

Abstract
Atrial fibrillation (AF) is the most common cardiac arrhythmia, and it causes substantial mortality. The autonomic nervous system, and particularly the adrenergic/cholinergic balance, has a profound influence on the occurrence of AF. Adrenergic stimulation from catecholamines can cause AF in patients. In human atrium, catecholamines can affect each of the electrophysiological mechanisms of AF initiation and/or maintenance. Catecholamines may produce membrane potential oscillations characteristic of afterdepolarisations, by increasing Ca2+ current, [Ca2+]i and consequent Na+-Ca2+ exchange, and may also enhance automaticity. Catecholamines might affect reentry, by altering excitability or conduction, rather than action potential terminal repolarisation or refractory period. However, which arrhythmia mechanisms predominate is unclear, and likely depends on cardiac pathology and adrenergic tone. Heart failure (HF), a major cause of AF, causes adrenergic activation and adaptational changes, remodelling, of atrial electrophysiology, Ca2+ homeostasis and adrenergic responses. Chronic AF also remodels these, but differently to HF. Myocardial infarction, and AF, cause neural remodelling that also may promote AF. β-adrenoceptor antagonists (β-blockers) are used in the treatment of AF, mainly to control the ventricular rate, by slowing AV conduction. β-blockers also reduce the incidence of AF, particularly in HF or after cardiac surgery, when adrenergic tone is high. Furthermore, the chronic treatment of patients with β-blockers remodels the atria, with a potentially anti-arrhythmic increase in the refractory period. Therefore, the suppression of AF by β-blocker treatment may involve an attenuation of arrhythmic activity that is caused by increased [Ca2+]i, coupled with effects of adaptation to the treatment. An improved understanding of the involvement of the adrenergic system and its control in basic mechanisms of AF under differing cardiac pathologies might lead to better treatments.
Keywords: Atrial fibrillation, Adrenergic, Catecholamine, Beta-blocker, Arrhythmia mechanism, Calcium, Action potential, Remodeling
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia, with an estimated prevalence in the general population of 0.4-1% (Fuster et al. 2006). AF affects primarily the elderly, e.g., 4.6% of people aged >65 years, and 7.1% of those >85 years (Murphy et al. 2007). Symptoms include palpitations, dizziness, fatigue, chest pain and dyspnoea. Moreover, AF substantially increases the risk of stroke, heart failure (HF) and death (Fuster et al. 2006). Pharmacological therapy is the mainstay of treatment. However, current anti-arrhythmic drugs for AF prevention have limited efficacy and considerable potential for adverse effects. The autonomic nervous system has a profound influence on the occurrence of AF, and a predominance of activity of either the adrenergic (sympathetic) (Dimmer et al. 1998) or cholinergic (parasympathetic) (Bettoni et al. 2002) branches can promote AF. Furthermore, AF may be generated and maintained by a variety of electrophysiological mechanisms (Workman et al. 2008), and a change in autonomic activity is expected to affect each differently. This review focuses on the effects of adrenergic stimulation and antagonism on the electrophysiological mechanisms of AF, and on their modulation by atrial remodelling from cardiac disease and drug treatment. The effects of adrenergic modulation are wide ranging, complex and species- and cardiac chamber-dependent, so studies of human atrium are highlighted where available.
The cardiac adrenergic system
The cardiac adrenergic system comprises adrenergic nerves, hormones and receptors. Pre-ganglionic adrenergic neurons of the spinal cord, sympathetic trunk, cervical ganglia and cardiac plexuses synapse with post-ganglionic neurons. Those penetrate the myocardium along coronary arterial pathways, terminating on cardiac myocytes and vessels of all chambers (Kawashima 2005). Adrenergic stimulation results in the release of the adrenergic hormones, catecholamines, including noradrenaline (norepinephrine) from post-ganglionic nerve terminals, and adrenaline (epinephrine) from the adrenal medulla. Catecholamines bind to and activate cell surface adrenoceptors, including the α, β1, β2 and β3 subtypes, all of which are present in human atrium (Mary-Rabine et al. 1978; Hedberg et al. 1985; Chamberlain et al. 1999). Their independent stimulation has complex and sometimes opposing effects on atrial function, and the net response to catecholamine stimulation depends on catecholamine type and relative adrenoceptor subtype density and sensitivity, which may vary with disease.
What is the evidence for involvement of the adrenergic system in AF?
In patients, heart rate variability studies indicated an increased level of adrenergic, relative to cholinergic, activity in the minutes preceding the onset of AF (Dimmer et al. 1998; Coccagna et al. 1997). However, increased cholinergic activity, perhaps following increased adrenergic activity (Bettoni et al. 2002) also generated AF. Infusion of isoprenaline (isoproterenol, ISO), a mixed β-receptor agonist, produced AF in 5% of patients with no history of AF, and in 84% of patients with paroxysmal AF, in a dose-dependent manner (Oral et al. 2008). In dogs, adrenaline (Sharifov et al. 2004) or ISO (Kiss et al. 2004; Sharifov et al. 2004) produced AF, and ISO facilitated acetylcholine-induced AF (Sharifov et al. 2004). Electrical stimulation of pulmonary vein (PV) autonomic nerves produced rapid PV arrhythmic activation, abolished by the β1-antagonist (β1-blocker) atenolol (Patterson et al. 2005). Furthermore, increased atrial adrenergic innervation was associated with chronic AF in patients (Gould et al. 2006). Propranolol (a mixed β-blocker) reduced the incidence of burst pacing-induced sustained atrial tachyarrhythmia (AT) associated with HF in dogs (Stambler et al. 2003). Moreover, several β-blockers are effective in suppressing AF in patients with various heart diseases (Lopez-Sendon et al. 2004). It is reasonable to infer, therefore, that adrenergic stimulation may be involved in the initiation and/or maintenance of AF. However, it is unclear how adrenergic stimulation or antagonism affect the various electrophysiological mechanisms of AF or, indeed, which of these mechanisms predominate.
Electrophysiological mechanisms of AF initiation and maintenance
The majority of atrial premature beats that trigger (initiate) AF originate from focal (focussed) ectopic (away from the sinoatrial (SA) node) electrical activity in or near the PVs. For example, in patients with drug-resistant paroxysmal AF, 94% of ectopic foci that initiated spontaneous AF were located, using multi-electrode catheter mapping, to the PVs (Haissaguerre et al. 1998). The electrophysiological mechanism of the trigger is often considered to be either abnormal automaticity (AA) or triggered activity. AA is the premature firing of action potentials due to increased pacemaking, i.e. accelerated diastolic, phase 4, membrane depolarisation. It is favoured by an increase in diastolic inward, depolarising, ion currents such as the funny current (If) and potentially the L-type Ca2+ (ICaL) and Na+-Ca2+ exchanger (INa/Ca) currents. It is also favoured by a decrease in outward, hyperpolarising, currents such as the delayed rectifier K+ currents, of which there are various types, having either ultra-rapid (IKur), rapid (IKr) or slow (IKs) activation kinetics. Triggered activity is premature firing due to oscillations in membrane potential following an action potential upstroke, i.e. afterdepolarisations. Those may be delayed (DADs), which occur after full repolarisation. They are favoured by high heart rates and excessive intracellular Ca2+ (Ca2+i) loading. That can result in a transient inward current carried by INa/Ca, as 1 Ca2+ is extruded in exchange for 3 Na+. Alternatively, they may be early afterdepolarisations (EADs), which occur during repolarisation. Those are favoured by low heart rates, increases in the action potential duration (APD) in the voltage range at which ICaL can reactivate, or by abnormalities in INa activation or inactivation. Once initiated, AF may be maintained by single or multiple wavelet intra-mural reentry. Reentry is rapid circuitous activation caused by unidirectional conduction block. It is favoured by premature beats, increased spatial heterogeneity of electrical activity, and a decrease in the circuit length, the wavelength. For example, a premature impulse may divide at a functional or anatomical obstacle, with one wavefront being blocked at refractory, i.e., inexcitable tissue. The other may activate adjacent tissue with a short effective refractory period (ERP), circulate the obstacle and reenter the previously inexcitable tissue provided that the wavelength, the product of ERP and conduction velocity, is shorter than the available conduction path (Workman et al. 2008). A reduced ERP may result from early availability of the Na+ channel current (INa) due, e.g., to shortening of the terminal phase of repolarisation of the action potential. Slowed conduction may result from decreased excitability, increased intercellular fibrosis or reduced intercellular gap junction current (Igap). Myocardial tissue capable of supporting reentry is sometimes termed a reentrant “substrate”, i.e., having structural or functional characteristics predisposing to reentry, perhaps resulting from pathological remodelling. It should be noted, however, that a pathological predisposition to sustained non-reentrant activity (AA and/or triggered activity) could also constitute, or contribute to, a substrate for AF. Furthermore, the focal trigger of AF in the PVs could be either micro-reentrant, triggered or abnormally automatic (Haissaguerre et al. 1998). Whether reentrant or non-reentrant mechanisms predominate is under debate (Wit et al. 2007), and likely depends on, e.g., the underlying cardiac pathology and autonomic tone. The effects of changes in adrenergic activity on each of the reentrant and non-reentrant mechanisms should, therefore, be considered when attempting to understand how adrenergic modulation affects the initiation and maintenance of AF.
Effects of adrenergic stimulation on pacemaking
In the natural pacemaker, the SA node, adrenergic stimulation accelerates phase 4, shortening diastole, thus increasing heart rate. Multiple ion current mechanisms have been proposed. In rabbit SA node, β-stimulation increased If, associated with a positive shift in its activation voltage (Barbuti et al. 2007), and accelerated the deactivation of IKr and IKS (Lei et al. 2000); each potentially accelerating phase 4. β-stimulation also increases SA nodal ICaL and peak [Ca2+]i, potentially enhancing phase 4 via increased INa/Ca (Bers 2008). The atrium is not normally a pacemaker, although isolated tissues obtained from patients undergoing cardiac surgery may activate spontaneously. Right atrial (RA) pectinate muscles sometimes featured a “single pacemaker”, and adrenaline transiently decreased, then markedly increased, its rate (Trautwein et al. 1962). Specialised conducting fibres from human RA were identified by their pacemaker activity after ceasing stimulation, and adrenaline increased phase 4 and automatic rate (Mary-Rabine et al. 1978). A similar effect of adrenergic stimulation was observed in other human studies (Mary-Rabine et al. 1980; Levi et al. 1981), as well as in healthy animal atria (Davis 1975; Ono et al. 1995). Specialised pacemaker-type cells, similar to those normally found in SA node, have been identified in patients’ PVs (Perez-Lugones et al. 2003). Pacemaker activity was found in rabbit PV isolated cells, which was enhanced by ISO (Chen et al. 2002). However, it is unclear whether pacemaking or AA occur in either PV or atrium in-vivo, or whether such activity could arise from adrenergic stimulation.
Can adrenergic stimulation cause afterdepolarisations in human atrium?
Adrenergic stimulation produces several types of arrhythmic electrical activity in human atrial isolated tissues or cells, that are distinct from the production of regular automaticity or the acceleration of automatic rate. In unstimulated, quiescent RA muscle strips, adrenaline produced “bizarre” action potentials of “various shapes, durations and frequencies”, some with “distinct second spikes” (Sleator et al. 1964) (Figure 1a). ISO produced “DADs” or “triggered activity” in unstimulated RA “specialized fibers” (Wang et al. 2006). Adrenaline produced “DADs” immediately after cessation of physiological-rate stimulation in RA trabeculae (Yeh et al. 1992). In RA isolated cells stimulated at physiological rate, ISO caused action potentials to be interrupted frequently by low amplitude, usually sub-threshold, transient depolarisations, termed “cellular arrhythmic depolarisations” (CADs) (Redpath et al. 2006) (Figure 1b). They could occur after repolarisation, during repolarisation or after a CAD, and may represent DADs, EADs or AA. Adrenergic stimulation also produced “arrhythmic contractions” in human RA appendages (Kaumann et al. 1993), although electrical activity was not studied. Therefore, adrenergic stimulation produced arrhythmic electrical activity in each of four studies that could be found on human atrial isolated tissues or cells. For such activity to qualify as a DAD, its amplitude should increase and its coupling interval decrease in proportion with an increasing stimulation rate (Wit et al. 2007), as demonstrated in canine atria with adrenaline (Johnson et al. 1986). However, in none of these human atrial studies were potential DADs qualified in that way. Which type of non-reentrant activity is produced by adrenergic stimulation in human atrium remains unclear.
Figure 1. Arrhythmic electrical activity produced by adrenergic stimulation in human atrium.
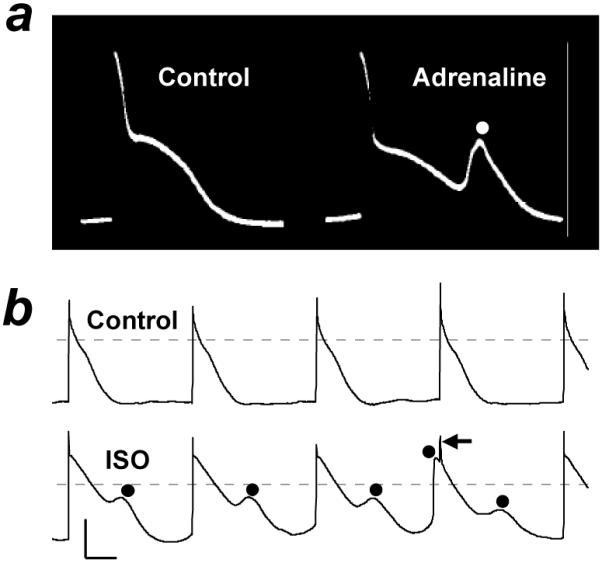
Action potentials recorded from human right atrial isolated tissue (a) and myocyte (b) in the absence (control) or presence of adrenaline or isoprenaline (ISO). Myocyte stimulated at 75 beats/min. Circles: spontaneous depolarisations; one occurring just before stimulus spike (←). Calibration bars: 50 mV (vertical), 200 ms (horizontal). Based on data in (Sleator et al. 1964) (a) and (Redpath et al. 2006) (b) with permission from American Physiological Society, and Elsevier, respectively.
Can adrenergic stimulation affect atrial reentry?
Reported effects of adrenergic stimulation on atrial reentry wavelength are rather discordant. In the absence of arrhythmias, wavelength change has been assessed by measuring ERP and conduction velocity. In patients, intravenous ISO increased wavelength, associated with suppression of extrastimuli-induced repetitive atrial firing, by causing a marked increase in conduction velocity and only a small change in ERP (Shimizu et al. 1994a). However, an absence of change in atrial wavelength in dogs (Rensma et al. 1988), or an increase due to ERP-prolongation in rabbits (Smeets et al. 1986) also were reported. In patients with atrial flutter, ISO decreased the arrhythmia cycle length, primarily by slowing conduction, thus potentially promoting reentry (Stambler et al. 1996). ISO also sustained reentrant circuits in PVs, but without changing conduction velocity (Arora et al. 2003). In diseased atria that are partially depolarised, adrenergic stimulation might inhibit reentry, by increasing excitability and overcoming conduction block. For example, in tissues from patients with RA enlargement, slow-rate ectopic foci fired independently of the pacing stimuli, revealing conduction block between driven and ectopic cells. Adrenaline hyperpolarised the maximum diastolic potential (MDP), and abolished the ectopic foci by electrically connecting them to the driven cells (Gelband et al. 1977). However, since adrenergic stimulation increases ICaL, this could potentially promote reentry in tissues which remain sufficiently depolarised to keep INa inactive, by generating slowly conducted Ca2+-dependent action potentials. Atrial reentry and AF can result from wavelength decrease associated with reduction of ERP, as demonstrated with acetylcholine (Rensma et al. 1988). There are few studies of effects of adrenergic stimulation on human atrial ERP, with a report of a small (5%) decrease (Shimizu et al. 1994a) and of no change (Redpath et al. 2006). There are several studies, however, of effects of adrenergic stimulation on human atrial action potential terminal repolarisation, a major determinant of the ERP.
Effects of adrenergic stimulation on human atrial action potentials
Adrenergic stimulation is generally considered to shorten myocardial terminal repolarisation, as shown in canine ventricle, with a reduction in the APD at 95% repolarisation (APD95) in isolated tissues (Volders et al. 2003) and cells (Stengl et al. 2006) (Figure 2ai). In human ventricle, however, adrenergic stimulation may lengthen, rather than shorten, terminal repolarisation, since in isolated tissues or cells, APD95 was markedly prolonged by noradrenaline or ISO in three of four available studies (Eckel et al. 1982; Mitchell et al. 1986; Koumi et al. 1995) (Figure 2aii), and by adrenaline in two of three tissues in the other study (Yeh et al. 1992). It should be noted, however, that these preparations were from patients who were undergoing cardiac surgery, and the associated myocardial diseases and drug treatments could affect the responses to adrenergic stimulation. In undiseased human ventricular tissues, adrenaline had no effect on repolarisation already prolonged by dofetilide (Jost et al. 2005), but effects of adrenergic stimulation alone were not studied. In human atrium, by contrast with ventricle, terminal repolarisation is usually unaffected by adrenergic stimulation. Of the six available reports in isolated tissues or cells, five showed no significant effect of adrenaline or ISO on APD90 (Sleator et al. 1964; Gelband et al. 1977; Kecskemeti et al. 1985; Yeh et al. 1992; Redpath et al. 2006) (e.g. Figure 2aiii & iv), and one showed a marked increase in APD90 (Li et al. 1997). Species differences in effects of adrenergic stimulation on atrial terminal repolarisation should be noted, however, with shortening in dog (Liang et al. 1985; Yeh et al. 2007), and lengthening in guinea pig (Yoshimoto et al. 1998) and rat (Webb et al. 1956). The MDP and action potential maximum upstroke velocity, which can affect conduction velocity and hence reentry, also were unaffected by adrenergic stimulation, in normally polarised human atrial tissues (Gelband et al. 1977; Kecskemeti et al. 1985). However, the action potential plateau, phase 2, was markedly elevated by adrenergic stimulation in several reports, reflected by increased APD50 (Kecskemeti et al. 1985; Li et al. 1997; Redpath et al. 2006). For example, 50 nM ISO approximately doubled APD50 in human atrial cells (Redpath et al. 2006) (Figure 2aiv). The available human atrial studies, taken together, do not support an effect of adrenergic stimulation that could substantially affect reentry via action potential terminal repolarisation. However, the plateau elevation has the potential to promote afterdepolarisations, either by increasing APD in the voltage range at which ICaL can reactivate and thus cause EADs (Bers 2008), or by its association with increased Ca2+i loading and DADs. To understand the mechanisms by which adrenergic stimulation elevates the action potential plateau, without affecting terminal repolarisation, we need to know which ion currents are affected in human atrium by adrenergic stimulation, and also how these currents interact.
Figure 2. Species- and cardiac chamber-dependent effects of adrenergic stimulation on action potentials and ion currents.

a, Action potentials from dog (i) and human (ii) left ventricular myocyte, and from human right atrial tissue (iii) and myocyte (iv). C: control, ISO: isoprenaline, A: adrenaline, W: washout, HMR: HMR1556 (IKS blocker), ET: endothelin. Calibrations: 25 mV, 100 ms. Based on data in (Stengl et al. 2006) (i), (Koumi et al. 1995) (ii), (Yeh et al. 1992) (iii), and (Redpath et al. 2006) (iv) with permission from Oxford University Press, American Society for Clinical Investigation, S. Karger AG, Basel, and Elsevier, respectively. b, Simulated time courses of main human atrial ion currents (defined in text) determining action potential (top trace) shape. Ordinate scales equalised for all currents <0.5 pA/pF. All traces derived from mathematical model (Courtemanche et al. 1998) using CESE Pro 1.4.8 software (Simulogic Inc., Halifax, Canada). Arrows indicate reported effect of ISO on currents.
Effects of adrenergic stimulation on human atrial ion currents
The atrial action potential plateau amplitude is determined largely by the magnitude of ICaL (Figure 2b). This is an inward cationic, i.e. depolarising, current and its agonism and blockade elevate and lower the plateau, respectively, as shown using Bay K 8644 (Yue et al. 1997) or nifedipine (Workman et al. 2001). Adrenergic stimulation has consistently been shown to increase human atrial ICaL, often more than 2-fold, e.g. (Ouadid et al. 1995; Pelzmann et al. 1995; Li et al. 1997; Van Wagoner et al. 1999; Christ et al. 2004; Redpath et al. 2006; Greiser et al. 2007) (Figure 2b). The biophysical mechanism is an increase in the open probability of available ICaL channels, resulting from an increase in their open time and a decrease in their closed time. The signalling mechanism involves phosphorylation of ICaL channels by protein kinase A (PKA). That is activated by increased adenylyl cyclase-dependent cyclic AMP (cAMP), which results from the stimulation of GTP regulatory proteins due to the binding of catecholamines to β-receptors. ICaL may be moderately increased by high stimulation rates, due to slowing of current decay: so-called high frequency-induced upregulation (HFIUR). Since HFIUR was potentiated by ISO in human atrial cells (Piot et al. 1996), the increase in ICaL by adrenergic stimulation may be potentiated at the high atrial rates encountered during AF. The influence of ICaL on the action potential is normally balanced partly by the transient outward K+ current (ITO). However, ITO was unaffected by ISO in human atrium (Su et al. 1994) and should not, therefore, alter the influence of ICaL to elevate the plateau. Chloride currents (ICl) also may contribute to early repolarisation. However, ISO had inconsistent effects on human atrial ICl, with either an increase (Tsai et al. 2001) or no change (Sakai et al. 1995) in ICl.cAMP, or no change in ICl.swell (Sato et al. 1998). Reports of effects of adrenergic stimulation on other mechanosensitive currents (Stiber et al. 2009) in human atrium could not be found. The increased voltage resulting from the increase in ICaL may enhance the activation of delayed rectifier K+ currents, with a potential shortening influence on terminal repolarisation. However, the relative magnitudes, and contributions to repolarisation, of IKur, IKr and IKS in human atrium are incompletely understood. IKr and IKS were relatively small or present in only a minority of cells (Wang et al. 1994; Schaffer et al. 1998), but this might reflect disruption of these currents by the requisite “chunk” method of cell isolation (Yue et al. 1996), and in intact atrial tissue, IKr blockade may prolong APD90 (Wettwer et al. 2004). Adrenergic stimulation may increase both IKr (Heath et al. 2000) and IKS (Volders et al. 2003), but human atrial data are lacking. Altered plateau voltages resulting from changes in IKur also may affect terminal repolarisation via IKr and IKS (Wettwer et al. 2004), and adrenergic stimulation increased IKur in human atrium (Li et al. 1996; Su et al. 1994). Therefore, the net contribution of adrenergic stimulation-induced changes in IKur, IKr and IKS to human atrial terminal repolarisation is unclear. Several additional currents contribute to terminal repolarisation in human atrium, including the inward rectifier K+ currents IK1 and IKACh, the Na+, K+ pump current (Ip), INa/Ca, and possibly ATP-sensitive K+ current (IKATP) (Workman et al. 2008). ISO increased IKACh in canine atrium (Yeh et al. 2007), yet it may affect neither IK1 nor IKACh in human atrium (Voigt et al. 2009). Human atrial Ip has been measured directly in only a single study (Workman et al. 2003a), but adrenergic stimulation was not studied. However, the hyperpolarisation caused by rewarming of human atrial tissue, considered to be due to Ip reactivation, was unaffected by adrenaline (Rasmussen et al. 1985). INa/Ca was increased by ISO in cat atrium (Zhou et al. 1993), but human atrial data are lacking. IKATP was increased by ISO in cat ventricle, from ATPi depletion rather than a direct effect (Schackow et al. 1994), but atrial data are lacking. INa was reduced by ISO in human atrium (Lee et al. 1990). However, any conduction-slowing influence of that might be outweighed by an opposing increase in Igap (Salameh et al. 2006). If is consistently increased by β-stimulation in human atrium, associated with a positive shift in its activation voltage, e.g. (Hoppe et al. 1998; Lonardo et al. 2005). However, whether such an increase could lead to atrial arrhythmias from AA is uncertain (Workman et al. 1998), because of the rather negative activation voltage of If relative to atrial MDP, and the small current size under physiological [K+]o. Mathematical modelling should help to clarify the relative contribution of these ion current changes to the altered action potential shape under adrenergic stimulation. However, the weight of currently available evidence, summarised in Figure 2b, suggests that in human atrium the plateau is elevated primarily by ICaL increase, and that an associated increase in IKur, with little or no direct effect on other repolarising currents, results in no net change in terminal repolarisation. The contrasting changes in terminal repolarisation seen in some other species and in ventricle may result in part from differing magnitudes of the various delayed rectifier current components. Moreover, the prominent increase in ICaL has important implications for Ca2+i homeostasis and arrhythmogenesis.
Effects of adrenergic stimulation on atrial Ca2+i and its contribution to arrhythmogenesis
The flow of Ca2+ into myocardial cells via ICaL during systole causes contraction by triggering a marked increase in [Ca2+]i, by the opening of sarcoplasmic reticulum (SR) Ca2+ release channels (ryanodine receptors, RyR); so-called Ca2+-induced Ca2+-release (CICR). During relaxation, Ca2+ is returned to the SR by the SR Ca2+ pump (SERCA). The amplitude of the transient rise in [Ca2+]i is influenced in part by the amplitude of ICaL. Adrenergic stimulation increases the Ca2+i transient and accelerates its decline, by causing the phosphorylation of various proteins including ICaL channels and phospholamban (PLB) (Bers 2002). There is probably only a minor contribution in the steady state from phosphorylation of RyR (Eisner et al. 2009). The phosphorylation of PLB reduces its inhibitory effect on SERCA, thus increasing Ca2+SR uptake, and is the dominant cause of accelerated Ca2+i decline under β-stimulation. The faster Ca2+SR uptake also contributes to an increased Ca2+SR content, thus increasing Ca2+SR availability for CICR, and coupled with increased ICaL, is the main determinant of the increased Ca2+i transient under adrenergic stimulation (Bers 2002). Ca2+i homeostasis differs between atrium and ventricle (Dobrev et al. 2008). Atrial cells lack a fully developed transverse tubule network and, in contrast to ventricular cells, in which Ca2+ influx increases [Ca2+]i uniformly, the Ca2+ wave arises in the cell’s periphery, propagating to the centre. The Ca2+i transient was also smaller in atrium, and Ca2+SR content substantially larger (Walden et al. 2009). In human atrial cells, β-stimulation increased the Ca2+i transient amplitude (Hatem et al. 1995), as reported in atria of other species (Mackenzie et al. 2004; Danson et al. 2005; Coutu et al. 2006). In rat atrial cells which displayed primarily peripheral Ca2+i transients, β-stimulation produced large transients in central regions (Mackenzie et al. 2004). β-stimulation also increased atrial diastolic [Ca2+]i (Danson et al. 2005), and might enhance a diastolic Ca2+SR leak, as shown in rabbit ventricle (Curran et al. 2007). β-stimulation could elevate Ca2+SR content and [Ca2+]i sufficiently to cause propagating waves of CICR, which may increase Ca2+ extrusion via INa/Ca sufficiently to produce DADs (Eisner et al. 2009). Furthermore, increased [Ca2+]i may enhance pacemaker activity, by increasing INa/Ca (Bers 2008), thus potentially causing AA. Adrenergic stimulation caused DADs in canine atrial fibres, but Ca2+i was not studied (Johnson et al. 1986). ISO promoted spontaneous Ca2+i transients in canine atrial and PV cells, but action potentials were not studied (Coutu et al. 2006). Focal ectopic beats that were preceded by rises in [Ca2+]i were produced by perfusing canine atria with ryanodine plus ISO, but ISO alone was not studied (Chou et al. 2005). It is conceivable that adrenergic stimulation in human atrium could increase the Ca2+i transient, Ca2+SR content and diastolic [Ca2+]i sufficiently to cause DADs or AA, but more studies are required to clarify that.
Independent stimulation of adrenoceptor subtypes
The effects of adrenergic stimulation on atrial rhythm, pacemaking, propensity to spontaneous depolar-isations, reentry, and action potentials were demonstrated using both combined β- and α-stimulation with adrenaline, and β-stimulation only, with ISO. Effects of adrenaline and ISO, sometimes compared within studies, largely conformed, suggesting a prominent involvement of β. However, for human atrial ion currents or Ca2+i, β-stimulation only was used, except in one study (Christ et al. 2004). Moreover, adrenoceptor subtypes can mediate different, sometimes opposing (Li et al. 1996; Yeh et al. 2007) effects, that may also vary with cardiac disease. Independent β1- and β2-stimulation has been studied using ISO plus selective β-subtype blockers. In canine atrium, whilst only ~25% of β-receptors were β2, ~50% of the effect of ISO to decrease APD75 was via β2 (Liang et al. 1985). In human atrium, however, although up to 55% of β-receptors are β2 (Hedberg et al. 1985), ISO did not affect APD75 (Redpath et al. 2006). β2-stimulation increased human atrial ICaL (Skeberdis et al. 1997) and If (Lonardo et al. 2005), although the If increase was greater with β1-than β2-stimulation. Nevertheless, the electrophysiological effects of independent β2-, or indeed β1-, stimulation in human atrium are largely unknown. β3-stimulation had no effect on mouse atrial contraction (Oostendorp et al. 2000), but no human atrial studies were found. In ventricle, β3-stimulation moderately affected APD (Gauthier et al. 1996; Bosch et al. 2002), probably via IKS (Bosch et al. 2002) which may have limited involvement in human atrium. However, since β3-stimulation can affect IK1 (Scherer et al. 2007), ICaL and Ca2+i (Cheng et al. 2001), its potential to contribute to atrial adrenergic electrophysiological responses should not be overlooked. Myocardial effects of α-stimulation are multiple, complex and species-, chamber- and α-subtype-dependent. In human atrium, an α-agonist increased APD (Sato et al. 1995). That is consistent with effects of phenylephrine to inhibit IK1 (Su et al. 1994; Voigt et al. 2009), IKACh (Voigt et al. 2009) and IKur (Li et al. 1996). However, the APD increase could also involve an increase in the Ca2+i transient (Jahnel et al. 1992), although data are lacking in human atrium. Furthermore, α-stimulation may increase inositol (1,4,5)-trisphosphate (InsP3), and InsP3 can cause Ca2+i release, particularly in atrium, in which InsP3 receptors are prevalent (Bers 2002). However, the rate and extent of Ca2+i release is much lower via InsP3 than CICR (Bers 2002). α-, by contrast with β-, stimulation, did not produce DADs or EADs in canine ventricle (Priori et al. 1990), but whether it would in human atrium is unknown.
Heart failure causes AF, atrial remodelling and sympathetic activation
AF is usually associated with cardiac disease, such as coronary artery disease, valve disease, hypertension, myocardial infarction (MI), or HF. These, and AF, can cause chronic adaptational changes, i.e. remodelling, of atrial structure and function, including adrenergic responses, as well as increasing adrenergic tone. Each disease may, therefore, influence an involvement of the adrenergic system in the development of AF in a variety of ways. HF is a major cause of AF, and both systolic and diastolic ventricular dysfunction were independently associated with increased risk of AF (Tsang et al. 2002). Severe HF was associated with an ~70% increase in plasma noradrenaline (Bolger et al. 2002). However, the maximum concentration reached, 2.7 nM, was ~75-fold lower than was half-maximally effective at increasing ICaL (Christ et al. 2004). Therefore, a promotion by HF of adrenergically-mediated atrial arrhythmic activity involving, e.g. ICaL increase, may require a localised catecholamine accumulation. HF can predispose to atrial arrhythmic activity by remodelling atrial electrophysiology and Ca2+i homeostasis. In dogs, chronic ventricular tachypacing-induced congestive HF (CHF) increased atrial ERP and APD and produced spontaneous depolarisations (Stambler et al. 2003; Yeh et al. 2008). This was associated with various ion current changes including decreased ITO, ICaL and IKS and increased INa/Ca, whilst IK1, IKr and IKur were unaffected (Li et al. 2000). CHF also increased atrial diastolic [Ca2+]i, the Ca2+i transient, and Ca2+SR content, associated with “spontaneous Ca2+ transient events” (Yeh et al. 2008). Ca2+i changes could be caused partly by the APD increase, and partly by increased CaMKII-phosphorylation of PLB potentially enhancing Ca2+SR uptake (Yeh et al. 2008). It has been suggested (Lehnart et al. 2004) that chronic adrenergic activation in HF may cause hyperphosphorylation of RyRs and a consequent increased diastolic leak of Ca2+SR. However, leaky RyRs are unlikely to be sufficient, in the steady state, to cause Ca2+i waves and DADs via INa/Ca, because of compensatory changes of Ca2+SR content (Eisner et al. 2009). Nevertheless, CHF-induced atrial arrhythmic activity may involve RyRs, since it was abolished by RyR blockade in dogs (Stambler et al. 2003; Yeh et al. 2008). Such arrhythmic activity might be potentiated by increased adrenergic tone, e.g., via increased phosphorylation of ICaL and PLB, although this awaits investigation. Human atrial electrophysiological changes have been associated with CHF or left ventricular systolic dysfunction (LVSD). Whilst the data are often compounded by variability in patients’ disease states and drug treatments (Workman et al. 2008), LVSD has been independently associated with a reduction in the atrial cellular ERP (Workman et al. 2009). In that study the ERP correlated positively with the LV ejection fraction (Workman et al. 2009), which is a significant predictor of AF (Tsang et al. 2002). Such ERP decrease, which contrasts with the ERP increase in canine CHF, might predispose to reentry by decreasing the wavelength. The ionic mechanisms are unclear, although an associated reduction in ITO (Workman et al. 2009) might be involved. IK1, ICaL and the sustained outward current (ISUS; predominantly IKur) were unaffected. IKS, a likely major contributor to the APD increase in canine CHF, may be minimally involved in human atrium. Other atrial currents such as INa/Ca, constitutively active (CA) IKACh, INa, Ip, and stretch-activated currents, as well as Ca2+i, and afterdepolarisations, remain to studied in human HF (Workman et al. 2008). The expression of atrial HCN2 and HCN4 (encode If) was increased in human HF (Stillitano et al. 2008), but atrial If was not measured. Some human atrial responses to adrenergic stimulation have been studied, although the data were equivocal. The effect of ISO to increase ICaL or HFIUR was attenuated (Ouadid et al. 1995; Piot et al. 1996), unchanged (Workman et al. 2009) or potentiated (Dinanian et al. 2008) in patients with HF or LVSD, and contractile responses to ISO were attenuated (Harding et al. 1990). An attenuated β-response may involve impaired β-receptor function or signalling, or a reduction in receptor density, as shown in atria of patients with end stage HF (Steinfath et al. 1992) or pigs with CHF (Roth et al. 1993). Furthermore, HF may preferentially downregulate β1, in atrium, depending on the cause of HF (Steinfath et al. 1992), and that could result from chronic adrenergic stimulation (Brown et al. 1992). β3 density was not studied in atrium, although this was increased in ventricle (Moniotte et al. 2001). The multiple, complex and interacting influences of HF on the involvement of the adrenergic system in the development of AF are, therefore, yet to be resolved.
AF causes AF and atrial remodeling
Once AF occurs, a persistence of the rapid atrial activation causes atrial electrophysiological remodelling that promotes AF. This includes a reduction in the atrial ERP, AF cycle length and reentry wavelength (Workman et al. 2008). Chronic AF in patients was associated with a decreased atrial ERP and RA conduction velocity, increased ERP dispersion (Kojodjojo et al. 2007) and decreased atrial isolated tissue or cellular ERP (Workman et al. 2001) or APD90 (Bosch et al. 1999; Workman et al. 2001; Wettwer et al. 2004; Pau et al. 2007). In dogs, chronic rapid atrial rate also decreased APD90 in isolated PVs (Chen et al. 2000; Cha et al. 2005), and either enhanced (Chen et al. 2000) or did not enhance (Cha et al. 2005) the ability of PVs to produce arrhythmic activity. The atrial ion current changes are different in chronic AF than in HF or LVSD (Workman et al. 2008; Workman et al. 2009), with an increased IK1 and CA IKACh, and a markedly decreased ICaL and ITO. Ip was unchanged (Workman et al. 2003a); data on IKur and IKATP are equivocal; and several other currents remain to be studied (Workman et al. 2008). These include IKr and IKS, whose activation could be enhanced by action potential-triangulation in chronic AF (Workman et al. 2001). Rapid atrial rate also remodels Ca2+i homeostasis. In canine cultured atrial cells, chronic rapid stimulation reversed the elevation in both diastolic [Ca2+]i and Ca2+i transient that had resulted from shorter periods of stimulation. This was probably via decreased CICR, due to transcriptional downregulation of ICaL (Qi et al. 2008). AF-remodelling of Ca2+i in human atrium is only poorly understood. Ca2+i sparks, which reflect Ca2+ release from single clusters of RyRs, were either increased (Hove-Madsen et al. 2004) or unchanged (Liang et al. 2008) in frequency, and increased (Liang et al. 2008) or unchanged (Hove-Madsen et al. 2004) in duration. The frequency of Ca2+i waves was increased in both studies, and Ca2+SR content was unchanged. Chronic AF also increased PKA phosphorylation of RyR (Vest et al. 2005). The effects of adrenergic stimulation on Ca2+i were not studied in AF-remodelled atrium. Moreover, AF without HF may not change catecholamine levels in patients (Berglund et al. 1990). Nevertheless, chronic AF has consistently been shown to be associated with a potentiated effect of ISO to increase human atrial ICaL (Van Wagoner et al. 1999; Skasa et al. 2001; Christ et al. 2004; Greiser et al. 2007). This contrasts with an attenuated effect of 5-hydroxytryptamine (5-HT) on ICaL (Pau et al. 2007), despite both acting via cAMP. The kinase/phosphatase balance can affect ICaL in human AF without altered channel transcription or translation (Christ et al. 2004). Since β- (unlike 5-HT-) receptor expression was unchanged in AF (Grammer et al. 2001), the potentiated ICaL response to adrenergic stimulation might involve altered signalling, perhaps via CaMKII (Christ et al. 2004). Chronic AF was also associated with an attenuated effect of α-stimulation to decrease human atrial IK1 and IKACh (Voigt et al. 2009). The lack of change in atrial Ca2+SR content associated with chronic AF, in contrast to the increase by HF, might suggest a lower susceptibility to any adrenergically-mediated arrhythmic activity than in HF. However, that remains to be investigated. Furthermore, AF-remodelling may interact with HF-remodelling (Workman et al. 2008), but no studies of adrenergic stimulation in that setting could be found.
Atrial neural remodelling by MI and by chronic AF
Atrial adrenergic nerves can be modified by myocardial diseases associated with AF, and also by persistent rapid atrial activation; so-called neural remodelling. For example, ventricular MI in dogs increased the atrial density and spatial heterogeneity of both tyrosine hydroxylase, which stains adrenergic nerves, and growth-associated protein, which stains for nerve sprouting; axonal regeneration. This may have resulted from injury of atrial nerves passing through the ventricle (Miyauchi et al. 2003). Chronic AT in dogs caused a spatially heterogeneous atrial adrenergic hyperinnervation, an increased atrial tissue noradrenaline concentration, and nerve sprouting (Jayachandran et al. 2000; Chang et al. 2001). Comparable changes were associated with chronic AF in humans, including hyperinnervation and increased tissue noradrenaline (Gould et al. 2006). Increases in atrial tissue adrenergic innervation and associated catecholamines, particularly of a spatially heterogeneous nature, may favour reentry and/or non-reentry.
Clinical use of β-blockers in AF: when and how is adrenergic control effective?
In patients with AF, β-blockers are used mainly to prevent the rapidly activating atria from stimulating the ventricles at excessively high rates. The mechanism is a slowing of atrioventricular (AV) nodal conduction, associated with increased AV nodal ERP and AH interval (Prystowsky 1988). This probably involves an anti-adrenergic effect on ICaL, which is the main inward current in AV node (Workman et al. 1999). β-blockers can also prevent AF, convert it to sinus rhythm, or maintain sinus rhythm after it is restored, depending on β-blocker type and cardiac pathology (Lopez-Sendon et al. 2004). For example, in patients with CHF, β1-blockade substantially reduced the incidence of AF (Van Veldhuisen et al. 2006). In patients with LVSD following MI, carvedilol, a β1-, β2- and α1-antagonist, also substantially reduced the incidence of AF/atrial flutter (McMurray et al. 2005). β-blockers reduced the incidence of AF following cardiac surgery (Mathew et al. 2004; Workman et al. 2006), and post-surgery β-blocker withdrawal increased it (Mathew et al. 2004). Carvedilol was more effective than a β1-blocker (Haghjoo et al. 2007), perhaps by decreasing IKur and ITO (Deng et al. 2007). A β1-blocker also caused a moderate reduction in the incidence of paroxysmal AF (Steeds et al. 1999) or of relapse into AF after electrical or pharmacological cardioversion (Kuhlkamp et al. 2000). Acute β-selective blockade has little or no effect on atrial electrophysiology in the absence of catecholamines (Wit et al. 1975), and catecholamine levels were not affected by β-blockade in patients (Bolger et al. 2002). Therefore, the relatively high efficacy of β-blockers to prevent AF associated with elevated adrenergic tone, as occurs in, e.g. CHF (Bolger et al. 2002) or post-surgery (Engelman et al. 1983), suggests an anti-adrenergic suppression of pro-arrhythmic effects of catecholamines. The electrophysiological mechanisms are likely to be multiple. For example, a reduction by atenolol of cAMP-stimulated ICaL in human atrial cells (Mewes et al. 1993) might suppress arrhythmic activity that involves increased CICR. A similar mechanism might account for an acute effect of propranolol to suppress either noradrenaline-induced AA in canine atrial fibres (Davis 1975), or AT in a CHF-remodelled atrium that produced spontaneous depolarisations (Stambler et al. 2003). A reduction of ISO-stimulated IKur by propranolol in human atrial cells (Li et al. 1996) is consistent with its acute effect to moderately increase atrial APD90 and ERP in patients (Shimizu et al. 1994b). That might moderately increase wavelength, although acute propranolol did not affect wavelength in canine atrium (Rensma et al. 1988), or ERP in AF-remodelled goat atrium (Wijffels et al. 1997). Furthermore, an acute suppression by atenolol of autonomic nerve stimulation-induced PV arrhythmic activity in dogs, might involve inhibition of AA or reentry, as well as afterdepolarisations (Patterson et al. 2005). Moreover, atrial anti-arrhythmic effects of β-blockers may also involve another type of electrophysiological remodelling: that from the long term drug treatment.
Atrial remodelling by chronic β-blocker treatment
The long term use of β-blockers causes adaptive changes in, e.g., atrial electrophysiology, contraction and adrenoceptors, which may contribute to the effects of these drugs on AF. Such “pharmacological remodelling” (Workman et al. 2003b) was originally demonstrated in rabbits (Raine et al. 1981). Treatment with either a β1-or a mixed β-blocker increased APD in atrium (Figure 3a) and ventricle; maximally after six days in atrium (Raine et al. 1981). This was an adaptation to the treatment, not effects due to the presence of the drugs, because the APD was recorded at a sufficient time after the last dose for the drugs to have been eliminated from the body. That was confirmed in plasma and tissue samples (Raine et al. 1980). Subsequent studies in atrial cells from patients in sinus rhythm showed that β1-blocker treatment for ≥ 7 days was associated with an increased APD90 and ERP (Workman et al. 2003b; Redpath et al. 2006; Workman et al. 2006) (Figure 3b), and a decreased ITO (Workman et al. 2003b; Marshall et al. 2009) and IK1 (Marshall et al. 2006). The atrial ERP correlated with β-blocker dose (Workman et al. 2008). The ITO reduction, as found also in atrial cells from rabbits treated with carvedilol (Cao et al. 2006), did not involve altered voltage-dependency, kinetics or channel expression (Marshall et al. 2009). ICaL did not change, either in amplitude (Workman et al. 2003b; Pau et al. 2003; Redpath et al. 2006; Workman et al. 2006; Pau et al. 2007), activation voltage-dependence (Redpath et al. 2006), single channel gating (Klein et al. 2003) or expression (Grammer et al. 2001). That is consistent with a lack of change in APD50 (Workman et al. 2003b; Redpath et al. 2006). ISUS also was unaffected (Workman et al. 2003b; Marshall et al. 2006). The human atrial electrophysiological changes most likely reflect an adaptation to the treatment also, because recordings were made in the presumed absence of residual β-blocker (cells had been isolated and washed) and the APD90 and ERP increases were independent of various clinical covariables (Workman et al. 2003b; Workman et al. 2006). No reports on atrial Ca2+i were found, but chronic β1-blockade in mice increased a HF-impaired ventricular Ca2+i transient (Bartholomeu et al. 2008). Catecholamine-induced arrhythmic contractions in human atrial muscles were exacerbated by β-blocker therapy (Kaumann et al. 1993), probably not involving any altered ICaL response to β-stimulation (Redpath et al. 2006). Nevertheless, human atrial β-receptors, perhaps predominantly β1, were upregulated by β-blocker therapy (Hedberg et al. 1985; Michel et al. 1988), possibly related to a reduction in adrenergic nerve activity (Raine et al. 1977). The β-blocker-remodelling of atrial APD90 and ERP might be expected to increase reentry wavelength and thus contribute to anti-fibrillatory actions of β-blockers. However, the net effect of β-blockers in patients undergoing treatment is likely to include anti-adrenergic effects in an atrium remodelled by the chronic treatment, as well as by underlying cardiac disease.
Figure 3. Remodelling of atrial action potentials by chronic β-blocker treatment.
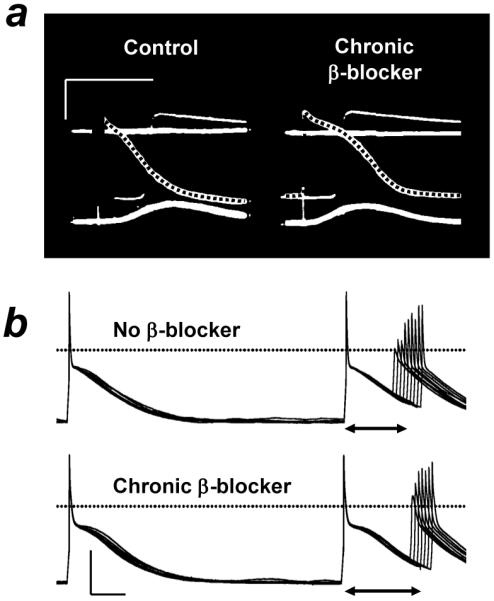
a, Action potentials (dotted) in right atrium isolated from a rabbit not treated (control) or treated (chronic β-blocker) for 24 days with metoprolol. b, Action potentials and effective refractory period (↔) recorded in an atrial cell obtained from a patient not treated (upper panel) or treated (lower) with a β-blocker. Calibrations: 50 mV, 100 ms. Based on data in (Raine et al. 1981) (a) and (Workman et al. 2003b) (b) with permission from Wolters Kluwer Health/Lippincott, Williams & Wilkins, and Oxford University Press, respectively.
Concluding remarks, and potential future directions
The evidence in this review suggests that AF that is caused by adrenergic stimulation results, in large part, from a promotion of atrial arrhythmic activity involving increased ICaL and Ca2+i, rather than from any effect on action potential terminal repolarisation or ERP. The suppression of AF by treatment with β-blockers is likely to involve attenuation of such arrhythmic activity, potentially coupled with an ERP-prolonging adaptation to the treatment; so-called pharmacological remodelling. It is hoped that an improved understanding of the involvement of the adrenergic system and its control in basic mechanisms of AF under differing cardiac pathologies will lead to better pharmacological treatments. However, there are numerous and wide gaps in our knowledge, particularly pertaining to human atrium, which suggest avenues for further research. For example, what are the predominant PV and atrial electrophysiological mechanisms of AF initiation and maintenance in different cardiac diseases? Could adrenergic stimulation increase atrial Ca2+i sufficiently to cause AA or DADs in patients, and how would HF and chronic AF affect such responses? Does adrenergic stimulation substantially influence human atrial IKr or IKS? What is the relative involvement of α- and β-subtypes in human atrial arrhythmogenic responses to adrenergic stimulation, and does carvedilol’s marked ability to suppress AF post-MI involve α1-antagonism? What are the molecular mechanisms of atrial pharmacological remodelling by chronic β-blockade, and how are they affected by cardiac diseases? Future therapeutic strategies for adrenergic control of AF may include non-pharmacological interventions. However, radiofrequency ablation of atrial ganglionated plexi, sites rich in adrenergic and cholinergic neurons, may not be as efficacious as other ablation procedures (Katritsis 2008). Despite continuous improvement of ablation techniques, pharmacological therapy is the mainstay of treatment for AF. New anti-AF drugs in development include “atrial-selective compounds”, and “multi-channel blockers” such as dronedarone (Ehrlich et al. 2009). This drug’s anti-adrenergic activity may be expected to contribute to its anti-arrhythmic efficacy.
Acknowledgements
British Heart Foundation for financial support (Basic Science Lectureship Award, renewal: BS/06/003), and Dr John Dempster, University of Strathclyde, for helpful discussions about CESE Pro software.
References
- Arora R, Verheule S, Scott L, et al. Arrhythmogenic substrate of the pulmonary veins assessed by high-resolution optical mapping. Circulation. 2003;107:1816–1821. doi: 10.1161/01.CIR.0000058461.86339.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbuti A, Terragni B, Brioschi C, et al. Localization of f-channels to caveolae mediates specific β2-adrenergic receptor modulation of rate in sinoatrial myocytes. J Mol Cell Cardiol. 2007;42:71–78. doi: 10.1016/j.yjmcc.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bartholomeu JB, Vanzelli AS, Rolim NPL, et al. Intracellular mechanisms of specific beta-adrenoceptor antagonists involved in improved cardiac function and survival in a genetic model of heart failure. J Mol Cell Cardiol. 2008;45:240–249. doi: 10.1016/j.yjmcc.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Berglund H, Boukter S, Theodorsson E, et al. Raised plasma concentrations of atrial natriuretic peptide are independent of left atrial dimensions in patients with chronic atrial fibrillation. Br Heart J. 1990;64:9–13. doi: 10.1136/hrt.64.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- Bettoni M, Zimmermann M. Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation. 2002;105:2753–2759. doi: 10.1161/01.cir.0000018443.44005.d8. [DOI] [PubMed] [Google Scholar]
- Bolger AP, Sharma R, Li W, et al. Neurohormonal activation and the chronic heart failure syndrome in adults with congenital heart disease. Circulation. 2002;106:92–99. doi: 10.1161/01.cir.0000020009.30736.3f. [DOI] [PubMed] [Google Scholar]
- Bosch RF, Zeng X, Grammer JB, et al. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–131. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- Bosch RF, Schneck AC, Kiehn J, et al. β3-adrenergic regulation of an ion channel in the heart-inhibition of the slow delayed rectifier potassium current IKs in guinea pig ventricular myocytes. Cardiovasc Res. 2002;56:393–403. doi: 10.1016/s0008-6363(02)00601-6. [DOI] [PubMed] [Google Scholar]
- Brown L, Sernia C, Newling R, et al. Cardiac responses after norepinephrine-induced ventricular hypertrophy in rats. J Cardiovasc Pharmacol. 1992;20:316–323. doi: 10.1097/00005344-199208000-00019. [DOI] [PubMed] [Google Scholar]
- Cao F, Huang CX, Wang T, et al. Effects of carvedilol on rabbit atrial cell electrophysiology. Heart Rhythm. 2006;3(Suppl 1):S178. Abstract. [Google Scholar]
- Cha TJ, Ehrlich JR, Zhang L, et al. Atrial tachycardia remodeling of pulmonary vein cardiomyocytes: comparison with left atrium and potential relation to arrhythmogenesis. Circulation. 2005;111:728–735. doi: 10.1161/01.CIR.0000155240.05251.D0. [DOI] [PubMed] [Google Scholar]
- Chamberlain PD, Jennings KH, Paul F, et al. The tissue distribution of the human β3-adrenoceptor studied using a monoclonal antibody: direct evidence of the β3-adrenoceptor in human adipose tissue, atrium and skeletal muscle. Int J Obesity. 1999;23:1057–1065. doi: 10.1038/sj.ijo.0801039. [DOI] [PubMed] [Google Scholar]
- Chang CM, Wu TJ, Zhou S, et al. Nerve sprouting and sympathetic hyperinnervation in a canine model of atrial fibrillation produced by prolonged right atrial pacing. Circulation. 2001;103:22–25. doi: 10.1161/01.cir.103.1.22. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Chen SA, Chang MS, et al. Arrhythmogenic activity of cardiac muscle in pulmonary veins of the dog: implication for the genesis of atrial fibrillation. Cardiovasc Res. 2000;48:265–273. doi: 10.1016/s0008-6363(00)00179-6. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Chen SA, Chen YC, et al. Electrophysiology of single cardiomyocytes isolated from rabbit pulmonary veins: implication in initiation of focal atrial fibrillation. Basic Res Cardiol. 2002;97:26–34. doi: 10.1007/s395-002-8384-6. [DOI] [PubMed] [Google Scholar]
- Cheng HJ, Zhang ZS, Onishi K, et al. Upregulation of functional β3-adrenergic receptor in the failing canine myocardium. Circ Res. 2001;89:599–606. doi: 10.1161/hh1901.098042. [DOI] [PubMed] [Google Scholar]
- Chou CC, Nihei M, Zhou S, et al. Intracellular calcium dynamics and anisotropic reentry in isolated canine pulmonary veins and left atrium. Circulation. 2005;111:2889–2897. doi: 10.1161/CIRCULATIONAHA.104.498758. [DOI] [PubMed] [Google Scholar]
- Christ T, Boknik P, Wohrl S, et al. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004;110:2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- Coccagna G, Capucci A, Bauleo S, et al. Paroxysmal atrial fibrillation in sleep. Sleep. 1997;20:396–398. doi: 10.1093/sleep/20.6.396. [DOI] [PubMed] [Google Scholar]
- Courtemanche M, Ramirez RJ, Nattel S. Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am J Physiol. 1998;275:H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301. [DOI] [PubMed] [Google Scholar]
- Coutu P, Chartier D, Nattel S. Comparison of Ca2+-handling properties of canine pulmonary vein and left atrial cardiomyocytes. Am J Physiol. 2006;291:H2290–H2300. doi: 10.1152/ajpheart.00730.2005. [DOI] [PubMed] [Google Scholar]
- Curran J, Hinton MJ, Rios E, et al. β-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- Danson EJF, Zhang YH, Sears CE, et al. Disruption of inhibitory G-proteins mediates a reduction in atrial β-adrenergic signaling by enhancing eNOS expression. Cardiovasc Res. 2005;67:613–623. doi: 10.1016/j.cardiores.2005.04.034. [DOI] [PubMed] [Google Scholar]
- Davis LD. Effects of autonomic neurohumors on transmembrane potentials of atrial plateau fibers. Am J Physiol. 1975;229:1351–1356. doi: 10.1152/ajplegacy.1975.229.5.1351. [DOI] [PubMed] [Google Scholar]
- Deng C, Yu X, Kuang S, et al. Effects of carvedilol on transient outward and ultra-rapid delayed rectifier potassium currents in human atrial myocytes. Life Sci. 2007;80:665–671. doi: 10.1016/j.lfs.2006.10.012. [DOI] [PubMed] [Google Scholar]
- Dimmer C, Tavernier R, Gjorgov N, et al. Variations of autonomic tone preceding onset of atrial fibrillation after coronary artery bypass grafting. Am J Cardiol. 1998;82:22–25. doi: 10.1016/s0002-9149(98)00231-8. [DOI] [PubMed] [Google Scholar]
- Dinanian S, Boixel C, Juin C, et al. Downregulation of the calcium current in human right atrial myocytes from patients in sinus rhythm but with a high risk of atrial fibrillation. Eur Heart J. 2008;29:1190–1197. doi: 10.1093/eurheartj/ehn140. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Nattel S. Calcium handling abnormalities in atrial fibrillation as a target for innovative therapeutics. J Cardiovasc Pharmacol. 2008;52:293–299. doi: 10.1097/FJC.0b013e318171924d. [DOI] [PubMed] [Google Scholar]
- Eckel L, Gristwood RW, Nawrath H, et al. Inotropic and electrophysiological effects of histamine on human ventricular heart muscle. J Physiol. 1982;330:111–123. doi: 10.1113/jphysiol.1982.sp014332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich JR, Nattel S. Novel approaches for pharmacological management of atrial fibrillation. Drugs. 2009;69:757–774. doi: 10.2165/00003495-200969070-00001. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Kashimura T, O’Neill SC, et al. What role does modulation of the ryanodine receptor play in cardiac inotropy and arrhythmogenesis? J Mol Cell Cardiol. 2009;46:474–481. doi: 10.1016/j.yjmcc.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Engelman RM, Haag B, Lemeshow S, et al. Mechanism of plasma catecholamine increases during coronary artery bypass and valve procedures. J Thorac Cardiovasc Surg. 1983;86:608–615. [PubMed] [Google Scholar]
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation. Executive summary. J Am Coll Cardiol. 2006;48:854–906. doi: 10.1016/j.jacc.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Gauthier C, Tavernier G, Charpentier F, et al. Functional β3-adrenoceptor in the human heart. J Clin Invest. 1996;98:556–562. doi: 10.1172/JCI118823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelband H, Rosen MR, Myerburg RJ, et al. Restorative effect of epinephrine on the electrophysiologic properties of depressed human atrial tissue. J Electrocardiol. 1977;10:313–320. doi: 10.1016/s0022-0736(77)80003-4. [DOI] [PubMed] [Google Scholar]
- Gould PA, Yii M, McLean C, et al. Evidence for increased atrial sympathetic innervation in persistent human atrial fibrillation. PACE-Pacing Clin Electrophysiol. 2006;29:821–829. doi: 10.1111/j.1540-8159.2006.00447.x. [DOI] [PubMed] [Google Scholar]
- Grammer JB, Zeng X, Bosch RF, et al. Atrial L-type Ca2+-channel, β-adrenoreceptor, and 5-hydroxytryptamine type 4 receptor mRNAs in human atrial fibrillation. Basic Res Cardiol. 2001;96:82–90. doi: 10.1007/s003950170081. [DOI] [PubMed] [Google Scholar]
- Greiser M, Halaszovich CR, Frechen D, et al. Pharmacological evidence for altered src kinase regulation of ICa,L in patients with chronic atrial fibrillation. Naunyn-Schmiedeberg’s Arch Pharmacol. 2007;375:383–392. doi: 10.1007/s00210-007-0174-6. [DOI] [PubMed] [Google Scholar]
- Haghjoo M, Saravi M, Hashemi MJ, et al. Optimal β-blocker for prevention of atrial fibrillation after on-pump coronary artery bypass graft surgery: carvedilol versus metoprolol. Heart Rhythm. 2007;4:1170–1174. doi: 10.1016/j.hrthm.2007.04.022. [DOI] [PubMed] [Google Scholar]
- Haissaguerre M, Jais P, Shah DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- Harding SE, Jones SM, O’Gara P, et al. Reduced β-agonist sensitivity in single atrial cells from failing human hearts. Am J Physiol. 1990;259:H1009–H1014. doi: 10.1152/ajpheart.1990.259.4.H1009. [DOI] [PubMed] [Google Scholar]
- Hatem SN, Sweeten T, Vetter V, et al. Evidence for presence of Ca2+ channel-gated Ca2+ stores in neonatal human atrial myocytes. Am J Physiol. 1995;268:H1195–H1201. doi: 10.1152/ajpheart.1995.268.3.H1195. [DOI] [PubMed] [Google Scholar]
- Heath BM, Terrar DA. Protein kinase C enhances the rapidly activating delayed rectifier potassium current, IKr, through a reduction in C-type inactivation in guinea-pig ventricular myocytes. J Physiol. 2000;522:391–402. doi: 10.1111/j.1469-7793.2000.t01-2-00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedberg A, Kempf F, Jr., Josephson ME, et al. Coexistence of Beta-1 and Beta-2 adrenergic receptors in the human heart: effects of treatment with receptor antagonists or calcium entry blockers. J Pharmacol Exp Ther. 1985;234:561–568. [PubMed] [Google Scholar]
- Hoppe UC, Beuckelmann DJ. Characterization of the hyperpolarization-activated inward current in isolated human atrial myocytes. Cardiovasc Res. 1998;38:788–801. doi: 10.1016/s0008-6363(98)00047-9. [DOI] [PubMed] [Google Scholar]
- Hove-Madsen L, Llach A, Bayes-Genis A, et al. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87. [DOI] [PubMed] [Google Scholar]
- Jahnel U, Nawrath H, Shieh RC, et al. Modulation of cytosolic free calcium concentration by α1-adrenoceptors in rat atrial cells. Naunyn-Schmiedeberg’s Arch Pharmacol. 1992;346:88–93. doi: 10.1007/BF00167576. [DOI] [PubMed] [Google Scholar]
- Jayachandran JV, Sih HJ, Winkle W, et al. Atrial fibrillation produced by prolonged rapid atrial pacing is associated with heterogeneous changes in atrial sympathetic innervation. Circulation. 2000;101:1185–1191. doi: 10.1161/01.cir.101.10.1185. [DOI] [PubMed] [Google Scholar]
- Johnson N, Danilo P, Jr., Wit AL, et al. Characteristics of initiation and termination of catecholamine-induced triggered activity in atrial fibers of the coronary sinus. Circulation. 1986;74:1168–1179. doi: 10.1161/01.cir.74.5.1168. [DOI] [PubMed] [Google Scholar]
- Jost N, Virag L, Bitay M, et al. Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation. 2005;112:1392–1399. doi: 10.1161/CIRCULATIONAHA.105.550111. [DOI] [PubMed] [Google Scholar]
- Katritsis DG. Catheter ablation of atrial fibrillation: for whom and how? Angiology. 2008;59:103S–106S. doi: 10.1177/0003319708318581. [DOI] [PubMed] [Google Scholar]
- Kaumann AJ, Sanders L. Both β1- and β2-adrenoceptors mediate catecholamine-evoked arrhythmias in isolated human right atrium. Naunyn-Schmiedeberg’s Arch Pharmacol. 1993;348:536–540. doi: 10.1007/BF00173215. [DOI] [PubMed] [Google Scholar]
- Kawashima T. The autonomic nervous system of the human heart with special reference to its origin, course, and peripheral distribution. Anat Embryol. 2005;209:425–438. doi: 10.1007/s00429-005-0462-1. [DOI] [PubMed] [Google Scholar]
- Kecskemeti V, Kelemen K, Solti F, et al. Physiological and pharmacological analysis of transmembrane action potentials of human atrial fibers. Adv Myocardiol. 1985;6:37–47. [PubMed] [Google Scholar]
- Kiss O, Zima E, Soos P, et al. Intracoronary endothelin-1 infusion combined with systemic isoproterenol treatment: antagonistic arrhythmogenic effects. Life Sci. 2004;75:537–548. doi: 10.1016/j.lfs.2003.11.033. [DOI] [PubMed] [Google Scholar]
- Klein G, Schroder F, Vogler D, et al. Increased open probability of single cardiac L-type calcium channels in patients with chronic atrial fibrillation: role of phosphatase 2A. Cardiovasc Res. 2003;59:37–45. doi: 10.1016/s0008-6363(03)00357-2. [DOI] [PubMed] [Google Scholar]
- Kojodjojo P, Peters NS, Davies DW, et al. Characterization of the electroanatomical substrate in human atrial fibrillation: the relationship between changes in atrial volume, refractoriness, wavefront propagation velocities, and AF burden. J Cardiovasc Electrophysiol. 2007;18:269–275. doi: 10.1111/j.1540-8167.2007.00723.x. [DOI] [PubMed] [Google Scholar]
- Koumi S, Backer CL, Arentzen CE, et al. β-adrenergic modulation of the inwardly rectifying potassium channel in isolated human ventricular myocytes. Alteration in channel response to β-adrenergic stimulation in failing human hearts. J Clin Invest. 1995;96:2870–2881. doi: 10.1172/JCI118358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlkamp V, Schirdewan A, Stangl K, et al. Use of metoprolol CR/XL to maintain sinus rhythm after conversion from persistent atrial fibrillation: a randomized, double-blind, placebo-controlled study. J Am Coll Cardiol. 2000;36:139–146. doi: 10.1016/s0735-1097(00)00693-8. [DOI] [PubMed] [Google Scholar]
- Lee HC, Matsuda JJ, Lemmer JH, et al. Regulation of sodium currents by β-adrenergic stimulation in isolated cardiac myocytes from rabbit and human. J Mol Cell Cardiol. 1990;22(Suppl I):S14. Abstract. [Google Scholar]
- Lehnart SE, Wehrens XHT, Marks AR. Calstabin deficiency, ryanodine receptors, and sudden cardiac death. Biochem Biophys Res Commun. 2004;322:1267–1279. doi: 10.1016/j.bbrc.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Lei M, Brown HF, Terrar DA. Modulation of delayed rectifier potassium current, iK, by isoprenaline in rabbit isolated pacemaker cells. Exp Physiol. 2000;85:27–35. [PubMed] [Google Scholar]
- Levi R, Malm JR, Bowman FO, et al. The arrhythmogenic actions of histamine on human atrial fibers. Circ Res. 1981;49:545–550. doi: 10.1161/01.res.49.2.545. [DOI] [PubMed] [Google Scholar]
- Li D, Melnyk P, Feng J, et al. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101:2631–2638. doi: 10.1161/01.cir.101.22.2631. [DOI] [PubMed] [Google Scholar]
- Li GR, Feng J, Wang Z, et al. Adrenergic modulation of ultrarapid delayed rectifier K+ current in human atrial myocytes. Circ Res. 1996;78:903–915. doi: 10.1161/01.res.78.5.903. [DOI] [PubMed] [Google Scholar]
- Li GR, Nattel S. Properties of human atrial ICa at physiological temperatures and relevance to action potential. Am J Physiol. 1997;272:H227–H235. doi: 10.1152/ajpheart.1997.272.1.H227. [DOI] [PubMed] [Google Scholar]
- Liang BT, Frame LH, Molinoff PB. β2-adrenergic receptors contribute to catecholamine-stimulated shortening of action potential duration in dog atrial muscle. Proc Natl Acad Sci USA. 1985;82:4521–4525. doi: 10.1073/pnas.82.13.4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Xie H, Zhu PH, et al. Ryanodine receptor-mediated Ca2+ events in atrial myocytes of patients with atrial fibrillation. Cardiology. 2008;111:102–110. doi: 10.1159/000119697. [DOI] [PubMed] [Google Scholar]
- Lonardo G, Cerbai E, Casini S, et al. Pharmacological modulation of the hyperpolarization-activated current (If) in human atrial myocytes: focus on G protein-coupled receptors. J Mol Cell Cardiol. 2005;38:453–460. doi: 10.1016/j.yjmcc.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Lopez-Sendon J, Swedberg K, McMurray J, et al. Expert consensus document on β-adrenergic receptor blockers. The task force on beta-blockers of the European Society of Cardiology. Eur Heart J. 2004;25:1341–1362. doi: 10.1016/j.ehj.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Mackenzie L, Roderick HL, Berridge MJ, et al. The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction. J Cell Sci. 2004;117:6327–6337. doi: 10.1242/jcs.01559. [DOI] [PubMed] [Google Scholar]
- Marshall G, Rankin AC, Kane KA, et al. Pharmacological remodelling of human atrial K+ currents by chronic beta-blockade. Eur Heart J. 2006;27:30. Abstract. [Google Scholar]
- Marshall GE, Tellez JO, Russell JA, et al. Remodelling of human atrial ITO current but not ion channel expression by chronic beta-blockade. Heart Rhythm. 2009;6(Suppl 1):S230. doi: 10.1007/s00424-011-1061-z. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary-Rabine L, Hordof AJ, Bowman FO, et al. Alpha and beta adrenergic effects on human atrial specialized conducting fibers. Circulation. 1978;57:84–90. doi: 10.1161/01.cir.57.1.84. [DOI] [PubMed] [Google Scholar]
- Mary-Rabine L, Hordof AJ, Danilo P, Jr., et al. Mechanisms for impulse initiation in isolated human atrial fibers. Circ Res. 1980;47:267–277. doi: 10.1161/01.res.47.2.267. [DOI] [PubMed] [Google Scholar]
- Mathew JP, Fontes ML, Tudor IC, et al. A multicenter risk index for atrial fibrillation after cardiac surgery. JAMA-J Am Med Assoc. 2004;291:1720–1729. doi: 10.1001/jama.291.14.1720. [DOI] [PubMed] [Google Scholar]
- McMurray J, Kober L, Robertson M, et al. Antiarrhythmic effect of carvedilol after acute myocardial infarction: results of the carvedilol post-infarct survival control in left ventricular dysfunction (CAPRICORN) trial. J Am Coll Cardiol. 2005;45:525–530. doi: 10.1016/j.jacc.2004.09.076. [DOI] [PubMed] [Google Scholar]
- Mewes T, Dutz S, Ravens U, et al. Activation of calcium currents in cardiac myocytes by empty β-adrenoceptors. Circulation. 1993;88:2916–2922. doi: 10.1161/01.cir.88.6.2916. [DOI] [PubMed] [Google Scholar]
- Michel MC, Pingsmann A, Beckeringh JJ, et al. Selective regulation of β1- and β2-adrenoceptors in the human heart by chronic β-adrenoceptor antagonist treatment. Br J Pharmacol. 1988;94:685–692. doi: 10.1111/j.1476-5381.1988.tb11576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell MR, Powell T, Sturridge MF, et al. Electrical properties and response to noradrenaline of individual heart cells isolated from human ventricular tissue. Cardiovasc Res. 1986;20:869–876. doi: 10.1093/cvr/20.12.869. [DOI] [PubMed] [Google Scholar]
- Miyauchi Y, Zhou S, Okuyama Y, et al. Altered atrial electrical restitution and heterogeneous sympathetic hyperinnervation in hearts with chronic left ventricular myocardial infarction. Implications for atrial fibrillation. Circulation. 2003;108:360–366. doi: 10.1161/01.CIR.0000080327.32573.7C. [DOI] [PubMed] [Google Scholar]
- Moniotte S, Kobzik L, Feron O, et al. Upregulation of β3-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001;103:1649–1655. doi: 10.1161/01.cir.103.12.1649. [DOI] [PubMed] [Google Scholar]
- Murphy NF, Simpson CR, Jhund PS, et al. A national survey of the prevalence, incidence, primary care burden and treatment of atrial fibrillation in Scotland. Heart. 2007;93:606–612. doi: 10.1136/hrt.2006.107573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K, Eto K, Sakamoto A, et al. Negative chronotropic effect of endothelin 1 mediated through ETA receptors in guinea pig atria. Circ Res. 1995;76:284–292. doi: 10.1161/01.res.76.2.284. [DOI] [PubMed] [Google Scholar]
- Oostendorp J, Kaumann AJ. Pertussis toxin suppresses carbachol-evoked cardiodepression but does not modify cardiostimulation mediated through β1- and putative β4-adrenoceptors in mouse left atria: no evidence for β2- and β3-adrenoreceptor function. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;361:134–145. doi: 10.1007/s002109900156. [DOI] [PubMed] [Google Scholar]
- Oral H, Crawford T, Frederick M, et al. Inducibility of paroxysmal atrial fibrillation by isoproterenol and its relation to the mode of onset of atrial fibrillation. J Cardiovasc Electrophysiol. 2008;19:466–470. doi: 10.1111/j.1540-8167.2007.01089.x. [DOI] [PubMed] [Google Scholar]
- Ouadid H, Albat B, Nargeot J. Calcium currents in diseased human cardiac cells. J Cardiovasc Pharmacol. 1995;25:282–291. doi: 10.1097/00005344-199502000-00014. [DOI] [PubMed] [Google Scholar]
- Patterson E, Po SS, Scherlag BJ, et al. Triggered firing in pulmonary veins initiated by in vitro autonomic nerve stimulation. Heart Rhythm. 2005;2:624–631. doi: 10.1016/j.hrthm.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Pau D, Workman AJ, Kane KA, et al. Electrophysiological effects of 5-hydroxytryptamine on isolated human atrial myocytes, and the influence of chronic β-adrenoceptor blockade. Br J Pharmacol. 2003;140:1434–1441. doi: 10.1038/sj.bjp.0705553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pau D, Workman AJ, Kane KA, et al. Electrophysiological and arrhythmogenic effects of 5-hydroxytryptamine on human atrial cells are reduced in atrial fibrillation. J Mol Cell Cardiol. 2007;42:54–62. doi: 10.1016/j.yjmcc.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelzmann B, Schaffer P, Machler H, et al. Adenosine inhibits the L-type calcium current in human atrial myocytes. Naunyn-Schmiedeberg’s Arch Pharmacol. 1995;351:293–297. doi: 10.1007/BF00233249. [DOI] [PubMed] [Google Scholar]
- Perez-Lugones A, McMahon JT, Ratliff NB, et al. Evidence of specialized conduction cells in human pulmonary veins of patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2003;14:803–809. doi: 10.1046/j.1540-8167.2003.03075.x. [DOI] [PubMed] [Google Scholar]
- Piot C, Lemaire S, Albat B, et al. High frequency-induced upregulation of human cardiac calcium currents. Circulation. 1996;93:120–128. doi: 10.1161/01.cir.93.1.120. [DOI] [PubMed] [Google Scholar]
- Priori SG, Corr PB. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol. 1990;258:H1796–H1805. doi: 10.1152/ajpheart.1990.258.6.H1796. [DOI] [PubMed] [Google Scholar]
- Prystowsky EN. The effects of slow channel blockers and beta blockers on atrioventricular nodal conduction. J Clin Pharmacol. 1988;28:6–21. doi: 10.1002/j.1552-4604.1988.tb03095.x. [DOI] [PubMed] [Google Scholar]
- Qi XY, Yeh YH, Xiao L, et al. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res. 2008;103:845–854. doi: 10.1161/CIRCRESAHA.108.175463. [DOI] [PubMed] [Google Scholar]
- Raine AEG, Chubb IW. Long term β-adrenergic blockade reduces tyrosine hydroxylase and dopamine β-hydroxylase activities in sympathetic ganglia. Nature. 1977;267:265–267. doi: 10.1038/267265a0. [DOI] [PubMed] [Google Scholar]
- Raine AEG, Vaughan Williams EM. Adaptational responses to prolonged β-adrenoceptor blockade in adult rabbits. Br J Pharmacol. 1980;70:205–218. doi: 10.1111/j.1476-5381.1980.tb07926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine AEG, Vaughan Williams EM. Adaptation to prolonged β-blockade of rabbit atrial, Purkinje, and ventricular potentials, and of papillary muscle contraction. Time-course of development of and recovery from adaptation. Circ Res. 1981;48:804–812. doi: 10.1161/01.res.48.6.804. [DOI] [PubMed] [Google Scholar]
- Rasmussen HH, ten Eick RE, Okita GT, et al. Inhibition of electrogenic Na-pumping attributable to binding of cardiac steroids to high-affinity pump sites in human atrium. J Pharmacol Exp Ther. 1985;235:629–635. [PubMed] [Google Scholar]
- Redpath CJ, Rankin AC, Kane KA, et al. Anti-adrenergic effects of endothelin on human atrial action potentials are potentially anti-arrhythmic. J Mol Cell Cardiol. 2006;40:717–724. doi: 10.1016/j.yjmcc.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Rensma PL, Allessie MA, Lammers WJEP, et al. Length of excitation wave and susceptibility to reentrant atrial arrhythmias in normal conscious dogs. Circ Res. 1988;62:395–410. doi: 10.1161/01.res.62.2.395. [DOI] [PubMed] [Google Scholar]
- Roth DA, Urasawa K, Helmer GA, et al. Downregulation of cardiac guanosine 5′-triphosphate-binding proteins in right atrium and left ventricle in pacing-induced congestive heart failure. J Clin Invest. 1993;91:939–949. doi: 10.1172/JCI116315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai R, Hagiwara N, Kasanuki H, et al. Chloride conductance in human atrial cells. J Mol Cell Cardiol. 1995;27:2403–2408. doi: 10.1016/s0022-2828(95)92199-0. [DOI] [PubMed] [Google Scholar]
- Salameh A, Frenzel C, Boldt A, et al. Subchronic alpha- and beta-adrenergic regulation of cardiac gap junction protein expression. Faseb J. 2006;20:365–367. doi: 10.1096/fj.05-4871fje. [DOI] [PubMed] [Google Scholar]
- Sato R, Koumi SI. Modulation of the inwardly rectifying K+ channel in isolated human atrial myocytes by α1-adrenergic stimulation. J Membr Biol. 1995;148:185–191. doi: 10.1007/BF00207274. [DOI] [PubMed] [Google Scholar]
- Sato R, Koumi SI. Characterization of the stretch-activated chloride channel in isolated human atrial myocytes. J Membr Biol. 1998;163:67–76. doi: 10.1007/s002329900371. [DOI] [PubMed] [Google Scholar]
- Schackow TE, ten Eick RE. Enhancement of ATP-sensitive potassium current in cat ventricular myocytes by β-adrenoreceptor stimulation. J Physiol. 1994;474:131–145. doi: 10.1113/jphysiol.1994.sp020008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer P, Pelzmann B, Bernhart E, et al. Estimation of outward currents in isolated human atrial myocytes using inactivation time course analysis. Pflugers Arch. 1998;436:457–468. doi: 10.1007/s004240050657. [DOI] [PubMed] [Google Scholar]
- Scherer D, Kiesecker C, Kulzer M, et al. Activation of inwardly rectifying Kir2.x potassium channels by β3-adrenoceptors is mediated via different signaling pathways with a predominant role of PKC for Kir2.1 and of PKA for Kir2.2. Naunyn-Schmiedeberg’s Arch Pharmacol. 2007;375:311–322. doi: 10.1007/s00210-007-0167-5. [DOI] [PubMed] [Google Scholar]
- Sharifov OF, Fedorov VV, Beloshapko GG, et al. Roles of adrenergic and cholinergic stimulation in spontaneous atrial fibrillation in dogs. J Am Coll Cardiol. 2004;43:483–490. doi: 10.1016/j.jacc.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Shimizu A, Fukatani M, Tanigawa M, et al. Mechanism of the suppression of repetitive atrial firing by isoproterenol-comparison with disopyramide. Int J Cardiol. 1994a;43:175–183. doi: 10.1016/0167-5273(94)90006-x. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Tsuchioka Y, Karakawa S, et al. Differential effect of pharmacological autonomic blockade on some electrophysiological properties of the human ventricle and atrium. Br Heart J. 1994b;71:34–37. doi: 10.1136/hrt.71.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skasa M, Jungling E, Picht E, et al. L-type calcium currents in atrial myocytes from patients with persistent and non-persistent atrial fibrillation. Basic Res Cardiol. 2001;96:151–159. doi: 10.1007/s003950170065. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Jurevicius J, Fischmeister R. Beta-2 adrenergic activation of L-type Ca++ current in cardiac myocytes. J Pharmacol Exp Ther. 1997;283:452–461. [PubMed] [Google Scholar]
- Sleator W, Jr., De Gubareff T. Transmembrane action potentials and contractions of human atrial muscle. Am J Physiol. 1964;206:1000–1014. doi: 10.1152/ajplegacy.1964.206.5.1000. [DOI] [PubMed] [Google Scholar]
- Smeets JLRM, Allessie MA, Lammers WJEP, et al. The wavelength of the cardiac impulse and reentrant arrhythmias in isolated rabbit atrium. The role of heart rate, autonomic transmitters, temperature, and potassium. Circ Res. 1986;58:96–108. doi: 10.1161/01.res.58.1.96. [DOI] [PubMed] [Google Scholar]
- Stambler BS, Wood MA, Ellenbogen KA. Pharmacologic alterations in human type I atrial flutter cycle length and monophasic action potential duration. Evidence of a fully excitable gap in the reentrant circuit. J Am Coll Cardiol. 1996;27:453–461. doi: 10.1016/0735-1097(95)00459-9. [DOI] [PubMed] [Google Scholar]
- Stambler BS, Fenelon G, Shepard RK, et al. Characterization of sustained atrial tachycardia in dogs with rapid ventricular pacing-induced heart failure. J Cardiovasc Electrophysiol. 2003;14:499–507. doi: 10.1046/j.1540-8167.2003.02519.x. [DOI] [PubMed] [Google Scholar]
- Steeds RP, Birchall AS, Smith M, et al. An open label, randomised, crossover study comparing sotalol and atenolol in the treatment of symptomatic paroxysmal atrial fibrillation. Heart. 1999;82:170–175. doi: 10.1136/hrt.82.2.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinfath M, Lavicky J, Schmitz W, et al. Regional distribution of β1- and β2-adrenoceptors in the failing and nonfailing human heart. Eur J Clin Pharmacol. 1992;42:607–611. doi: 10.1007/BF00265923. [DOI] [PubMed] [Google Scholar]
- Stengl M, Ramakers C, Donker DW, et al. Temporal patterns of electrical remodeling in canine ventricular hypertrophy: focus on IKs downregulation and blunted β-adrenergic activation. Cardiovasc Res. 2006;72:90–100. doi: 10.1016/j.cardiores.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Stiber JA, Seth M, Rosenberg PB. Mechanosensitive channels in striated muscle and the cardiovascular system: not quite a stretch anymore. J Cardiovasc Pharmacol. 2009;54:116–122. doi: 10.1097/FJC.0b013e3181aa233f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillitano F, Lonardo G, Zicha S, et al. Molecular basis of funny current (If) in normal and failing human heart. J Mol Cell Cardiol. 2008;45:289–299. doi: 10.1016/j.yjmcc.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Su MJ, Chi JF, Chu SH. Adrenergic modulation of potassium currents in isolated human atrial myocytes. J Biomed Sci. 1994;1:193–200. doi: 10.1007/BF02253349. [DOI] [PubMed] [Google Scholar]
- Trautwein W, Kassebaum DG, Nelson RM, et al. Electrophysiological study of human heart muscle. Circ Res. 1962;10:306–312. doi: 10.1161/01.res.10.3.306. [DOI] [PubMed] [Google Scholar]
- Tsai CS, Cheng TH, Lin CI, et al. Inhibitory effect of endothelin-1 on the isoproterenol-induced chloride current in human cardiac myocytes. Eur J Pharmacol. 2001;424:97–105. doi: 10.1016/s0014-2999(01)01145-1. [DOI] [PubMed] [Google Scholar]
- Tsang TSM, Gersh BJ, Appleton CP, et al. Left ventricular diastolic dysfunction as a predictor of the first diagnosed nonvalvular atrial fibrillation in 840 elderly men and women. J Am Coll Cardiol. 2002;40:1636–1644. doi: 10.1016/s0735-1097(02)02373-2. [DOI] [PubMed] [Google Scholar]
- Van Veldhuisen DJ, Aass H, El Allaf D, et al. Experiences from the MERIT-HF study Presence and development of atrial fibrillation in chronic heart failure. Eur J Heart Fail. 2006;8:539–546. doi: 10.1016/j.ejheart.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Van Wagoner DR, Pond AL, Lamorgese M, et al. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ Res. 1999;85:428–436. doi: 10.1161/01.res.85.5.428. [DOI] [PubMed] [Google Scholar]
- Vest JA, Wehrens XHT, Reiken SR, et al. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005;111:2025–2032. doi: 10.1161/01.CIR.0000162461.67140.4C. [DOI] [PubMed] [Google Scholar]
- Voigt N, Bollman B, Wettwer E, et al. Alpha-adrenergic regulation of IK1 and ACh-gated IK,ACh is impaired in patients with atrial fibrillation. Naunyn-Schmiedeberg’s Arch Pharmacol. 2009;379(Suppl 1):52. Abstract. [Google Scholar]
- Volders PGA, Stengl M, van Opstal JM, et al. Probing the contribution of IKs to canine ventricular repolarization. Key role for β-adrenergic receptor stimulation. Circulation. 2003;107:2753–2760. doi: 10.1161/01.CIR.0000068344.54010.B3. [DOI] [PubMed] [Google Scholar]
- Walden AP, Dibb KM, Trafford AW. Differences in intracellular calcium homeostasis between atrial and ventricular myocytes. J Mol Cell Cardiol. 2009;46:463–473. doi: 10.1016/j.yjmcc.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Wang C, Zhang YJ, Wang YL, et al. Effect of dipfluzine on delayed afterdepolarizations and triggered activity induced by isoprenaline in human atrial fibers. Acta Pharmaceutica Sinica. 2006;41:184–187. [PubMed] [Google Scholar]
- Wang Z, Fermini B, Nattel S. Rapid and slow components of delayed rectifier current in human atrial myocytes. Cardiovasc Res. 1994;28:1540–1546. doi: 10.1093/cvr/28.10.1540. [DOI] [PubMed] [Google Scholar]
- Webb JL, Hollander P. The action of acetylcholine and epinephrine on the cellular membrane potentials and contractility of rat atrium. Circ Res. 1956;4:332–336. doi: 10.1161/01.res.4.3.332. [DOI] [PubMed] [Google Scholar]
- Wettwer E, Hala O, Christ T, et al. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110:2299–2306. doi: 10.1161/01.CIR.0000145155.60288.71. [DOI] [PubMed] [Google Scholar]
- Wijffels MCEF, Kirchhof C, Dorland R, et al. Electrical remodeling due to atrial fibrillation in chronically instrumented conscious goats. Roles of neurohumoral changes, ischemia, atrial stretch, and high rate of electrical activation. Circulation. 1997;96:3710–3720. doi: 10.1161/01.cir.96.10.3710. [DOI] [PubMed] [Google Scholar]
- Wit AL, Hoffman BF, Rosen MR. Electrophysiology and pharmacology of cardiac arrhythmias IX. Cardiac electrophysiologic effects of beta adrenergic receptor stimulation and blockade. Part C. Am Heart J. 1975;90:795–803. doi: 10.1016/0002-8703(75)90471-8. [DOI] [PubMed] [Google Scholar]
- Wit AL, Boyden PA. Triggered activity and atrial fibrillation. Heart Rhythm. 2007;4:S17–S23. doi: 10.1016/j.hrthm.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman AJ, Rankin AC. Serotonin, If and human atrial arrhythmia. Cardiovasc Res. 1998;40:436–437. doi: 10.1016/s0008-6363(98)00258-2. [DOI] [PubMed] [Google Scholar]
- Workman AJ, Kane KA, Rankin AC. Ionic basis of a differential effect of adenosine on refractoriness in rabbit AV nodal and atrial isolated myocytes. Cardiovasc Res. 1999;43:974–984. doi: 10.1016/s0008-6363(99)00166-2. [DOI] [PubMed] [Google Scholar]
- Workman AJ, Kane KA, Rankin AC. The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res. 2001;52:226–235. doi: 10.1016/s0008-6363(01)00380-7. [DOI] [PubMed] [Google Scholar]
- Workman AJ, Kane KA, Rankin AC. Characterisation of the Na, K pump current in atrial cells from patients with and without chronic atrial fibrillation. Cardiovasc Res. 2003a;59:593–602. doi: 10.1016/s0008-6363(03)00466-8. [DOI] [PubMed] [Google Scholar]
- Workman AJ, Kane KA, Russell JA, et al. Chronic beta-adrenoceptor blockade and human atrial cell electrophysiology: evidence of pharmacological remodelling. Cardiovasc Res. 2003b;58:518–525. doi: 10.1016/s0008-6363(03)00263-3. [DOI] [PubMed] [Google Scholar]
- Workman AJ, Pau D, Redpath CJ, et al. Post-operative atrial fibrillation is influenced by beta-blocker therapy but not by pre-operative atrial cellular electrophysiology. J Cardiovasc Electrophysiol. 2006;17:1230–1238. doi: 10.1111/j.1540-8167.2006.00592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman AJ, Kane KA, Rankin AC. Cellular bases for human atrial fibrillation. Heart Rhythm. 2008;5:S1–S6. doi: 10.1016/j.hrthm.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman AJ, Pau D, Redpath CJ, et al. Atrial cellular electrophysiological changes in patients with ventricular dysfunction may predispose to AF. Heart Rhythm. 2009;6:445–451. doi: 10.1016/j.hrthm.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh TC, Vassalle M, Lin CI. Arrhythmogenic mechanisms in human atrial and ventricular muscle fibers. Cardiology. 1992;80:205–214. doi: 10.1159/000175004. [DOI] [PubMed] [Google Scholar]
- Yeh YH, Ehrlich JR, Qi X, et al. Adrenergic control of a constitutively active acetylcholine-regulated potassium current in canine atrial cardiomyocytes. Cardiovasc Res. 2007;74:406–415. doi: 10.1016/j.cardiores.2007.01.020. [DOI] [PubMed] [Google Scholar]
- Yeh YH, Wakili R, Qi XY, et al. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythmia Electrophysiol. 2008;1:93–102. doi: 10.1161/CIRCEP.107.754788. [DOI] [PubMed] [Google Scholar]
- Yoshimoto K, Hattori Y, Houzen H, et al. Histamine H1-receptor-mediated increase in the Ca2+ transient without a change in the Ca2+ current in electrically stimulated guinea-pig atrial myocytes. Br J Pharmacol. 1998;124:1744–1750. doi: 10.1038/sj.bjp.0702008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Feng J, Li GR, et al. Transient outward and delayed rectifier currents in canine atrium: properties and role of isolation methods. Am J Physiol. 1996;270:H2157–H2168. doi: 10.1152/ajpheart.1996.270.6.H2157. [DOI] [PubMed] [Google Scholar]
- Yue L, Feng J, Gaspo R, et al. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res. 1997;81:512–525. doi: 10.1161/01.res.81.4.512. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Lipsius SL. Na+-Ca2+ exchange current in latent pacemaker cells isolated from cat right atrium. J Physiol. 1993;466:263–285. [PMC free article] [PubMed] [Google Scholar]