

Abstract
The differentiation of B cells into antibody-secreting plasma cells upon antigen stimulation, a crucial step in the humoral immune response, is disrupted by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Several key regulatory proteins in the B-cell transcriptional network have been identified, with two coupled mutually repressive feedback loops among the three transcription factors B-cell lymphoma 6 (Bcl-6), B lymphocyte–induced maturation protein 1(Blimp-1), and paired box 5 (Pax5) forming the core of the network. However, the precise mechanisms underlying B-cell differentiation and its disruption by TCDD are not fully understood. Here we show with a computational systems biology model that coupling of the two feedback loops at the Blimp-1 node, through parallel inhibition of Blimp-1 gene activation by Bcl-6 and repression of Blimp-1 gene deactivation by Pax5, can generate a bistable switch capable of directing B cells to differentiate into plasma cells. We also use bifurcation analysis to propose that TCDD may suppress the B-cell to plasma cell differentiation process by raising the threshold dose of antigens such as lipopolysaccharide required to trigger the bistable switch. Our model further predicts that high doses of TCDD may render the switch reversible, thus causing plasma cells to lose immune function and dedifferentiate to a B cell–like state. The immunotoxic implications of these predictions are twofold. First, TCDD and related compounds would disrupt the initiation of the humoral immune response by reducing the proportion of B cells that respond to antigen and differentiate into antibody-secreting plasma cells. Second, TCDD may also disrupt the maintenance of the immune response by depleting the pool of available plasma cells through dedifferentiation.
Keywords: TCDD, immunotoxicity, bistability, coupled feedback loops, dedifferentiation, cellular reprogramming
INTRODUCTION
The differentiation of B cells in lymphoid organs into antibody-secreting plasma cells upon antigen stimulation is a crucial step in the humoral immune response (Calame, 2001; Shapiro-Shelef and Calame, 2005). The initial rapid response to antigen consists of noncirculating naive B cells in the marginal zone of the spleen proliferating and differentiating mainly into short-lived plasma cells that secrete the low-affinity antibody immunoglobulin M (IgM) (Calame et al., 2003). Subsequently, stimulation by antigen and T helper cells causes naive follicular B cells to undergo the germinal center reaction, which produces plasma cells secreting high-affinity antibody (McHeyzer-Williams et al., 2001). Post–germinal center plasma cells that migrate to the bone marrow and receive survival signals from stromal cells therein can survive for several months as “long-lived plasma cells” (Shapiro-Shelef and Calame, 2004, 2005; Slifka and Ahmed, 1998; Slifka et al., 1998).
The environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) suppresses the humoral immune response by interfering with the antigen-induced differentiation of B cells into plasma cells (Dooley and Holsapple, 1988; Suh et al., 2002; Sulentic et al., 1998; Yoo et al., 2004). TCDD belongs to a class of contaminants known as halogenated aromatic hydrocarbons (HAHs) that produce toxic effects in mammals through binding to the aryl hydrocarbon receptor (AhR) (Poland and Knutson, 1982; Rowlands and Gustafsson, 1997; Schmidt and Bradfield, 1996). The toxic effects of AhR agonists, which feature prominently among the high-risk substances associated with hazardous sites on the National Priorities List http://www.epa.gov/superfund/sites/npl/ compiled by the U.S. Environmental Protection Agency and the Agency for Toxic Substances and Disease Registry (ATSDR), are of much interest to the environmental health research community.
Arguably, the two most sensitive target organs for the health effects of HAHs are the liver and the immune system (Andersen et al., 1997; Birnbaum, 1994; Dooley and Holsapple, 1988; Holsapple et al., 1991; Vorderstrasse et al., 2003). A review of the toxicological hazard of Superfund waste sites by scientists from ATSDR states that “immunotoxicity is the end point lacking for the greatest number of Superfund priority substances for which human exposure pathways have been identified” and describes this as “a serious deficiency in knowledge needed by health and risk assessors because of the essential role of the immune system for protecting one's health” (Johnson and DeRosa, 1997). B cells are sensitive target in the immunosuppressive effect of TCDD (Dooley and Holsapple, 1988). A mechanistic investigation of this effect requires a detailed understanding of the molecular machinery and signaling pathways responsible for the differentiation of B cells to antibody-secreting plasma cells and of the manner in which TCDD may interfere with these pathways.
The AhR-signaling pathway appears to play a dual role in the host as (1) a mediator of xenobiotic metabolism and toxicity and (2) a regulator of normal physiological processes, particularly in vascular and hematopoietic development. However, there is as yet no consensus on the endogenous developmental cue for AhR signaling (Nguyen and Bradfield, 2008; Stevens et al., 2009). Specifically, it remains unclear whether the AhR plays an endogenous role in B-cell differentiation. We and others have observed that activation of leukocytes in general, and primary B cells in particular, leads to a marked but transient increase in AhR messenger RNA (mRNA) and protein levels (Allan and Sherr, 2005; Crawford et al., 1997; Marcus et al., 1998), which makes it tempting to speculate that the AhR may play a critical function in B-cell differentiation. However, this speculation is tempered by the fact that AhR-null mice are capable of mounting primary humoral immune responses of a magnitude similar to wild-type mice (Vorderstrasse et al., 2001), suggesting that the AhR can influence but is not required for physiological B-cell differentiation and plasma cell formation.
To understand the AhR-mediated immunotoxic effects of TCDD from a dynamic systems perspective, we have developed a computational model of the biochemical pathways that regulate the B-cell differentiation program, with particular emphasis on the system-level mechanisms that underlie this process. We hypothesize that mutual regulation of three key transcription factors in the B-cell regulatory program, B lymphocyte–induced maturation protein 1 (Blimp-1), B-cell lymphoma 6 (Bcl-6), and paired box 5 (Pax5), forms the basis of a bistable switch allowing the system to choose from one of two discrete alternative states: undifferentiated B cells and fully differentiated plasma cells (Igarashi et al., 2007). A possible mechanism by which TCDD interferes with this process through the activation of AhR is also described. We propose that TCDD may suppress the differentiation of B cells into plasma cells by raising the threshold dose of antigen (e.g., lipopolysaccharide [LPS]) required to trigger the differentiation switch. Surprisingly, the model predicts that high doses of TCDD may also cause reprogramming of fully differentiated plasma cells back to a B cell–like phenotype.
METHODS
Computational Model
Key proteins in the B-cell transcriptional network regulate B-cell to plasma cell differentiation.
As with many cellular fate decisions, a transcriptional program involving a number of interacting transcription factors appears to underlie the terminal differentiation of B cells. Genetic and biochemical studies have implicated Blimp-1, Bcl-6, MTA3, Pax5, Bach2, MITF, IRF-4, and XBP-1 as important transcriptional regulators of the B-cell differentiation pathway (Igarashi et al., 2007; Shapiro-Shelef and Calame, 2005). A tightly controlled circuit comprising three of these transcription factors, Blimp-1, Pax5, and Bcl-6, forms the core of this pathway (Fig. 1).
FIG. 1.

The core transcriptional circuit underlying the terminal differentiation of B cells to antibody-secreting plasma cells and its disruption by TCDD. The circuit consists of coupled double-negative feedback loops involving the three transcription factors Pax5, Bcl-6, and Blimp-1. AP-1 triggers the differentiation switch by activating Blimp-1 under stimulation by LPS. TCDD interferes with the differentiation program by downregulating AP-1 activity in an AhR-dependent manner. AhR also suppresses antibody secretion directly by binding to dioxin response elements in the IgH 3′α enhancer [→ denotes activation and ––| denotes suppression].
Blimp-1, a zinc finger–containing transcriptional repressor, functions as a hub in the gene network underlying B-cell differentiation. It appears to act as a “master regulator” of this process by repressing the proteins Bcl-6 and Pax5 that are responsible for maintaining the B-cell phenotype (Sciammas and Davis, 2004; Shaffer et al., 2002). Blimp-1 is maintained at a low level in the B-cell state but is induced strongly in the plasma cell state (Calame et al., 2003). High levels of Blimp-1 repress transcription of the c-Myc gene to cause cell cycle arrest—a condition associated with terminal differentiation of B cells into plasma cells (Igarashi et al., 2007; Lin et al., 1997). Compatible with its plasma cell–promoting role, ectopic expression of Blimp-1 in B cells at the appropriate developmental state drives differentiation to antibody-secreting plasma cells (Sciammas and Davis, 2004; Shaffer et al., 2002; Shapiro-Shelef and Calame, 2005; Turner et al., 1994).
Pax5 expression is initiated in (precursor) pro-B cells and continued in mature B cells to establish B-cell identity. The repression of Pax5 by Blimp-1 induces B cells to commit to the plasma cell differentiation pathway (Igarashi et al., 2007; Lin et al., 2002). Pax5 can act both as an activator and as a repressor depending on the context. For genes responsible for the functioning of B-cell receptors, Pax5 acts as a transcriptional activator, although it represses genes incompatible with the B-cell phenotype (Delogu et al., 2006; Nutt et al., 1998)—in particular, those associated with progression from the B-cell to plasma cell state (Horcher et al., 2001; Reimold et al., 2001). Deletion of Pax5 in DT40 chicken B cells leads to strong upregulation of XBP-1, a critical plasma cell transcription factor (Nera et al., 2006). Importantly, Pax5 inhibits the expression of immunoglobulin heavy chain (IgH), immunoglobulin kappa light chain (Igκ), and the immunoglobulin joining (J) chain—all necessary components of the IgM molecule that mediates immune response (Rinkenberger et al., 1996; Shaffer et al., 1997; Singh and Birshtein, 1996). Further, ectopic expression of Pax5 results in suppression of IgM production (Schneider et al., 2008). While Blimp-1 represses Pax5 gene expression, it has recently been reported that Pax5 also represses Blimp-1 by directly binding to the promoter of the human PRDM1 gene, which encodes the Blimp-1 protein (Mora-Lopez et al., 2007). Thus, a motif of mutual inhibition arises between Blimp-1 and Pax5, forming a double-negative regulatory feedback loop.
The primary function of the transcription factor Bcl-6, which is present at high levels in B cells, is to repress Blimp-1 by direct binding to a response element in its gene (Tunyaplin et al., 2004), as well as by indirect inhibitory interactions with activator protein 1 (AP-1) (Vasanwala et al., 2002) and signal transducer and activator of transcription 3 (Reljic et al., 2000)—both transcriptional activators of Blimp-1. Reciprocally, Blimp-1 also represses Bcl-6 (Shaffer et al., 2002). Bcl-6 and Blimp-1 thus mutually inhibit each other to form a second double-negative feedback loop in addition to the Blimp-1-Pax5 loop. Blimp-1 is therefore topologically situated at the center of two “coupled” or interlinked double-negative feedback loops, affirming its role as a master regulator of plasma cell differentiation (Fig. 1). This coupled feedback circuit ensures that the transcriptional profiles of B-cell and plasma cell phenotypes are mutually exclusive—i.e., B cells are associated with high expression of Pax5 and Bcl-6 and low expression of Blimp-1, whereas plasma cells are associated with high expression of Blimp-1 and low expression of Pax5 and Bcl-6. The potential role of double-negative regulatory circuits as a “switch”, allowing a cell to toggle between two discrete states, has long been hypothesized (Ferrell, 2002; Monod and Jacob, 1961; Staudt, 2004). The B-cell to plasma cell differentiation switch can be turned “on” by the transcription factor AP-1 that is activated by antigens such as LPS and thereafter induces Blimp-1 (Ohkubo et al., 2005; Vasanwala et al., 2002) (Fig. 1).
TCDD suppresses B-cell to plasma cell differentiation through inhibition of AP-1 activity.
The primary target in the immunosuppressive effect of TCDD on IgM antibody secretion is the differentiation of B cells into plasma cells (Dooley and Holsapple, 1988). This effect is mediated by AhR—as evidenced by the suppression of LPS-induced IgM secretion by TCDD in the AhR-expressing CH12.LX B cell line but not in the AhR-deficient BCL-1 B cell line (Sulentic et al., 1998), as well as the suppression of the anti-sheep erythrocyte IgM antibody forming cell response in wild-type but not AhR-null mice (Vorderstrasse et al., 2001). IgH, Igκ, J chain, and XBP-1—all essential components in the assembly and secretion of IgM antibody—are persistently repressed by TCDD (Yoo et al., 2004). This is consistent with the observation that TCDD simultaneously causes enhanced expression of Pax5, which is a repressor of these protein molecules (Yoo et al., 2004). The mechanism of IgM suppression is at least partially transcription mediated. Dioxin response elements, which are specifically recognized by AhR, have been identified within the IgH 3′α enhancer (Sulentic et al., 2000). The upregulation of Pax5 itself may be explained by the TCDD-induced inhibition of Blimp-1 protein, an upstream negative regulator of Pax5, in light of the following observations: (1) LPS stimulation of B cells leads to upregulation of the AP-1 protein complex, which in turn positively regulates Blimp-1 expression and B-cell differentiation (Ohkubo et al., 2005) and (2) TCDD strongly inhibits LPS-induced DNA-binding and transcriptional activity of AP-1 (Suh et al., 2002). Together, these observations suggest that TCDD is likely to suppress Blimp-1 expression indirectly by inhibiting AP-1 activity (Fig. 1). Further confirmation of this hypothesis comes from our recent studies showing that while LPS stimulation upregulates DNA-binding activity of AP-1 protein at three response motifs within the Blimp-1 promoter, TCDD treatment suppresses AP-1 binding to these motifs between 24 and 72 h, in concordance with suppression of both Blimp-1 gene expression and protein activity by TCDD (Schneider et al., 2009). Thus, AhR-dependent downregulation of AP-1 activity is likely to be an important mechanism in the TCDD-induced inhibition of B-cell differentiation, although the particular mechanism by which ligand-activated AhR impairs AP-1 activity remains unclear.
Model development.
A schematic diagram of our model, based on the interactions summarized above, is given in Figure 2. We used ordinary differential equations (ODEs) to model reaction kinetics. The representation of transcriptional control of Blimp-1, Pax5, and Bcl-6 is based on our current understanding of eukaryotic gene regulation (Fiering et al., 2000; Kaern et al., 2005; Zhang et al., 2006). Specifically, at any given time, a gene could be in one of two discrete transcriptional states: inactive or active (GENE0 and GENE1, respectively, in Fig. 2), corresponding, respectively, to the compact and relaxed chromatin structures of the promoter. Once in the active state, the gene is transcribed at a constant rate, whereas in the inactive state, no transcription occurs. Transitions between the inactive and active states (i.e., gene activation and deactivation) are controlled by transcription activators and repressors specifically targeting the promoter. The effect of a repressor on a gene can be exerted in one of two ways: either actively or passively. In the active mode, it represses the target gene by recruiting corepressors (which catalyze chromatin condensation) to promote deactivation of the gene. In the passive mode, it represses the target gene by blocking transcription activators or coactivators (which catalyze chromatin relaxation) to inhibit activation of the gene (HannaRose and Hansen, 1996; Thiel et al., 2004). Incorporation of gene activation and deactivation steps allows, as described below, explicit modeling of the distinct modes of mutual transcriptional repression among Blimp-1, Bcl-6, and Pax5, in accordance with the literature.
FIG. 2.

A detailed network representation of all interactions in the model. The symbol Φ represents mRNA and protein degradation; GENE0 and GENE1 represent, respectively, the on- and off-states of a gene.
Blimp-1 has been shown to repress both Bcl-6 and Pax5 gene expressions (Lin et al., 2002; Sciammas and Davis, 2004; Shaffer et al., 2002). As a transcriptional repressor, Blimp-1 is able to suppress target genes by recruiting corepressors such as histone deacetylase, histone methyltranferase, and members of the Groucho family (Gyory et al., 2004; Ren et al., 1999; Yu et al., 2000). These corepressors function as chromatin-modifying enzymes to alter the local chromatin structure to a transcriptionally repressed (inactive) state. We implemented this active mode of transcriptional repression in our model by having Blimp-1 promote the deactivation step of the Bcl-6 and Pax5 genes, as opposed to inhibiting the activation step (Fig. 2). Pax5 represses Blimp-1 gene expression directly (Mora-Lopez et al., 2007), likely by an active mechanism mediated through recruitment of corepressors from the Groucho family (Eberhard et al., 2000; Milili et al., 2002). Thus, similar to the repressive action exerted by Blimp-1, repression of Blimp-1 by Pax5 was implemented in the model by having Pax5 promote the deactivation step of the Blimp-1 gene. Despite the possibility that the Bcl-6 protein may directly bind to target genes as a repressor, its suppressive effect on Blimp-1 appears to be primarily AP-1 dependent. Bcl-6 can bind to AP-1 and block its transcriptional activity (Vasanwala et al., 2002), thus functioning as a passive repressor of Blimp-1. Since AP-1 positively regulates Blimp-1 gene expression, Bcl-6 exerts its repression on Blimp-1 by impinging upon its activation step, thereby suppressing the maximal induction of Blimp-1 by AP-1 (Fig. 2).
Through these specific transcriptional regulatory actions, the coupled double-negative feedback loops between Bcl-6, Blimp-1, and Pax5 make up the core transcriptional switch in our model. The distinct modes of repression exerted by Bcl-6 and Pax5 on Blimp-1 (passive vs. active) are essential to establish robust bistability in the absence of other explicit ultrasensitive motifs, as discussed in the “Results”.
The endotoxin LPS is recognized by the cell surface Toll-like receptor 4 (TLR4), which through a complex signaling cascade activates mitogen-activated protein (MAP) kinases, leading to subsequent phosphorylation and activation of the AP-1 protein (Chang and Karin, 2001; Kawai and Akira, 2007; Lu et al., 2008; Shaulian and Karin, 2002). In our model we simplify this sequence of events by having LPS directly phosphorylate AP-1 to AP-1p—the active form of the AP-1 transcription factor. Ligand activation of AhR by TCDD leads to dimerization with aryl hydrocarbon receptor nuclear translocator (ARNT) and subsequent inhibition of AP-1 activity (Suh et al., 2002). The precise mechanism underlying the inhibition being unclear, we model this inhibitory step as a dephosphorylation of AP-1p by the TCDD-AhR-ARNT complex (Fig. 2). This ensures that the model reflects the dose-dependent suppression of AP-1–binding activity by TCDD (Schneider et al., 2009), irrespective of the actual mechanism. In the absence of TCDD, the active transcriptional factor AP-1p induces Blimp-1 transcription by promoting its gene activation.
The ODE-based computational model was implemented using the PathwayLab software (InNetics, Inc., Linköping, Sweden), which automatically converts a directed-graph representation of a cellular signaling network into a system of ODEs. The model was then exported to MATLAB (The MathWorks, Inc., Natick, MA) for more complex analyses including dose-response surface simulation. Appendix 1 lists the ODEs representing the interactions among all species in the model. Note that we account for both alleles of each gene in the model, where either allele can be in an on- or off-state: hence the number of off-states and on-states of each gene add up to 2. For example, in Equation 1 (2 − Bcl6_gene1) ≡ Bcl6_gene0, where Bcl6_gene1 represents the on-state, and Bcl6_gene0 the off-state, of the Bcl-6 gene. The variables TA and TAA in Appendix 1 represent the TCDD-AhR and TCDD-AhR-ARNT complexes, respectively. Supplementary table 1 in the “Supplementary Data” lists the values of all reaction parameters kij and kdij in the model equations. Total numbers of molecules of AP-1, AhR, and ARNT (AP1total, AhRtotal, and ARNTtotal, respectively) are treated as parameters in the model. The steady-state behavior of the model was analyzed using bifurcation analysis, a common method for exploring multi-stable switching phenomena in cellular decision-making processes (Chickarmane et al., 2006; Ferrell and Xiong, 2001; Huang et al., 2007; Roeder and Glauche, 2006; Tyson and Novak, 2001; Tyson et al., 2001, 2002; Xiong and Ferrell, 2003). Bifurcation diagrams were generated using the XPP-AUT program (http://www.math.pitt.edu/∼bard/xpp/xpp.html).
RESULTS
A Bistable Switch Underlying B-cell to plasma cell differentiation
As outlined above, the core of the transcriptional regulatory circuit underlying B-cell to plasma cell differentiation is formed by the coupled double-negative feedback loops emerging from the interactions among the three transcription factors Bcl-6, Blimp-1, and Pax5. Under appropriate parameter conditions this circuit exhibits bistability, which unambiguously separates the undifferentiated and differentiated cellular states.
Figure 3A shows the time-course of Blimp-1 protein level predicted by the model for varying levels of the LPS stimulus (protein levels in all figures are expressed in number of molecules). There is a sharp transition from the low Blimp-1 state associated with the B-cell phenotype to the high Blimp-1 state characterizing the plasma cell phenotype. The system shows no significant response for doses of LPS < 0.04 μg/ml, which supports the idea of a threshold dose of LPS required to trigger the B-cell to plasma cell differentiation switch. To illustrate this threshold-dependent switch, the steady-state Blimp-1 versus LPS dose-response curve is plotted in the form of a bifurcation diagram in Figure 3B. The system as described in the model exhibits a “true discontinuity” at the threshold (LPS ≅ 0.05 μg/ml) that precludes an intermediate-level response (Ferrell, 1998). The bistable switching behavior arises from the nonlinearity and positive feedback inherent in the system (Angeli et al., 2004; Bhattacharya et al., forthcoming; Ferrell, 2002; Ferrell and Xiong, 2001; Tyson et al., 2003) and has been demonstrated in a number of biological systems (Bagowski and Ferrell, 2001; Chang et al., 2006; Xiong and Ferrell, 2003) as well as in synthetic molecular circuits (Becskei et al., 2001; Gardner et al., 2000; Ozbudak et al., 2004). A bistable system exhibits hysteresis: a characteristic feature of any bistable system, whereby different dose-response curves are obtained depending on whether the system is subjected to increasing or decreasing dose, as emphasized by the arrows in Figure 3B (Bagowski and Ferrell, 2001; Ferrell, 2002).
FIG. 3.

Bistability in the B-cell to plasma cell differentiation model. (A) Time-course of Blimp-1 protein for increasing doses of antigen LPS (dose values in micrograms per milliliter indicated on figure). A sharp transition occurs from the B-cell state (low Blimp-1) to the plasma cell state (high Blimp-1) for doses of LPS higher than 0.04 μg/ml. (B) Blimp-1 versus LPS bifurcation diagram illustrating a threshold-dependent bistable switch. The solid lines represent stable steady states; the dashed line represents unstable steady states. The arrows show the direction of dosing and the discontinuity at the switching threshold.
The other notable feature of the switch portrayed in the bifurcation diagram is that it is irreversible—another property characteristic of cellular differentiation processes. The system stays in the “on” (differentiated) state with high Blimp-1 levels even after the triggering stimulus (LPS) is removed, as indicated by the leftward-pointing arrow in Figure 3B. The irreversibility in the system can also be illustrated by its predicted response to a pulse of LPS at 1.0 μg/ml (Figs. 4A–C). Once the LPS pulse is removed, the phosphorylated form of AP-1 (AP-1p) immediately drops to its basal unstimulated level (Fig. 4A), but the level of the Blimp-1 protein remains high (Fig. 4B) and the level of the Pax5 protein (as well as that of Bcl-6: result not shown) remains low (Fig. 4C). Given a sufficient strength and duration of LPS dose, the system thus remains in the differentiated plasma cell state even after the antigen is removed. These predictions are qualitatively similar to the short-term transient expression of AP-1 family genes, and long-term induction and suppression of Blimp-1 and Bcl-6 genes, respectively, observed in murine splenic B cells stimulated with an LPS dose of 3 μg/ml (Ohkubo et al., 2005). Our previous studies have also indicated a peak 25-fold induction of Blimp-1 mRNA transcripts 72 h after LPS activation of mouse splenocytes (Schneider et al., 2009).
FIG. 4.

Effect of applying a pulse dose of LPS on (A) AP-1p, (B) Blimp-1, and (C) Pax5. Strength of LPS dose: 1.0 μg/ml; dosing period: 60 h.
Coupled Double-Negative Feedback Loops Produce Bistability in the B-Cell Transcription Network
The bistable behavior of the system does not derive from any external input in the form of antigen but rather is an innate property of the system arising from the kinetic interactions among various molecular components of the B-cell transcriptional network. This can be illustrated by plotting in three-dimensional phase space the three steady-state stimulus-response curves obtained by using, in turn, the amount of Blimp-1, Bcl-6, and Pax5 proteins as the independent variable (stimulus) and the steady-state levels attained by the other two proteins as the response (Fig. 5A). Note that the three stimulus-response curves all intersect at the same three fixed points in three-dimensional phase space, indicating the innate bistability of the system in the absence of any LPS or TCDD input (for a theoretical explanation, see Angeli et al., 2004). The three points of intersection correspond to three steady states of the system. The point of intersection with low Blimp-1 and high Bcl-6/Pax5 levels (labeled “A” in Fig. 5A) is a stable steady state representing the B-cell phenotype; the point with intermediate values of all three proteins (labeled “B”) is an unstable steady state; and the point with high Blimp-1 and low Bcl-6/Pax5 levels (labeled “C”) is a stable steady state representing the plasma cell phenotype.
FIG. 5.

Steady-state stimulus-response curves among the transcriptional regulators Blimp-1, Bcl-6, and Pax5. (A) The three individual steady-state stimulus-response curves among Blimp-1, Bcl-6, and Pax5, obtained by using, in turn, the amount of Blimp-1 (Curve 1), Bcl-6 (Curve 2), and Pax5 (Curve 3) proteins as the independent parameter (stimulus) and the steady-state levels attained by the other two proteins as the response. Intersection points A, B, and C correspond to, respectively, a stable steady state representing the B-cell phenotype, an unstable steady state, and a stable steady state representing the plasma cell phenotype. (B) The Blimp-1 versus Bcl-6 stimulus-response curve (Curve 2) is highly ultrasensitive, with a maximum slope > 4, while the Bcl-6 versus Blimp-1 curve (Curve 1) is not (maximum slope < 1). This is emphasized by the two dashed lines drawn in the figure with slopes of 4 and 1. These two stimulus-response curves are essentially the projection onto the Blimp-1-Bcl-6 plane of the three-dimensional stimulus-response curves among Bcl-6, Blimp-1, and Pax5 plotted in (A).
For a system to be bistable, the underlying molecular circuit must incorporate positive feedback, together with step(s) that can transfer signal in an ultrasensitive fashion with effective Hill coefficient > 1 (Angeli et al., 2004; Ferrell, 2002; Ferrell and Xiong, 2001; Tyson and Novak, 2001). Usually such sharp transitions are achieved directly via ultrasensitive signaling motifs such as cooperative binding, homodimerization, multistep signaling, or a zero-order switch (Zhang et al., forthcoming). None of these motifs is known to be present in the gene transcriptional circuit for B-cell terminal differentiation. Without ultrasensitivity, neither of the two double-negative feedback loops (i.e., the Bcl-6-Blimp-1 loop or the Blimp-1-Pax5 loop) could generate bistability by itself (result not shown). However, as illustrated above, the full system containing the two monostable double-negative feedback loops coupled at the Blimp-1 node does exhibit bistability.
What makes bistable behavior possible in this case is the fact that when the two double-negative feedback loops are coupled together, each loop in effect serves as an ultrasensitive motif for one of the two arms of the other loop. This is illustrated by the steady-state stimulus-response curves between Blimp-1 and Bcl-6 (Fig. 5B). Although Blimp-1 represses Bcl-6 in an inverse Michaelis-Menten manner with a slope less than 1, the reciprocal process, i.e., the repression of Blimp-1 by Bcl-6, is strongly ultrasensitive with a much steeper slope (> 4). This ultrasensitivity originates from the mutual inhibition loop formed between Pax5 and Blimp-1, which functions as an ultrasensitive motif that is terminally attached to the Bcl-6 → Blimp-1 arm of the other loop.
The specific manner in which the two double-negative feedback loops are coupled is critically important also for generating ultrasensitivity strong enough to support robust bistability. Blimp-1 is the nodal point of the coupling and is subject to repressive regulation by both Bcl-6 and Pax5. Repression of Blimp-1 can be effected either by inhibiting gene activation or by promoting gene deactivation. As detailed above in “Methods and Materials,” the literature suggests that Bcl-6 represses Blimp-1 primarily by inhibiting the gene activation step, whereas Pax5 represses Blimp-1 by promoting the gene deactivation step. By regulating Blimp-1 gene activity in these two distinct modes, the repressive effects of Bcl-6 and Pax5 act in a synergistic fashion. This synergy makes it possible for the two double-negative feedback loops to serve as highly ultrasensitive motifs for each other, thereby allowing bistability to emerge in the system. Had both Bcl-6 and Pax5 repressed Blimp-1 in a similar mode (i.e., either both promoting deactivation or both inhibiting activation of Blimp-1), their repressive effects would be simply additive, making the emergence of bistability unlikely in the absence of ultrasensitivity in the individual feedback loops.
Gene Deletion or Overexpression Leads to Transition Between B-Cell and Plasma Cell Phenotypes
Our model is able to qualitatively reproduce several experimental observations on the effect of deletion or overexpression of the individual regulatory proteins Blimp-1, Bcl-6, and Pax5. We simulated these effects by altering the transcription rates of the corresponding genes in the model. Overexpression of Blimp-1 causes B cells to differentiate into antibody-secreting plasma cells (Piskurich et al., 2000; Schliephake and Schimpl, 1996; Turner et al., 1994). This effect can be reproduced in our model by a bifurcation diagram of Blimp-1 protein level versus the Blimp-1 transcription rate, k13 (Fig. 6A). In the absence of the LPS antigenic stimulus, a sufficiently high rate of expression of the Blimp-1 protein can cause the system to switch from the B-cell state (low Blimp-1) to the plasma cell state (high Blimp-1). On the other hand, Blimp-1–deficient B cells are not able to undergo differentiation to the plasma cell state (Shapiro-Shelef et al., 2003). Consistent with this experimental observation, our simulation illustrates that a reduced Blimp-1 transcription rate raises the threshold dose of LPS required to trigger the B-to-plasma cell switch (Fig. 6B), making differentiation more difficult.
FIG. 6.

Dependence of the B-cell differentiation switch on the Blimp-1 transcription rate, k13. (A) Blimp-1 versus k13 bifurcation diagram with LPS dose = 0. Overexpression of Blimp-1 is sufficient to switch the system from the B-cell state (low Blimp-1) to the plasma cell state (high Blimp-1), as indicated by the arrows. (The default value of k13 in the model is 3.47e-4/s) (B) Blimp-1 versus LPS bifurcation diagrams for k13 (Blimp-1 transcription rate) = 3.47e-4/s (upper curve; default value of k13) and 2.0e-4/s (lower curve). A lower Blimp-1 transcription rate raises the threshold dose of LPS required to trigger B-cell to plasma cell differentiation from 0.05 to 0.16 μg/ml.
Ectopic expression of Bcl-6 in a plasma cell line resulted in the repression of plasma cell–specific transcripts and reactivation of the B-cell transcriptional program and phenotype. Simultaneous ectopic expression of the associated corepressor MTA3 further enhances this Bcl-6–induced dedifferentiation (Fujita et al., 2004). In agreement with this observation, simulated overexpression of Bcl-6 in the plasma cell state caused the system to revert back to the B-cell state (Fig. 7).
FIG. 7.

Blimp-1 versus k03 (Bcl-6 transcription rate) bifurcation diagram with LPS dose = 0. Overexpression of Bcl-6 is sufficient to switch the system from the plasma cell state (high Blimp-1) to the B-cell state (low Blimp-1), as indicated by the arrows. The default value of k03 in the model is 3.47 × 104/s.
Loss of Pax5 in the chicken B cell line DT40 led to upregulation of Blimp-1 and acquisition of plasma cell characteristics, while restoration of Pax5 expression normalized Blimp-1 levels to that seen in wild-type B cells (Nera et al., 2006). Both of these effects can be simulated by a bifurcation diagram of Blimp-1 level versus the Pax5 transcription rate, k23 (Fig. 8A), where the leftward- and rightward-pointing arrows indicate, respectively, the effects of loss and restoration of Pax5 expression. Conversely, ectopic expression of Pax5 in murine splenic B cells inhibited formation of plasma cells after LPS treatment (Lin et al., 2002). This result is explained by the effect of increasing the Pax5 transcription rate, which raises the threshold dose of LPS required to trigger the B-to-plasma cell switch (Fig. 8B), thus inhibiting the differentiation process.
FIG. 8.

Dependence of the B-cell differentiation switch on the Pax5 transcription rate k23. (A) Blimp-1 versus k23 bifurcation diagram with LPS dose = 0. Downregulation of Pax5 is sufficient to switch the system from the B-cell state (low Blimp-1) to the plasma cell state (high Blimp-1), whereas upregulation of Pax5 can reverse the differentiation. (B) Blimp-1 versus LPS bifurcation diagrams for k23 (Pax5 transcription rate) = 3.47 × 104/s (upper curve; default value) and 8.0 × 104/s (lower curve). The increase in Pax5 transcription rate raises the threshold dose of LPS required to trigger B-cell to plasma cell differentiation from 0.05 to 0.16 μg/ml.
TCDD Suppresses B-Cell Differentiation by Raising the Dose Threshold of the Bistable Switch
To examine the suppressive effect of the contaminant TCDD on the differentiation of B cells into plasma cells, we compare the Blimp-1 versus LPS bifurcation diagrams for doses of TCDD = 0nM and TCDD = 0.5nM (Fig. 9). The addition of TCDD shifts the bifurcation curve to the right, consistent with our observation of dose-dependent suppression by TCDD of Blimp-1 mRNA transcripts in mouse splenocytes 72 h after LPS activation (Schneider et al., 2009). There are two mechanistic implications of this effect. First, the threshold dose of LPS required to activate the differentiation switch is raised due to the presence of TCDD (from about 0.05 μg/ml LPS for TCDD = 0nM to 0.35 μg/ml LPS for TCDD = 0.5nM). We propose that this may be a likely mechanism for the observed suppression of antigen-induced differentiation of B cells into plasma cells by TCDD (Dooley and Holsapple, 1988; Suh et al., 2002; Sulentic et al., 1998; Yoo et al., 2004). Second, the model predicts that the addition of sufficiently high doses of TCDD may convert the system from an irreversible to a reversible bistable switch. This would imply that in the presence of sufficient amount of TCDD, reducing the applied LPS dose may cause differentiated plasma cells to dedifferentiate back into a B-cell or B cell–like state, thereby losing immune function. Thus, in addition to reducing the proportion of B cells that differentiate into plasma cells under antigen stimulation, the immunotoxic effect of TCDD may include a reduction of the pool of existing antibody-secreting plasma cells by dedifferentiation.
FIG. 9.

Blimp-1 versus LPS bifurcation diagrams for TCDD = 0nM (upper curve) and TCDD = 0.5nM (lower curve). The effect of TCDD on the bistable switch is manifested as a higher “on” threshold for the LPS dose. TCDD also renders the switch reversible, as indicated by the arrows in the figure.
For a more detailed investigation of the fate of B cells in the presence of both the stimulant LPS and the suppressor TCDD, we have generated steady-state “dose-response surfaces.” In the discussion below, we describe simulations initialized at the undifferentiated B-cell state (low Blimp-1, high Bcl-6, and high Pax5) as “forward” dosing and simulations initialized at the fully differentiated plasma cell state (high Blimp-1, low Bcl-6, and low Pax5) as “backward” dosing. The abrupt transition in the forward dose-response surface (Fig. 10A) from the “off” (B cell) state to the “on” (plasma cell) state reflects the discontinuity in the differentiation switch discussed above in the context of the Blimp-1 bifurcation diagrams. A backward dose-response surface, obtained by starting from the plasma cell state, is overlaid on the forward surface (Fig. 10B). The two distinct surfaces together serve as a visual representation of the bistability and hysteresis inherent in the differentiation switch, in the parameter space of LPS and TCDD doses. While the forward dose-response surface indicates that the LPS threshold required to turn the switch on is raised as the TCDD dose is increased, the backward dose-response surface shows that a sufficiently high dose of TCDD may flip the switch from the on-state back to the off-state, rendering it reversible. Note that the two dose-response surfaces in Figure 10B together comprise the portion of the “cusp-catastrophe” surface (Strogatz, 2001) that would be generated by plotting only the stable steady-state values of Blimp-1 protein level as a function of LPS and TCDD. The switching behavior of the system can also be visualized in a “phase diagram” by plotting the monostable (B or plasma cell) and bistable regimes for a range of LPS and TCDD doses (Fig. 10C). The two-parameter bifurcation diagram illustrates the higher threshold of LPS dose required with increasing TCDD dose to induce the differentiation, as well as the reversibility of the switch for high doses of TCDD.
FIG. 10.

Dose-response behavior of the model. (A) The “forward” dose-response surface, with “off” (B-cell) and “on” (plasma cell) states of the switch marked out. (B) The “backward” dose-response surface overlaid on the “forward” surface: the distinctness of the two surfaces represents the bistability and hysteresis in the model. (C) Phase diagram of the model generated by two-parameter bifurcation analysis, showing the bistable and monostable regimes of the system in the parameter space of LPS and TCDD doses.
DISCUSSION
The idea that double-negative feedback loops play a vital role in cellular differentiation was first proposed by Jacques Monod and Francois Jacob nearly 50 years ago (Monod and Jacob, 1961). Perturbation of one of the regulators in these circuits can drive the system toward one of two discrete cellular states, thus creating a toggle switch (Staudt, 2004). Such circuits can also give rise to the associated phenomena of bistability and hysteresis, where for a range of values of the input stimulus, either cellular state may be permissible depending on the initial condition. While bistability has been observed in small gene regulatory circuits in simple organisms (Becskei et al., 2001; Gardner et al., 2000; Ozbudak et al., 2004) and in signal transduction modules in Xenopus oocytes (Bagowski and Ferrell, 2001; Xiong and Ferrell, 2003), there have been few demonstrations to date of bistability in mammalian cell differentiation. Chang et al. (2006) have recently shown that the differentiation of human HL60 promyelocytic precursor cells to the neutrophil cell lineage after stimulation with dimethyl sulfoxide exhibits hysteresis. In another study, human bone marrow stromal cell–derived myogenic cells were reprogrammed into an osteogenic phenotype by inhibition of MAP kinase signaling and stimulation with bone morphogenic protein 2 (BMP2) in a reversible bistable manner (Wang et al., 2009). Also, double-negative feedback loops have been identified in the interactions of key transcription factors regulating lineage choice in several branches of the hematopoietic stem cell lineage (Orkin and Zon, 2008), suggesting that bistability may play a key role in cellular fate determination in mammals.
As the details of the molecular networks operating in cells are mapped out, it is becoming increasingly clear that cells often make use of multiple linked feedback loops rather than a single loop to accomplish discrete switch-like transitions. Coupled positive and double-negative feedback loops have been found in a variety of molecular circuits underlying such transitions, e.g., in stem cell differentiation, lineage specification, cell cycle, and oocyte maturation (Chickarmane et al., 2006, 2009; Palani and Sarkar, 2008; Sveiczer et al., 2004; Xiong and Ferrell, 2003). In addition to generating multiple stable steady states (Huang et al., 2007), coupled feedback loops can also change the dynamics of switching behavior by modulating the response time (Brandman et al., 2005; Choi et al., 2007; Huang et al., 2007). More importantly, coupled feedback loops are believed to increase the robustness of bistable behavior by expanding the parameter space over which bistability can occur (Ferrell, 2008; Kim et al., 2008).
In the current study, we have identified from the primary literature a coupled double-negative feedback-loop regulatory structure involving three transcription factors, Bcl-6, Blimp-1, and Pax5, that underlies antigen-induced differentiation of mammalian B lymphocytes into antibody-secreting plasma cells. The idea that a small number of master regulators control secondary regulators that in turn control a suite of target genes is important in the context of developmental decision points: alternative developmental outcomes can then be determined simply by triggering these master regulators (Lin et al., 2003). For instance, the three transcription factors Oct4, Sox2, and Nanog have recently emerged as key players in deciding the developmental fate of embryonic stem cells (Boyer et al., 2005; Chickarmane et al., 2006; Loh et al., 2006; Niwa, 2007).
In the B-cell transcriptional network, Bcl-6 and Pax5 repress Blimp-1 by regulating the activation and deactivation, respectively, of the Blimp-1 gene. The coupling of the Bcl-6-Blimp-1 and Pax5-Blimp-1 feedback loops thus achieved is similar to the control of the cell cycle regulator cyclin-dependent kinase CDK1 through two feedback loops involving the kinase Wee1 and phosphatase Cdc25 (Ferrell, 2008). In that case, a positive feedback loop is formed in which CDK1 activates Cdc25, which in turn phosphorylates and thus activates CDK1, while a double-negative feedback loop is formed by the inhibition of Wee1 by CDK and the dephosphorylation and subsequent inactivation of CDK1 by Wee1. This coupling of regulatory interactions at the CDK1 node allows the two feedback loops to function in synergy and facilitates the generation of bistability in the presence of ultrasensitivity. Our model, on the other hand, suggests that the coupling of the feedback loops among Blimp-1, Bcl-6, and Pax5 may be sufficient to generate bistable behavior in the absence of any ultrasensitivity in the individual feedback loops. This result is potentially important for generation of bistability in biological networks lacking any conventional ultrasensitive motif such as positive cooperativity, homomultimerization, multistep signaling, or zero-order ultrasensitivity.
Computational analysis suggests that the coupled feedback-loop circuit behaves as a bistable switch that exhibits hysteresis and irreversibility (Fig. 3). In the context of B-cell differentiation, the irreversibility in the switch would help ensure the persistence of the antibody-secreting plasma cells after the initial antigen stimulus recedes and thus could serve as a “memory” mechanism in long-term immune response (Manz et al., 1998). The model qualitatively reproduced effects of LPS and TCDD stimulation on B cells, as well as observed effects of deletion or overexpression of the key regulatory proteins Blimp-1, Bcl-6, and Pax5 on the B-cell differentiation switch. Additional transcription factors, such as Bach2, IRF-4, MITF, MTA3 etc., also play important roles in the B-cell to plasma cell differentiation pathway (Igarashi et al., 2007; Shapiro-Shelef and Calame, 2005). There are possibly other regulatory feedback loops in the B-cell transcriptional network, for instance between Blimp-1 and IRF-4 (Sciammas et al., 2006). These additional loops are likely to reinforce the dynamics of the core circuitry rather than fundamentally altering the switching behavior of the network.
Our modeling effort has generated a likely explanation for the suppressive effect of TCDD on the B-cell differentiation program, wherein TCDD raises the threshold dose of the antigen (LPS) required to trigger the differentiation switch. Interestingly, the model suggests that high doses of TCDD may lead to loss of the plasma cell phenotype and dedifferentiation to a B-cell or B cell–like state. The immunotoxic implications of these predictions are twofold. First, TCDD and related compounds would suppress the “initiation” of the humoral immune response by reducing the proportion of B cells that respond to antigen and differentiate into antibody-secreting plasma cells. Second, TCDD may also disrupt the “maintenance” of humoral immunity by causing long-lived plasma cells to lose their immune function and revert to a B cell–like state.
While dedifferentiation of plasma cells under the influence of an exogenous toxicant may appear to be a surprising prediction, recent studies suggest considerable plasticity of cellular identity in the B-lymphocyte lineage (Carotta and Nutt, 2008; Cobaleda and Busslinger, 2008; Hanna et al., 2008; Nutt, 2008; Welner et al., 2008). Ectopic expression of Bcl-6 and associated corepressor protein MTA3 in malignant plasma cell lines led to the repression of plasma cell–specific transcripts and reactivation of the B-cell transcriptional program (Fujita et al., 2004; Staudt, 2004). While our model qualitatively reproduces this result (Fig. 7), it is also consistent with our prediction of TCDD-induced dedifferentiation from the plasma cell to B-cell phenotype, as TCDD exposure leads to forced upregulation of Bcl-6 and Pax5 through suppression of Blimp-1 activity. Further, conditional deletion of the Pax5 gene in adult mice caused mature B cells to dedifferentiate to an uncommitted progenitor state, followed by differentiation into the T-cell lineage (Cobaleda et al., 2007). Specific knockdown of Pax5 accompanied by forced expression of the four transcription factors Oct4, Sox2, Klf4, and c-Myc also reprogrammed mature mouse B cells to stem cell–like induced pluripotent cells (Hanna et al., 2008). Other investigators have shown that ectopic expression of the regulatory transcription factors CCAAT/enhancer binding protein α (C/EBPα) and C/EBPβ in differentiated B cells reprograms them into macrophages through inhibition of Pax5 (Xie et al., 2004).
Together, these observations suggest a fluid identity in the lymphocyte lineage, where specific genetic perturbations may lead to reprogramming of mature differentiated cells to a progenitor phenotype. They also provide a context to interpret our counter-intuitive prediction. TCDD may induce plasma cell dedifferentiation by suppressing the Blimp-1 expression program, which by virtue of the coupled double-negative feedback loops would lead in turn to upregulation of Bcl-6 and Pax5 expression and activation of the B-cell phenotype. If verified experimentally, this would be an instance of cellular reprogramming induced by an exogenous small-molecule compound rather than by direct manipulation of one or more transcription factors—a result with significant biological as well as therapeutic implications (Holden and Vogel, 2008; Scadden, 2007; Vogel and Holden, 2007).
System-level biological modeling is a necessary and powerful tool for environmental health researchers in analyzing biochemical pathways of toxicological relevance. A computational model can be thought of as a formal integrated statement of the current knowledge about a particular signaling system, incorporating information about the topology and dynamic behavior of the underlying pathway. Such “mechanistic” models are valuable in explaining both the normal functioning of cellular signaling pathways and their perturbation by toxic compounds (Andersen et al. 2002, 2005). Accordingly, our biologically based model of AhR-mediated suppression of the humoral immune response is presented as a case study of an approach that is an alternative (NAS/NRC, 2007) to traditional dose-response assessment techniques for TCDD and other toxicants based primarily on statistical curve fitting using empirical dose-response data (McGrath et al., 1995).
We should emphasize that as with most mechanistic biological modeling efforts, the quantitative predictions of our model are constrained by the available data. Important parts of the biochemical pathway underlying B-cell to plasma cell differentiation are not yet characterized with sufficient detail—e.g., the precise mechanism by which TCDD suppresses AP-1–binding activity and therefore Blimp-1 expression (Schneider et al., 2009; Suh et al., 2002). However, the key event simulated by our model—the differentiation of B cells to the plasma cell state—is fairly robust to fluctuations in the model parameters (Supplementary figure 1).
While the current work purports to provide a mechanistic basis for a fundamental immunosuppressive effect of TCDD, further work will be necessary to relate it to a rigorous risk assessment paradigm. In particular, since the model is based on in vitro studies, more detailed experimental data describing interactions between the AhR cascade and the signaling pathways controlling the B-cell differentiation program are required. As the mapping of these interactions becomes increasingly refined, we anticipate that our model will begin to provide critical insights into a number of important data gaps in the area of risk assessment, including enhanced capabilities in predicting the toxicity of complex mixtures of HAHs. Specifically, our deterministic ODE-based model can serve as a basis for more detailed stochastic multicell models (Gillespie, 1976; Ullah and Wolkenhauer, 2009; Wilkinson, 2009) of immune suppression by TCDD. Explicit multicellular spatial models of tissue-level response (An et al., 2009) and organ-level physiologically-based pharmacokinetic models will also be required to extrapolate model predictions from in vitro to in vivo exposure scenarios and thereafter to response in humans. Together, this suite of modeling tools will aid the assessment of low-dose HAH toxicity and can lead to a better understanding of the shape of the dose-response curve for this class of environmental contaminants.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
Superfund Research Program of the National Institute of Environmental Health Sciences (P42 ES04911).
Supplementary Material
Acknowledgments
The authors would like to thank Dina Schneider, Colin North, Haitian Lu, Barbara Kaplan, Jay Goodman, and John LaPres for helpful discussions.
This manuscript has been reviewed by the National Center for Computational Toxicology, U.S. Environmental Protection Agency, and approved for publication. Approval does not signify that the contents necessarily reflect the views and policies of the agency nor does the mention of trade names or commercial products constitute endorsement or recommendation for use.
APPENDIX 1
Differential Equations Representing the Deterministic Model of B-Cell Transcriptional Regulation and Its Disruption by TCDD (Parameter Values in Supplementary table 1)
 |
(1) |
| (2) |
| (3) |
 |
(4) |
| (5) |
| (6) |
 |
(7) |
| (8) |
| (9) |
| (10) |
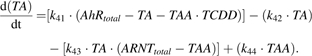 |
(11) |
| (12) |
References
- Allan LL, Sherr DH. Constitutive activation and environmental chemical induction of the aryl hydrocarbon receptor/transcription factor in activated human B lymphocytes. Mol. Pharmacol. 2005;67:1740–1750. doi: 10.1124/mol.104.009100. [DOI] [PubMed] [Google Scholar]
- An G, Mi Q, Dutta-Moscato J, Vodovotz Y. Agent-based models in translational systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009;1:159–171. doi: 10.1002/wsbm.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen ME, Birnbaum LS, Barton HA, Eklund CR. Regional hepatic CYP1A1 and CYP1A2 induction with 2,3,7,8-tetrachlorodibenzo-p-dioxin evaluated with a multicompartment geometric model of hepatic zonation. Toxicol. Appl. Pharmacol. 1997;144:145–155. doi: 10.1006/taap.1996.8067. [DOI] [PubMed] [Google Scholar]
- Andersen ME, Dennison JE, Thomas RS, Conolly RB. New directions in incidence-dose modeling. Trends Biotechnol. 2005;23:122–127. doi: 10.1016/j.tibtech.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Andersen ME, Yang RSH, French CT, Chubb LS, Dennison JE. Molecular circuits, biological switches, and nonlinear dose-response relationships. Environ. Health Perspect. 2002;110:971–978. doi: 10.1289/ehp.02110s6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli D, Ferrell JE, Sontag ED. Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feed back systems. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1822–1827. doi: 10.1073/pnas.0308265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagowski CP, Ferrell JE. Bistabillity in the JNK cascade. Curr. Biol. 2001;11:1176–1182. doi: 10.1016/s0960-9822(01)00330-x. [DOI] [PubMed] [Google Scholar]
- Becskei A, Seraphin B, Serrano L. Positive feedback in eukaryotic gene networks: cell differentiation by graded to binary response conversion. EMBO J. 2001;20:2528–2535. doi: 10.1093/emboj/20.10.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Zhang Q, Andersen ME. Bistable signaling motifs and cell fate decisions. In: Krishnan K, Andersen ME, editors. Quantitative Modeling in Toxicology. Hoboken, NJ: John Wiley & Sons; Forthcoming. [Google Scholar]
- Birnbaum LS. The mechanism of dioxin toxicity—relationship to risk assessment. Environ. Health Perspect. 1994;102:157–167. doi: 10.1289/ehp.94102s9157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JR, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Ferrell JE, Li R, Meyer T. Interlinked fast and slow positive feedback loops drive reliable cell decisions. Science. 2005;310:496–498. doi: 10.1126/science.1113834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calame KL. Plasma cells: Finding new light at the end of B cell development. Nat. Immunol. 2001;2:1103–1108. doi: 10.1038/ni1201-1103. [DOI] [PubMed] [Google Scholar]
- Calame KL, Lin KI, Tunyaplin C. Regulatory mechanisms that determine the development and function of plasma cells. Annu. Rev. Immunol. 2003;21:205–230. doi: 10.1146/annurev.immunol.21.120601.141138. [DOI] [PubMed] [Google Scholar]
- Carotta S, Nutt SL. Losing B cell identity. Bioessays. 2008;30:203–207. doi: 10.1002/bies.20725. [DOI] [PubMed] [Google Scholar]
- Chang HH, Oh PY, Ingber DE, Huang S. Multistable and multistep dynamics in neutrophil differentiation. BMC Cell Biol. 2006;7:11. doi: 10.1186/1471-2121-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LF, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Chickarmane V, Enver T, Peterson C. Computational modeling of the hematopoietic erythroid-myeloid switch reveals insights into cooperativity, priming, and irreversibility. PLoS Comput. Biol. 2009;5:e1000268. doi: 10.1371/journal.pcbi.1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chickarmane V, Troein C, Nuber UA, Sauro HM, Peterson C. Transcriptional dynamics of the embryonic stem cell switch. PLoS Comput. Biol. 2006;2:1080–1092. doi: 10.1371/journal.pcbi.0020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Han S, Yokota H, Cho KH. Coupled positive feedbacks provoke slow induction plus fast switching in apoptosis. FEBS Lett. 2007;581:2684–2690. doi: 10.1016/j.febslet.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Cobaleda C, Busslinger M. Developmental plasticity of lymphocytes. Curr. Opin. Immunol. 2008;20:139–148. doi: 10.1016/j.coi.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Cobaleda C, Jochum W, Busslinger M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature. 2007;449:473–477. doi: 10.1038/nature06159. [DOI] [PubMed] [Google Scholar]
- Crawford RB, Holsapple MP, Kaminski NE. Leukocyte activation induces aryl hydrocarbon receptor up-regulation, DNA binding, and increased Cyp1a1 expression in the absence of exogenous ligand. Mol. Pharmacol. 1997;52:921–927. doi: 10.1124/mol.52.6.921. [DOI] [PubMed] [Google Scholar]
- Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity. 2006;24:269–281. doi: 10.1016/j.immuni.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Dooley RK, Holsapple MP. Elucidation of cellular targets responsible for tetrachlorodibenzo-p-dioxin (TCDD)-induced suppression of antibody responses: I. The role of the B lymphocyte. Immunopharmacology. 1988;16:167–180. doi: 10.1016/0162-3109(88)90005-7. [DOI] [PubMed] [Google Scholar]
- Eberhard D, Jimenez G, Heavey B, Busslinger M. Transcriptional repression by Pax5 (BSAP) through interaction with corepressors of the Groucho family. EMBO J. 2000;19:2292–2303. doi: 10.1093/emboj/19.10.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE. How regulated protein translocation can produce switch-like responses. Trends Biochem. Sci. 1998;23:461–465. doi: 10.1016/s0968-0004(98)01316-4. [DOI] [PubMed] [Google Scholar]
- Ferrell JE. Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr. Opin. Cell Biol. 2002;14:140–148. doi: 10.1016/s0955-0674(02)00314-9. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr Feedback regulation of opposing enzymes generates robust, all-or-none bistable responses. Curr. Biol. 2008;18:R244–R245. doi: 10.1016/j.cub.2008.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE, Xiong W. Bistability in cell signaling: how to make continuous processes discontinuous, and reversible processes irreversible. Chaos. 2001;11:227–236. doi: 10.1063/1.1349894. [DOI] [PubMed] [Google Scholar]
- Fiering S, Whitelaw E, Martin DI. To be or not to be active: the stochastic nature of enhancer action. Bioessays. 2000;22:381–387. doi: 10.1002/(SICI)1521-1878(200004)22:4<381::AID-BIES8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Fujita N, Jaye DL, Geigerman C, Akyildiz A, Mooney MR, Boss JM, Wade PA. MTA3 and the Mi-2/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell. 2004;119:75–86. doi: 10.1016/j.cell.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- Gillespie DT. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 1976;22:403–434. [Google Scholar]
- Gyory I, Wu J, Fejer G, Seto E, Wright KL. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat. Immunol. 2004;5:299–308. doi: 10.1038/ni1046. [DOI] [PubMed] [Google Scholar]
- Hanna J, Markoulaki S, Schorderet P, Carey BW, Beard C, Wernig M, Creyghton MP, Steine EJ, Cassady JP, Foreman R, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133:250–264. doi: 10.1016/j.cell.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HannaRose W, Hansen U. Active repression mechanisms of eukaryotic transcription repressors. Trends Genet. 1996;12:229–234. doi: 10.1016/0168-9525(96)10022-6. [DOI] [PubMed] [Google Scholar]
- Holden C, Vogel G. Cell biology: a seismic shift for stem cell research. Science. 2008;319:560–563. doi: 10.1126/science.319.5863.560. [DOI] [PubMed] [Google Scholar]
- Holsapple MP, Snyder NK, Wood SC, Morris DL. A review of 2,3,7,8-tetrachlorodibenzo-para-dioxin-induced changes in immunocompetence—1991 update. Toxicology. 1991;69:219–255. doi: 10.1016/0300-483x(91)90184-3. [DOI] [PubMed] [Google Scholar]
- Horcher M, Souabni A, Busslinger M. Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity. 2001;14:779–790. doi: 10.1016/s1074-7613(01)00153-4. [DOI] [PubMed] [Google Scholar]
- Huang S, Guo YP, May G, Enver T. Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev. Biol. 2007;305:695–713. doi: 10.1016/j.ydbio.2007.02.036. [DOI] [PubMed] [Google Scholar]
- Igarashi K, Ochiai K, Muto A. Architecture and dynamics of the transcription factor network that regulates B-cell to plasma cell differentiation. J. Biochem. (Tokyo) 2007;141:783–789. doi: 10.1093/jb/mvm106. [DOI] [PubMed] [Google Scholar]
- Johnson BL, DeRosa C. The toxicologic hazard of superfund hazardous-waste sites. Rev. Environ. Health. 1997;12:235–251. doi: 10.1515/reveh.1997.12.4.235. [DOI] [PubMed] [Google Scholar]
- Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. TLR signaling. Semin. Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Kim J-R, Yoon Y, Cho K-H. Coupled feedback loops form dynamic motifs of cellular networks. Biophys. J. 2008;94:359–365. doi: 10.1529/biophysj.107.105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol. Cell. Biol. 2002;22:4771–4780. doi: 10.1128/MCB.22.13.4771-4780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KI, Tunyaplin C, Calame K. Transcriptional regulatory cascades controlling plasma cell differentiation. Immunol. Rev. 2003;194:19–28. doi: 10.1034/j.1600-065x.2003.00040.x. [DOI] [PubMed] [Google Scholar]
- Lin Y, Wong KK, Calame K. Repression of c-myc transcription by blimp-1, an inducer of terminal B cell differentiation. Science. 1997;276:596–599. doi: 10.1126/science.276.5312.596. [DOI] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang WW, Chen X, Bourque G, George J, Leong B, Liu J, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Manz RA, Lohning M, Cassese G, Thiel A, Radbruch A. Survival of long-lived plasma cells is independent of antigen. Int. Immunol. 1998;10:1703–1711. doi: 10.1093/intimm/10.11.1703. [DOI] [PubMed] [Google Scholar]
- Marcus RS, Holsapple MP, Kaminski NE. Lipopolysaccharide activation of murine splenocytes and splenic B cells increased the expression of aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator. J. Pharmacol. Exp. Ther. 1998;287:1113–1118. [PubMed] [Google Scholar]
- McGrath LF, Cooper KR, Georgopoulos P, Gallo MA. Alternative models for low dose-response analysis of biochemical and immunological end-points for tetrachlorodibenzo-p-dioxin. Regul. Toxicol. Pharmacol. 1995;21:382–396. doi: 10.1006/rtph.1995.1053. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams LJ, Driver DJ, McHeyzer-Williams MG. Germinal center reaction. Curr. Opin. Hematol. 2001;8:52–59. doi: 10.1097/00062752-200101000-00010. [DOI] [PubMed] [Google Scholar]
- Milili M, Gauthier L, Veran J, Mattei MG, Schiff C. A new Groucho TLE4 protein may regulate the repressive activity of Pax5 in human B lymphocytes. Immunology. 2002;106:447–455. doi: 10.1046/j.1365-2567.2002.01456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Jacob F. General conclusions—teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harbor Symp. Quant. Biol. 1961;26:389. doi: 10.1101/sqb.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- Mora-Lopez F, Reales E, Brieva JA, Campos-Caro A. Human BSAP and BLIMP1 conform an autoregulatory feedback loop. Blood. 2007;110:3150–3157. doi: 10.1182/blood-2007-05-092262. [DOI] [PubMed] [Google Scholar]
- NAS/NRC. Toxicity Testing in the 21st Century: A Vision and a Strategy. Washington, DC: NAS Press; 2007. [Google Scholar]
- Nera KP, Kohonen P, Narvi E, Peippo A, Mustonen L, Terho P, Koskela K, Buerstedde JM, Lassila O. Loss of Pax5 promotes plasma cell differentiation. Immunity. 2006;24:283–293. doi: 10.1016/j.immuni.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H. How is pluripotency determined and maintained? Development. 2007;134:635–646. doi: 10.1242/dev.02787. [DOI] [PubMed] [Google Scholar]
- Nutt SL. B-cell identity—commitment is not forever. N. Engl. J. Med. 2008;358:82–83. doi: 10.1056/NEJMcibr0707112. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Morrison AM, Dorfler P, Rolink A, Busslinger M. Identification of BSAP (Pax-5) target genes in early B-cell development by loss- and gain-of-function experiments. EMBO J. 1998;17:2319–2333. doi: 10.1093/emboj/17.8.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo Y, Arima M, Arguni E, Okada S, Yamashita K, Asari S, Obata S, Sakamoto A, Hatano M, O-Wang J, et al. A role for c-fos/activator protein 1 in B lymphocyte terminal differentiation. J. Immunol. 2005;174:7703–7710. doi: 10.4049/jimmunol.174.12.7703. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbudak EM, Thattai M, Lim HN, Shraiman BI, van Oudenaarden A. Multistability in the lactose utilization network of Escherichia coli. Nature. 2004;427:737–740. doi: 10.1038/nature02298. [DOI] [PubMed] [Google Scholar]
- Palani S, Sarkar CA. Positive receptor feedback during lineage commitment can generate ultrasensitivity to ligand and confer robustness to a bistable switch. Biophys. J. 2008;95:1575–1589. doi: 10.1529/biophysj.107.120600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskurich JF, Lin KI, Lin Y, Wang Y, Ting JPY, Calame K. BLIMP-I mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat. Immunol. 2000;1:526–532. doi: 10.1038/82788. [DOI] [PubMed] [Google Scholar]
- Poland A, Knutson JC. 2,3,7,8-Tetrachlorodibenzo-para-dioxin and related halogenated aromatic-hydrocarbons—examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- Reljic R, Wagner SD, Peakman LJ, Fearon DT. Suppression of signal transducer and activator of transcription 3-dependent B lymphocyte terminal differentiation by BCL-6. J. Exp. Med. 2000;192:1841–1847. doi: 10.1084/jem.192.12.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Chee KJ, Kim TH, Maniatis T. PRDI-BF1/Blimp-1 repression is mediated by corepressors of the Groucho family of proteins. Genes Dev. 1999;13:125–137. doi: 10.1101/gad.13.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkenberger JL, Wallin JJ, Johnson KW, Koshland ME. An interleukin-2 signal relieves BSAP (Pax5)-mediated repression of the immunoglobulin J chain gene. Immunity. 1996;5:377–386. doi: 10.1016/s1074-7613(00)80263-0. [DOI] [PubMed] [Google Scholar]
- Roeder I, Glauche I. Towards an understanding of lineage specification in hematopoietic stem cells: a mathematical model for the interaction of transcription factors GATA-1 and PU.1. J. Theor. Biol. 2006;241:852–865. doi: 10.1016/j.jtbi.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Rowlands JC, Gustafsson JA. Aryl hydrocarbon receptor-mediated signal transduction. Crit. Rev. Toxicol. 1997;27:109–134. doi: 10.3109/10408449709021615. [DOI] [PubMed] [Google Scholar]
- Scadden DT. The weight of cell identity. J. Clin. Invest. 2007;117:3653–3655. doi: 10.1172/JCI34181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliephake DE, Schimpl A. Blimp-1 overcomes the block in IgM secretion in lipopolysaccharide anti-mu F(ab′)(2)-co-stimulated B lymphocytes. Eur. J. Immunol. 1996;26:268–271. doi: 10.1002/eji.1830260142. [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu. Rev. Cell Dev. Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- Schneider D, Manzan MA, Crawford RB, Chen W, Kaminski NE. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated impairment of B cell differentiation involves dysregulation of paired box 5 (Pax5) isoform, Pax5a. J. Pharmacol. Exp. Ther. 2008;326:463–474. doi: 10.1124/jpet.108.139857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D, Manzan MA, Yoo BS, Crawford RB, Kaminski N. Involvement of Blimp-1 and AP-1 dysregulation in the 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated suppression of the IgM response by B Cells. Toxicol. Sci. 2009;108:377–388. doi: 10.1093/toxsci/kfp028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciammas R, Davis MM. Modular nature of Blimp-1 in the regulation of gene expression during B cell maturation. J. Immunol. 2004;172:5427–5440. doi: 10.4049/jimmunol.172.9.5427. [DOI] [PubMed] [Google Scholar]
- Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006;25:225–236. doi: 10.1016/j.immuni.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Peng A, Schlissel MS. In vivo occupancy of the kappa light chain enhancers in primary pro- and pre-B cells: a model for kappa locus activation. Immunity. 1997;6:131–143. doi: 10.1016/s1074-7613(00)80420-3. [DOI] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Calame K. Plasma cell differentiation and multiple myeloma. Curr. Opin. Immunol. 2004;16:226–234. doi: 10.1016/j.coi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005;5:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19:607–620. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Singh M, Birshtein BK. Concerted repression of an immunoglobulin heavy-chain enhancer, 3′alpha E(hs1,2) Proc. Natl. Acad. Sci. U.S.A. 1996;93:4392–4397. doi: 10.1073/pnas.93.9.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifka MK, Ahmed R. Long-lived plasma cells: A mechanism for maintaining persistent antibody production. Curr. Opin. Immunol. 1998;10:252–258. doi: 10.1016/s0952-7915(98)80162-3. [DOI] [PubMed] [Google Scholar]
- Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- Staudt LM. Negative feedback for B cells. Nature. 2004;431:919–920. doi: 10.1038/431919a. [DOI] [PubMed] [Google Scholar]
- Stevens EA, Mezrich JD, Bradfield CA. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology. 2009;127:299–311. doi: 10.1111/j.1365-2567.2009.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strogatz S. Nonlinear Dynamics and Chaos: With Applications to Physics, Biology, Chemistry, and Engineering (Studies in Nonlinearity) Cambridge, MA: Perseus Books Group; 2001. [Google Scholar]
- Suh J, Jeon YJ, Kim HM, Kang JS, Kaminski NE, Yang KH. Aryl hydrocarbon receptor-dependent inhibition of AP-1 activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin in activated B cells. Toxicol. Appl. Pharmacol. 2002;181:116–123. doi: 10.1006/taap.2002.9403. [DOI] [PubMed] [Google Scholar]
- Sulentic CE, Holsapple MP, Kaminski NE. Aryl hydrocarbon receptor-dependent suppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin of IgM secretion in activated B cells. Mol. Pharmacol. 1998;53:623–629. [PubMed] [Google Scholar]
- Sulentic CE, Holsapple MP, Kaminski NE. Putative link between transcriptional regulation of IgM expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin and the aryl hydrocarbon receptor/dioxin-responsive enhancer signaling pathway. J. Pharmacol. Exp. Ther. 2000;295:705–716. [PubMed] [Google Scholar]
- Sveiczer A, Tyson JJ, Novak B. Modelling the fission yeast cell cycle. Brief Funct. Genomic. Proteomic. 2004;2:298–307. doi: 10.1093/bfgp/2.4.298. [DOI] [PubMed] [Google Scholar]
- Thiel G, Lietz M, Hohl M. How mammalian transcriptional repressors work. Eur. J. Biochem. 2004;271:2855–2862. doi: 10.1111/j.1432-1033.2004.04174.x. [DOI] [PubMed] [Google Scholar]
- Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J. Immunol. 2004;173:1158–1165. doi: 10.4049/jimmunol.173.2.1158. [DOI] [PubMed] [Google Scholar]
- Turner CA, Jr, Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell. 1994;77:297–306. doi: 10.1016/0092-8674(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Tyson JJ, Chen K, Novak B. Network dynamics and cell physiology. Nat. Rev. Mol. Cell Biol. 2001;2:908–916. doi: 10.1038/35103078. [DOI] [PubMed] [Google Scholar]
- Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 2003;15:221–231. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- Tyson JJ, Csikasz-Nagy A, Novak B. The dynamics of cell cycle regulation. Bioessays. 2002;24:1095–1109. doi: 10.1002/bies.10191. [DOI] [PubMed] [Google Scholar]
- Tyson JJ, Novak B. Regulation of the eukaryotic cell cycle: molecular antagonism, hysteresis, and irreversible transitions. J. Theor. Biol. 2001;210:249–263. doi: 10.1006/jtbi.2001.2293. [DOI] [PubMed] [Google Scholar]
- Ullah M, Wolkenhauer O. Stochastic approaches in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009 doi: 10.1002/wsbm.78. Advance access published on December 10, 2009; doi:10.1002/wsbm.078. [DOI] [PubMed] [Google Scholar]
- Vasanwala FH, Kusam S, Toney LM, Dent AL. Repression of AP-1 function: a mechanism for the regulation of Blimp-1 expression and B lymphocyte differentiation by the B cell lymphoma-6 protooncogene. J. Immunol. 2002;169:1922–1929. doi: 10.4049/jimmunol.169.4.1922. [DOI] [PubMed] [Google Scholar]
- Vogel G, Holden C. Developmental biology—field leaps forward with new stem cell advances. Science. 2007;318:1224–1225. doi: 10.1126/science.318.5854.1224. [DOI] [PubMed] [Google Scholar]
- Vorderstrasse BA, Andrea ABB, Lawrence BP. Examining the relationship between impaired host resistance and altered immune function in mice treated with TCDD. Toxicology. 2003;188:15–28. doi: 10.1016/s0300-483x(02)00749-7. [DOI] [PubMed] [Google Scholar]
- Vorderstrasse BA, Steppan LB, Silverstone AE, Kerkvliet NI. Aryl hydrocarbon receptor-deficient mice generate normal immune responses to model antigens and are resistant to TCDD-induced immune suppression. Toxicol. Appl. Pharmacol. 2001;171:157–164. doi: 10.1006/taap.2000.9122. [DOI] [PubMed] [Google Scholar]
- Wang L, Walker BL, Iannaccone S, Bhatt D, Kennedy PJ, Tse WT. Bistable switches control memory and plasticity in cellular differentiation. Proc. Natl. Acad. Sci. U. S. A. 2009;106:6638–6643. doi: 10.1073/pnas.0806137106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welner RS, Pelayo R, Kincade PW. Evolving views on the genealogy of B cells. Nat. Rev. Immunol. 2008;8:95–106. doi: 10.1038/nri2234. [DOI] [PubMed] [Google Scholar]
- Wilkinson DJ. Stochastic modelling for quantitative description of heterogeneous biological systems. Nat. Rev. Genet. 2009;10:122–133. doi: 10.1038/nrg2509. [DOI] [PubMed] [Google Scholar]
- Xie HF, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- Xiong W, Ferrell JE. A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature. 2003;426:460–465. doi: 10.1038/nature02089. [DOI] [PubMed] [Google Scholar]
- Yoo BS, Boverhof DR, Shnaider D, Crawford RB, Zacharewski TR, Kaminski NE. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the regulation of Pax5 in lipopolysaccharide-activated B cells. Toxicol. Sci. 2004;77:272–279. doi: 10.1093/toxsci/kfh013. [DOI] [PubMed] [Google Scholar]
- Yu J, Angelin-Duclos C, Greenwood J, Liao J, Calame K. Transcriptional repression by Blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol. Cell. Biol. 2000;20:2592–2603. doi: 10.1128/mcb.20.7.2592-2603.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Andersen ME, Conolly RB. Binary gene induction and protein expression in individual cells. Theor. Biol. Med. Model. 2006;3:18. doi: 10.1186/1742-4682-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Bhattacharya S, Woods CG, Andersen ME. Ultrasensitive response motifs in biochemical networks. In: Krishnan K, Andersen ME, editors. Quantitative Modeling in Toxicology. Hoboken, NJ: John Wiley & Sons; Forthcoming. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.