntyre E. Radford I. Villeval J.L. Fraser C.C. Cavazzana-Calvo M. Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency [see comment] N. Engl. J. Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- Hollander M. Wolfe D.A. Nonparametric Statistical Methods. John Wiley & Sons; New York: 1973. [Google Scholar]
- Imai C. Mihara K. Andreansky M. Nicholson I.C. Pui C.H. Geiger T.L. Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- Imai C. Iwamoto S. Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–383. doi: 10.1182/blood-2004-12-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June C.H. Adoptive T cell therapy for cancer in the clinic. J. Clin. Invest. 2007;117:1466–1476. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw M.H. Teng M.W. Smyth M.J. Darcy P.K. Supernatural T cells: Genetic modification of T cells for cancer therapy. Nat. Rev. Immunol. 2005;5:928–940. doi: 10.1038/nri1729. [DOI] [PubMed] [Google Scholar]
- Kowolik C.M. Topp M.S. Gonzalez S. Pfeiffer T. Olivares S. Gonzalez N. Smith D.D. Forman S.J. Jensen M.C. Cooper L.J. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- Leen A.M. Rooney C.M. Foster A.E. Improving T cell therapy for cancer. Annu. Rev. Immunol. 2007;25:243–265. doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- Marin V. Kakuda H. Dander E. Imai C. Campana D. Biondi A. D'Amico G. Enhancement of the anti-leukemic activity of cytokine induced killer cells with an anti-CD19 chimeric receptor delivering a 4-1BB-ζ activating signal. Exp. Hematol. 2007;35:1388–1397. doi: 10.1016/j.exphem.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Modak S. Kramer K. Gultekin S.H. Guo H.F. Cheung N.K. Monoclonal antibody 8H9 targets a novel cell surface antigen expressed by a wide spectrum of human solid tumors. Cancer Res. 2001;61:4048–4054. [PubMed] [Google Scholar]
- Moeller M. Haynes N.M. Trapani J.A. Teng M.W. Jackson J.T. Tanner J.E. Cerutti L. Jane S.M. Kershaw M.H. Smyth M.J. Darcy P.K. A functional role for CD28 costimulation in tumor recognition by single-chain receptor-modified T cells. Cancer Gene Ther. 2004;11:371–379. doi: 10.1038/sj.cgt.7700710. [DOI] [PubMed] [Google Scholar]
- Onda M. Wang Q.C. Guo H.F. Cheung N.K. Pastan I. In vitro and in vivo cytotoxic activities of recombinant immunotoxin 8H9(Fv)-PE38 against breast cancer, osteosarcoma, and neuroblastoma. Cancer Res. 2004;64:1419–1424. doi: 10.1158/0008-5472.can-03-0570. [DOI] [PubMed] [Google Scholar]
- Press W.H. Teukolsky S. Vetterling W.T. Flannery B.P. Numerical Recipes in C: The Art of Scientific Computing. Cambridge University Press; Cambridge: 1992. [Google Scholar]
- Rabinovich P.M. Komarovskaya M.E. Ye Z.J. Imai C. Campana D. Bahceci E. Weissman S.M. Synthetic messenger RNA as a tool for gene therapy. Hum. Gene Ther. 2006;17:1027–1035. doi: 10.1089/hum.2006.17.1027. [DOI] [PubMed] [Google Scholar]
- Ren-Heidenreich L. Mordini R. Hayman G.T. Siebenlist R. Lefever A. Comparison of the TCR ζ-chain with the FcR γ-chain in chimeric TCR constructs for T cell activation and apoptosis. Cancer Immunol. Immunother. 2002;51:417–423. doi: 10.1007/s00262-002-0301-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultze J.L. Cardoso A.A. Freeman G.J. Seamon M.J. Daley J. Pinkus G.S. Gribben J.G. Nadler L.M. Follicular lymphomas can be induced to present alloantigen efficiently: A conceptual model to improve their tumor immunogenicity. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8200–8204. doi: 10.1073/pnas.92.18.8200. [erratum appears in Proc. Natl. Acad. Sci. U.S.A. 1995;92:10818]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt J.S. The lognormal frequency distribution and human cancer. J. Surg. Res. 1969;9:151–157. doi: 10.1016/0022-4804(69)90046-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.