

Abstract
Human chromosomal fragile sites are regions of the genome that are prone to DNA breakage, and are classified as common or rare, depending on their frequency in the population. Common fragile sites frequently coincide with the location of genes involved in carcinogenic chromosomal translocations, suggesting their role in cancer formation. However, there has been no direct evidence linking breakage at fragile sites to the formation of a cancer-specific translocation. Here, we studied the involvement of fragile sites in the formation of RET/PTC rearrangements, which are frequently found in papillary thyroid carcinoma (PTC). These rearrangements are commonly associated with radiation exposure; however most of the tumors found in adults are not linked to radiation. In this study, we provide structural and biochemical evidence that the RET, CCDC6, and NCOA4 genes participating in two major types of RET/PTC rearrangements, are located in common fragile sites FRA10C and FRA10G, and undergo DNA breakage after exposure to fragile site-inducing chemicals. Moreover, exposure of human thyroid cells to these chemicals results in the formation of cancer-specific RET/PTC rearrangements. These results provide the direct evidence for the involvement of chromosomal fragile sites in the generation of cancer-specific rearrangements in human cells.
Keywords: Fragile site, RET/PTC rearrangement, FRA10C/FRA10G, papillary thyroid carcinomas
Introduction
Cancer development can be initiated by the accumulation of various genetic abnormalities that lead to the disregulation of genes involved in various cellular processes. Chromosomal translocations are one of such abnormalities commonly seen in cancer cells. Translocations result in the rearrangement of genetic material, which typically leads to the expression of an oncogenic fusion protein contributing to the neoplastic process (Gasparini et al., 2007). To date, there are a total of 705 known recurrent translocations in cancer that involve 459 different gene pairs, and are present in many different types of cancer (Mitelman, 2008).
In all translocations, the development of breaks in DNA strands must occur. There are various ways in which a cell can acquire these breaks, such as ionizing radiation (Weterings and Chen, 2008). DNA breaks are commonly repaired by two pathways, homologous recombination or non-homologous end joining (Shrivastav et al., 2008), but dysfunction of these pathways can contribute to the formation of chromosomal translocations (Gasparini et al., 2007). Alternatively, an overwhelming accumulation of DNA breaks could prevent these normally functioning pathways from eliminating all of the breaks, leading to translocation events.
Chromosomal fragile sites are known to contribute to the formation of DNA breaks and are hotspots for sister chromatin exchange (Glover and Stein, 1987), chromosomal translocations, deletions (Glover and Stein, 1988), and viral integrations (Popescu, 2003). Fragile sites are non-random specific loci which are stable under normal conditions, but upon certain culture conditions can form visible gaps or breaks in metaphase chromosomes (Durkin and Glover, 2007). Depending on their frequency in the population, fragile sites can be divided into two classes: common and rare. Common fragile sites, which constitute the majority of the two classes, are present in all individuals, and are a normal component of chromosome structure (Glover, 2006). Common fragile sites can be further classified based on their mode of induction, as not all sites are induced by the same compounds, nor to the same extent. Aphidicolin (APH) induces expression of the majority of common fragile sites. Other known fragile site-inducing conditions include the addition of 5-bromodeoxyuridine (BrdU), 5-azacytidine, and distamycin A and the removal of folic acid (Sutherland, 1991). Also, certain dietary and environmental factors have been shown to contribute to fragile site expression, including caffeine (Yunis and Soreng, 1984), ethanol (Kuwano and Kajii, 1987), hypoxia (Coquelle et al., 1998), and pesticides (Musio and Sbrana, 1997). Together, genetic influences on fragile site instability, along with external influences from chemical, dietary and environmental factors, suggest a possible role for fragile sites in sporadic cancer formation.
Fragile sites are also known to be late replicating regions of the genome. Delayed DNA replication has been observed in all fragile sites examined to date (Handt et al., 2000; Hansen et al., 1997; Hellman et al., 2000; Hellman et al., 2002; Palakodeti et al., 2004; Pelliccia et al., 2008; Wang et al., 1999). Delayed replication at fragile sites is believed to be attributed to the high propensity of DNA sequences to form stable secondary DNA structures (Gacy et al., 1995; Hewett et al., 1998; Mishmar et al., 1998; Samadashwily et al., 1997; Usdin and Woodford, 1995; Zhang and Freudenreich, 2007; Zlotorynski et al., 2003). Difficulties in passing of the replication fork, caused by secondary DNA structure formed within the fragile DNA regions, could result in stalled replication. ATR, a major replication checkpoint protein, is crucial for maintaining fragile site stability (Casper et al., 2004), and its inhibition by 2-aminopurine (2-AP) in conjunction with fragile site inducing chemicals significantly increases common fragile site expression (Casper et al., 2002). Therefore, it is suggested that DNA breakage at fragile sites results from delayed replication forks that escape the ATR-mediated checkpoint pathway (Durkin and Glover, 2007).
Many studies point towards the association between fragile sites and formation of cancer-specific translocations (Arlt et al., 2006). In a comprehensive survey, we found that 52% of all known recurrent simple chromosomal translocations have at least one gene located within a fragile site, strongly suggesting a potential role for fragile sites in the initiation of translocations events (Burrow et al., 2009). Also, Glover and colleagues found that upon addition of APH, submicroscopic deletions within FHIT, located in the fragile site FRA3B and associated with various human cancers, were detected and resembled those seen in cancer cells (Durkin et al., 2008). However, there has been no direct evidence linking breakage at fragile sites to the formation of cancer-causing chromosomal aberrations.
Genes participating in the two main types of RET/PTC rearrangements, RET/PTC1 and RET/PTC3, have been mapped to known fragile sites (Burrow et al., 2009). RET/PTC rearrangements are commonly found in papillary thyroid carcinomas (PTC), and in all cases result in the fusion of the tyrosine kinase domain of RET to the 5′ portion of various unrelated genes (Nikiforov, 2008). In the case of the RET/PTC1 and RET/PTC3, RET is fused with CCDC6 and NCOA4 respectively (Santoro et al., 2006). These rearrangements result in the expression of a fusion protein possessing constitutive tyrosine kinase activity, which is tumorigenic in thyroid follicular cells (Nikiforov, 2008). Both genes involved in the RET/PTC3 rearrangement, RET and NCOA4, are located at 10q11.2 within fragile site FRA10G, a common fragile site induced by APH. The CCDC6 gene, involved in RET/PTC1, is located at 10q21.2 within fragile site FRA10C, a common fragile site induced by BrdU. Major breakpoint cluster regions for these genes have been identified, and are located within intron 11 of RET, intron 5 of NCOA4, and intron 1 of CCDC6 (Nikiforov et al., 1999; Smanik et al., 1995). RET/PTC rearrangements are known to be associated with radiation exposure, although most of adult tumors are sporadic and those patients lack the radiation exposure history (Nikiforova and Nikiforov, 2008), implying that other mechanisms should be responsible for DNA breakage and RET/PTC formation in most tumors. Clinical studies have shown that RET/PTC3 rearrangements are common in radiation-induced tumors (Fugazzola et al., 1995; Motomura et al., 1998; Nikiforov et al., 1997). In contrast, sporadic PTC tumors have shown a greater prevalence of RET/PTC1 rearrangements (Fenton et al., 2000), which account for 70% of all RET/PTC tumor types (Nikiforova and Nikiforov, 2008). Because the participating genes co-localize with fragile sites and there is a well-established association between RET/PTC rearrangements and DNA damage induced by ionizing radiation, these rearrangements offer an excellent model to examine directly the role of fragile sites in the formation of cancer-specific chromosomal translocations.
In this study, we demonstrate that fragile site-inducing chemicals can create DNA breaks within the RET/PTC partner genes and ultimately lead to the formation of RET/PTC rearrangements, offering direct evidence for the role of fragile sites in cancer-specific translocations.
Results
Chromosomal disruptions in RET/PTC gene partners upon fragile site induction
To examine whether chromosomal regions involved in RET/PTC rearrangements are part of fragile sites, HTori-3 human thyroid cells were exposed to APH, APH+2-AP, and BrdU+2-AP. Metaphase spreads of cultured HTori-3 cells were hybridized with fluorescently labeled BAC probes covering the entire genomic sequence of RET, NCOA4 and CCDC6 (Figure 1). Without exposure to fragile site-inducing chemicals, metaphase chromosomes of HTori-3 cells appeared normal with smooth contours and intact RET signal (Figure 1a). With exposure to fragile site-inducing chemicals, the morphology of metaphase chromosomes appeared distorted with irregular surfaces and loss of continuity. After treatment with 0.4 μM APH for 24 hours, RET was disrupted in 6 ± 0.35% of chromosomes (Figure 1b; Table 1), NCOA4 was disrupted in 0.62% of chromosomes and no breaks were identified in the CCDC6 gene (Table 1). The appearance of breaks in RET but not in CCDC6 is consistent with the characteristics of the fragile sites in which each of these genes are located (RET located at APH-induced FRA10G, and CCDC6 at BrdU-induced FRA10C). The frequency of breakage observed in RET is in agreement with the previously published levels at FRA10G obtained using Giemsa-stained chromosomes, which were found to be on average at 4.6% following treatment of human skin fibroblasts with 0.2 μM APH for 26 hours (Murano et al., 1989). After addition of APH and 2-AP, 5.93 ± 0.52% of chromosomes showed breaks in RET; 0.63 ± 0.08 % showed breaks in NCOA4 and 0.98 ± 0.58% showed breaks in CCDC6. 2-AP is a general inhibitor of ATR kinase and is known to increase fragile site expression with or without the addition of replication inhibitors like APH (Casper et al., 2002). While breakage in RET and NCOA4 did not change significantly, breakage was now seen in CCDC6, consistent with 2-AP action. Treatment with BrdU and 2-AP resulted in 2.72 ± 0.78% of chromosomes showing breaks in CCDC6 (Figure 1c). However, RET and NCOA4 were each disrupted in 0.6 ± 0.08% of chromosomes after BrdU and 2-AP treatment (Table 1). Increased breakage in CCDC6 is consistent with its fragile site mode of induction. Also, the level of breakage at CCDC6 is comparable with previous reports at FRA10C, with DNA breakage ranging from 4–20% following treatment of human blood lymphocytes from ten patients with 50 mg/L BrdU for 4–6 hours (Sutherland et al., 1985). The breakage frequency seen in RET and NCOA4 with BrdU and 2-AP treatment is similar to that observed in CCDC6 after treatment with APH and 2-AP, showing consistency with 2-AP induced breakage. In concert, these results demonstrate directly that chemicals known to result in fragile site breakage cause DNA breaks within genomic sequences of genes participating in RET/PTC rearrangements.
Figure 1.

FISH on metaphase chromosomes of HTori-3 cells after treatment with fragile site-inducing chemicals. (a) Negative control without treatment showing smooth chromosomes with intact RET (red) signal. (b) Exposure to APH resulting in irregular chromosome contours and one RET signal (red) showing split in the signal whereas four other RET signal are intact. Centromeric probe for chromosome 10 is labeled in green. (c) Exposure to BrdU+2-AP resulting in the disruption of CCDC6 (green) while NCOA4 is intact (red). (d) Exposure to APH+2-AP+BrdU resulting in split in RET (red).
Table 1.
Percentage of chromosomes showing disruption of RET, NCOA4 and CCDC6 after exposure to fragile site-inducing agents.
| APH | APH+2-AP | BrdU+2-AP | |
|---|---|---|---|
| RET | 6.00 ± 0.35 | 5.93 ± 0.52 | 0.60 ± 0.08 |
| NCOA4 | 0.62 | 0.63 ± 0.08 | 0.60 ± 0.08 |
| CCDC6 | 0 | 0.98 ± 0.58 | 2.72 ± 0.78 |
Induction of DNA breaks in intron 11 of the RET gene by APH treatment
All RET/PTC rearrangements involve the fusion of the tyrosine kinase domain of RET, and the major breakpoint cluster region identified in tumor cells is located within intron 11 (Smanik et al., 1995). While fluorescence in situ hybridization (FISH) experiments allowed us to detect breaks occurring within the RET gene sequence, whether or not the breaks are located in intron 11 was next examined using ligation-mediated PCR (LM-PCR). HTori-3 cells were treated with APH for 24 hours, and the genomic DNAs from both the treated and untreated cells were subjected to primer extension with biotinylated primers that are specific to the regions of interest (Materials & Methods; Supplementary Figure 2). The synthesis reaction terminated at a DNA break to produce a duplex with a blunt end, and the duplex was ligated to a linker. The linker-attached DNAs were then isolated by streptavidin beads, amplified by two rounds of PCR, and visualized by agarose gel electrophoresis (Figure 2). Each lane on the agarose gel represents the DNA breaks isolated from approximately 4000 cells, and each band observed on the gel corresponds to a break found within the region of interest. DNA breaks were observed within intron 11 of RET after treatment with APH (Figure 2a) with a frequency of 0.024 ± 0.015 breaks per 100 cells, which was significantly higher than that in the untreated cells (0.004 ± 0.009/100 cells, p = 0.010) (Figure 2b). DNA samples from lanes 1, and 3–6 in Figure 2a (marked with asterisks) were sequenced to determine the location of the induced breakpoints in the RET gene (Figure 3). DNA sequencing revealed the breakpoints to be located within intron 11, and at a distance from exon 12 that is consistent with the size of the PCR product observed on the agarose gel in Figure 2a. The locations of these breakpoints were compared to the location of known breakpoints found in PTC tumors containing RET/PTC rearrangements (Figure 3) (Bongarzone et al., 1997; Klugbauer et al., 2001). Each induced breakpoint was found to be located near a human tumor breakpoint, with distances ranging from 2–15 base pairs. It is important to note that these induced breakpoints were detected prior to a rearrangement event, while the breakpoints found in tumors have been identified after a rearrangement event has occurred. In most cases, small modifications, such as deletions and insertions of 1–18 nucleotides, have been observed surrounding the fusion points in human tumors. These results confirm that the exposure of thyroid cells to APH induces the formation of DNA breaks within the major breakpoint cluster region found in the RET gene, and these induced breakpoints are located close to known breakpoints found in human tumors.
Figure 2.
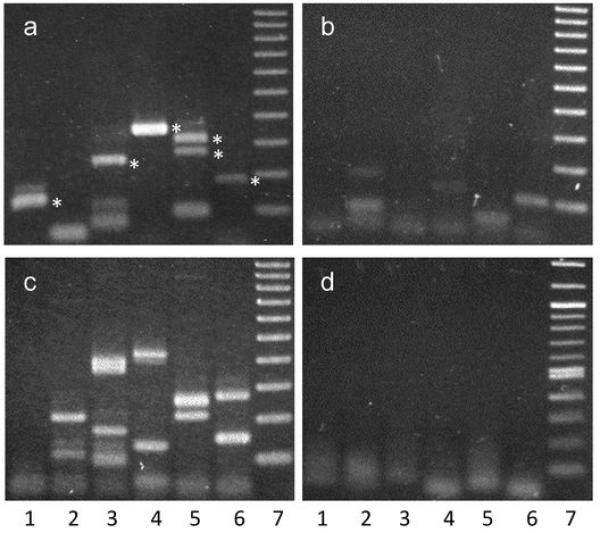
LM-PCR detection of breaks formed in HTori-3 cells after treatment with APH. LM-PCR detection of DNA breaks formed in HTori-3 cells at intron 11 of RET (a), the fragile site FRA3B (c), and the non-fragile 12p12.3 region (d) after treatment with APH. The same reaction was carried out as in (a) for intron 11 of RET, but using DNA from cells without APH treatment (b). Last lane of each gel is a 100 bp molecular weight ladder. Bands below 100bp correspond to primer dimers. Asterisks mark DNA fragments which were sequenced, and results are shown in Figure 3.
Figure 3.

Location of breakpoints within intron 11 of RET induced by treatment with APH. (a) DNA samples from lanes 1, and 3–6 in Figure 2a (marked with asterisks) were sequenced, and six breakpoints are identified and indicated by black arrowheads. The locations of known breakpoints found in tumors containing RET/PTC rearrangements are indicated by grey arrowheads (Bongarzone et al., 1997; Klugbauer et al., 2001). The grey arrow corresponds to the RET-7 primer with a dual biotin label (grey circles), which is annealed to exon 12 of the RET gene. The black solid and dashed arrows correspond to the RET-R1b and RET-R1 nested PCR primers, respectively. The sequence of intron 11 is italicized. (b) The distance of each induced breakpoint from the 5′ end of the RET-R1b primer and the nearest patient tumor breakpoint was listed.
DNA breaks were also examined within FRA3B after APH treatment. FRA3B is the most inducible fragile site in the human genome and contains FHIT, a gene involved in several cancers, where microscopic deletions have been observed after treatment with APH (Durkin et al., 2008; Wang et al., 1999). Intron 4 of the FHIT gene, a major region of high instability in various tumors and APH-treated cells (Boldog et al., 1997; Corbin et al., 2002), was examined here for DNA breaks. DNA breaks were detected within intron 4 of FHIT upon APH treatment (Figure 2c) at a frequency of 0.036 ± 0.020 breaks per 100 cells, confirming that indeed the APH treatment can induce fragile site breakage. An increased number of breaks were observed within FRA3B in comparison to RET, which is consistent with FRA3B being the most active fragile site in the genome. A non-fragile region, 12p12.3 (Zlotorynski et al., 2003) and the G6PD gene, within FRAXF (a rare folate-sensitive fragile site not induced by APH), were examined after treatment with APH, and in contrast to RET and FRA3B, no DNA breaks were observed within the 12p12.3 region (Figure 2d) or in exon 1 of G6PD (Supplementary Figure 3). The absence of breaks in 12p12.3 and G6PD suggests that the DNA breaks observed within RET and FRA3B after exposure to fragile site-inducing chemicals are due to their fragile nature in response to APH.
Generation of RET/PTC rearrangements after treatment with fragile site-inducing chemicals
To test for the induction of RET/PTC rearrangement after exposure to fragile site-inducing chemicals, HTori-3 cells were treated with APH and 2-AP for 24 hours with the addition of BrdU for the last 5 hours. These treatment conditions were chosen because they have been previously established to be optimal for the induction of fragile sites FRA10C and FRA10G (Murano et al., 1989; Sutherland et al., 1985). To confirm breakage in the genes after exposure, metaphase spreads were made and chromosomes were scored for disruption of the probe (Figure 1d). The breakage in the probes for RET, NCOA4 and CCDC6 were 7.47%, 1.15% and 2.87% respectively. The mRNA was then isolated and used in RT-PCR for detection of RET/PTC1 and RET/PTC3 formation. To assure that a cell with the rearrangement would be detected, 1 × 106 cells in a 10 cm culture dish were divided among 30 culture dishes 24 h post-exposure. Therefore, each well received no more than 3 × 104 cells, and if a dish contained only one cell with RET/PTC, it would constitute 1 part in 3 × 104, a fraction within the limit of detection (Caudill et al., 2005). No RET/PTC rearrangement was detected without any treatments in five independent experiments (Figure 4), indicating an extremely low level of spontaneous generation of RET/PTC in this human cell line and the absence of contamination. Similarly, no RET/PTC rearrangement was detected using the same experimental approach in HTori-3 cells in four independent experiments in a study reported by Caudill et al. (Caudill et al., 2005). Exposure to a combination of APH, 2-AP and BrdU resulted in the generation of RET/PTC1, with 5 total events identified in 5 independent experiments, each assaying 106 cells (incidence of 2, 1, 2, 0, 0 events per 106 cells) (Figure 4b). However, no RET/PTC3 rearrangements were identified. Representative RT-PCR blots are shown in Figure. 4a. Statistical analysis revealed a significant difference in the incidence of RET/PTC1 induction between untreated cells (zero events) and cells treated with fragile site-inducing agents (five total events) (p = 0.027). These results demonstrate that the exposure of thyroid cells to fragile site-inducing chemicals can lead to the formation of a carcinogenic RET/PTC rearrangement.
Figure 4.

Detection of RET/PTC rearrangements in HT-ori3 cells after treatment with fragile site-inducing chemicals. (a) Detection of RET/PTC rearrangements in representative RT-PCR experiment after exposure to APH+2-AP+BrdU. PC, Positive control. (b) Number of rearrangement events detected in untreated cells and cells exposed to APH+2-AP+BrdU. Five independent experiments were carried out for each treatment, and, each experiment analyzed 106 cells.
Discussion
Chromosomal rearrangements contribute to the development of many types of human tumors. Therefore, it is critical to understand the mechanisms of chromosomal rearrangements in cancer cells. Here, we demonstrated that DNA breakage at fragile sites FRA10C and FRA10G under fragile site-inducing conditions initiates and leads to the generation of RET/PTC1 rearrangement, which is known to contribute to PTC development. To our knowledge, this is the first demonstration that a cancer-specific rearrangement can be produced in human cells by inducing DNA breaks at fragile sites. Interestingly, only RET/PTC1 rearrangements were observed, and no RET/PTC3 rearrangements were identified. While breakage was seen within NCOA4, the RET/PTC3 partner gene, the frequency of breakage was lower when compared to RET and CCDC6. NCOA4 breakage remained relatively constant with each combination of fragile site-inducing chemicals, and was about 10-fold lower than the breakage observed within RET, and about 4.5-fold below the level found in CCDC6. The lower incidence of breakage within NCOA4 could contribute to the lack of RET/PTC3 rearrangement events. Also, clinical studies have revealed that RET/PTC3 rearrangements are frequent in radiation-induced tumors (Fugazzola et al., 1995; Motomura et al., 1998; Nikiforov et al., 1997), while RET/PTC1 rearrangements are more commonly seen in sporadic tumors (Fenton et al., 2000). Our observation of RET/PTC1 rearrangement, but not RET/PTC3 rearrangement, generated by fragile site induction, further supports the idea that sporadic PTC tumors may result from breakage at fragile sites. It is known that specific environmental and food toxins (such as caffeine, alcohol, tobacco) (Kuwano and Kajii, 1987; Yunis and Soreng, 1984), and other stress factors (such as hypoxia) (Coquelle et al., 1998) can induce fragile sites. Therefore, our results suggest that these exogenous factors may contribute to the occurrence of chromosomal rearrangements, and therefore cancer initiation in human populations, by a mechanism of DNA breakage at fragile sites.
To demonstrate that fragile site-inducing chemicals can cause DNA breaks at RET/PTC participating genes, FISH analysis of chromosome 10, and LM-PCR analysis at the nucleotide level of the RET gene were performed. Using FISH, we showed that upon exposure of human thyroid cells to fragile site-inducing chemicals, chromosomal breaks are formed within the RET and CCDC6 genes. RET and CCDC6 are located respectively within the APH and BrdU-induced fragile sites, and display breakage only after the addition of APH or BrdU, accordingly. These results demonstrate not only that the fragility is indeed present within the genes involved in RET/PTC rearrangements, but also underline the specificity of fragile site induction that was observed in these regions. While 2-AP addition is known to overall increase chromosomal breakage and fragile site FRA3B expression (Casper et al., 2002), no significant increase in breakage at RET and NCOA4 genes was noted in HTori-3 cells, indicating its weaker influence on the FRA10G site. Furthermore, the addition of 2-AP in combination with APH resulted in the appearance of breaks within CCDC6, while its combination with BrdU resulted in breaks within RET and NCOA4. This nonspecific effect of 2-AP on induction of DNA breaks at fragile sites is in agreement with its ability to inhibit ATR protein, which provides a key maintenance role in fragile site stability.
The DNA breaks generated in RET after exposure to APH were confirmed to be located within intron 11, which is the breakpoint cluster region identified in thyroid tumors, while untreated cells showed little to no breaks. These breaks are further confirmed to be fragile in nature, when comparing the formation of breaks within FRA3B, 12p12.3 and G6PD regions. FRA3B, the most inducible fragile site in the human genome (Durkin et al., 2008; Wang et al., 1999), displayed DNA breaks after treatment with APH (Figure 2c); while 12p12.3, a non-fragile region, and the G6PD gene, located within a rare folate-sensitive fragile site, showed no DNA breakage with the same treatment (Figure 2d and Supplementary Figure 3b). Together with cytogenetic analysis, these results demonstrate that fragile site-inducing chemicals can generate breaks within RET and CCDC6 genes, which could result in the formation of cancer-causing RET/PTC1 rearrangement.
The induction rate of RET/PTC rearrangement by fragile site-inducing chemicals was four magnitudes lower than the frequency of chromosomal breaks observed in RET and CCDC6 genes. DNA breaks, a serious threat to genome stability and cell viability, can trigger DNA repair pathways, including homologous recombination or non-homologous end joining (Shrivastav et al., 2008). The action of these pathways ensures proper repair of DNA breaks, and prevents the deleterious consequences of such breakage. However, some (small number of) DNA breaks escaping the repair pathways will ultimately result in large-scale chromosomal changes, such as RET/PTC rearrangement.
This study provides important information about the mechanisms of formation of carcinogenic chromosomal rearrangements in human cells. In addition, it establishes an experimental system that will allow for testing the role of specific environmental substances, dietary toxins, and other stress factors in the generation of chromosomal rearrangements and tumor initiation.
Materials and Methods
Cell line and culture conditions
The experiments were performed on HTori-3 cells, which are human thyroid epithelial cells transfected with an origin-defective SV40 genome. They are characterized as immortalized, partially transformed, differentiated cells having three copies of chromosome 10 with intact RET, NCOA4 and CCDC6 loci and preserve the expression of thyroid differentiation markers such as thyroglobulin production and sodium iodide symporter, as we reported previously (Caudill et al., 2005). The cells were purchased from the European Tissue Culture Collection and grown in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum.
Fragile site induction
HTori-3 cells (1 × 106) were plated in 10-cm culture dishes and 16 h later exposed for 24 h to APH (0.4 μM) or APH and 2-AP (2 mM) (Casper et al., 2002). When desired, cells were treated with BrdU (50 mg/L) for the last 5 h to in addition to 2-AP and/or APH for 24 h. For DNA breaksite detection 5 × 105 cells were plated in 10-cm culture dishes and treated the same as above with 0.4 μM APH.
Metaphase chromosome preparation
HTori-3 cells exposed to various chemicals were treated with 0.1 μg/ml of Colcemide for the last 2 hours before harvesting. Cells were incubated in hypotonic solution (0.075 M KCL), fixed in multiple changes of methanol:acetic acid (3:1) and dropped onto moistened slides in order to obtain metaphase spreads. Slides were aged overnight and pretreated with RNase before proceeding for hybridization.
Probes for FISH
BAC clones RP11-351D16 (RET), RP11-481A12 (NCOA4), RP11-435G3 and RP11-369L1 (CCDC6) were obtained from BAC/PAC Resources, Children's Hospital, Oakland. BAC clone RP11-481A12 containing the NCOA4 gene was subcloned into fosmid vector after cutting with restriction enzymes (Epicentre). A mixture of subcloned probes (SC10, SC19) containing 70 kb of the NCOA4 gene and its flanking regions was used as a probe for NCOA4. The probes were labeled by nick translation using Spectrum Green-dUTP, Spectrum Orange-dUTP or Spectrum Red-dUTP (Vysis Inc.). Hybridization was performed as previously described (Ciampi et al., 2005). On average 150 chromosomes were scored for breaks in the RET, NCOA4 and CCDC6 probes for each condition.
DNA breaksite mapping by LM-PCR
To detect DNA breaks within intron 11 of RET induced by APH, a 5′-biotinylated primer RET-7 corresponding to the RET at the 5′ end of exon 12 (the grey arrow in Figure 3a) was used to extend into intron 11. For the first and second rounds of nested PCR primers RET-R1b and RET-R1 were used, respectively. To isolate the DNA breaks, a duplex DNA linker LL3/LP2 was used as described (Kong and Maizels, 2001) as well as the corresponding linker specific primers LL4 and LL2 (Supplementary Figure 2). For FRA3B, the biotinylated primer FRA3B-20 was used to allow identification of break sites occurring at intron 4 of the FHIT gene, which contains major clusters of APH-induced breakpoints in FRA3B (Boldog et al., 1997; Corbin et al., 2002), and primers FRA3B-9 and FRA3B-23 were used in the first and second rounds of nested PCR, respectively. For detection of breaks within the 12p12.3 region, the biotinylated primer 12p12.3-1 and primers 12p12.3-2 and 12p12.3-3 were used. For detection of breaks within exon 1 of G6PD, the biotinylated primer G6PDF3 and primers G6PDF and G6PDF2 were used. Sequence of linkers and PCR primers is described in the Supplementary Figure 1.
DNA breaksite mapping was performed as described (Kong and Maizels, 2001) with modifications (Supplementary Figure 2). Genomic DNA was isolated from HTori-3 cells with or without APH treatment. Primer extension was performed using 200 ng of DNA at 45°C, and the DNA breaks were isolated through ligation of the LL3/LP2 linker, and then using streptavidin beads. Amplification of these DNA breaks was achieved by nested PCR of the extension-ligation products. The final PCR products were resolved by electrophoresis on a 1.3% agarose gel. Each band observed on the gel corresponds to a break isolated within the region of interest. To confirm the bands observed were located within intron 11 of RET, the PCR products were sequenced. The exact breakpoint sites were determined from the sequencing results by identifying the nucleotide adjacent to the LL3/LP2 linker sequence.
Detection of RET/PTC rearrangements
Upon treatment with fragile site-inducing agents for 24 hours, cells were split into 30 6-cm culture dishes at a density of approximately 3 × 104 cells per dish and grown for 3–4 days. To sustain growth for 9 days, cells were transferred to 10-cm culture dishes 4–5 days after seeding into 6-cm dishes. RNA was extracted from each culture dish using a Trizol reagent (Invitrogen). Then, mRNA was purified using the Oligotex mRNA minikit (QIAGEN). RT-PCR was performed using a Superscript first strand synthesis system kit and random hexamer priming (Invitrogen). PCR was performed to simultaneously detect RET/PTC1 and RET/PTC3 rearrangement using primers RET/PTC1 forward, RET/PTC3 forward, and common reverse (Supplementary Figure 1). As positive controls, cDNA from RET/PTC1-positive TPC-1 cells and RET/PTC3 positive tumor sample were used. Ten μl of each PCR product was electrophoresed in a 1.5% agarose gel, transferred to the nylon membrane, and hybridized with 32P-labeled oligonucleotide probes specific for RET/PTC1 and RET/PTC3 (Supplementary Figure 1). Evidence of RET/PTC rearrangement in the cells from a given flask was scored as one RET/PTC event. All statistics performed using one-tailed Student's t-test.
Supplementary Material
Acknowledgements
This work was supported by the National Cancer Institute (CA113863 to Y.-H. W and Y. E. N.).
Footnotes
The authors declare no conflict of interest.
References
- Arlt MF, Durkin SG, Ragland RL, Glover TW. Common fragile sites as targets for chromosome rearrangements. DNA Repair (Amst) 2006;5:1126–35. doi: 10.1016/j.dnarep.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Boldog F, Gemmill RM, West J, Robinson M, Robinson L, Li E, et al. Chromosome 3p14 homozygous deletions and sequence analysis of FRA3B. Hum Mol Genet. 1997;6:193–203. doi: 10.1093/hmg/6.2.193. [DOI] [PubMed] [Google Scholar]
- Bongarzone I, Butti MG, Fugazzola L, Pacini F, Pinchera A, Vorontsova TV, et al. Comparison of the breakpoint regions of ELE1 and RET genes involved in the generation of RET/PTC3 oncogene in sporadic and in radiation-associated papillary thyroid carcinomas. Genomics. 1997;42:252–9. doi: 10.1006/geno.1997.4685. [DOI] [PubMed] [Google Scholar]
- Burrow AA, Williams LE, Pierce LC, Wang YH. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics. 2009;10:59. doi: 10.1186/1471-2164-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper AM, Durkin SG, Arlt MF, Glover TW. Chromosomal instability at common fragile sites in Seckel syndrome. Am.J.Hum.Genet. 2004;75:654–660. doi: 10.1086/422701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–89. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- Caudill CM, Zhu Z, Ciampi R, Stringer JR, Nikiforov YE. Dose-dependent generation of RET/PTC in human thyroid cells after in vitro exposure to gamma-radiation: a model of carcinogenic chromosomal rearrangement induced by ionizing radiation. J Clin Endocrinol Metab. 2005;90:2364–9. doi: 10.1210/jc.2004-1811. [DOI] [PubMed] [Google Scholar]
- Ciampi R, Zhu Z, Nikiforov YE. BRAF copy number gains in thyroid tumors detected by fluorescence in situ hybridization. Endocr Pathol. 2005;16:99–105. doi: 10.1385/ep:16:2:099. [DOI] [PubMed] [Google Scholar]
- Coquelle A, Toledo F, Stern S, Bieth A, Debatisse M. A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol Cell. 1998;2:259–65. doi: 10.1016/s1097-2765(00)80137-9. [DOI] [PubMed] [Google Scholar]
- Corbin S, Neilly ME, Espinosa R, 3rd, Davis EM, McKeithan TW, Le Beau MM. Identification of unstable sequences within the common fragile site at 3p14.2: implications for the mechanism of deletions within fragile histidine triad gene/common fragile site at 3p14.2 in tumors. Cancer Res. 2002;62:3477–84. [PubMed] [Google Scholar]
- Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–92. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- Durkin SG, Ragland RL, Arlt MF, Mulle JG, Warren ST, Glover TW. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc Natl Acad Sci U S A. 2008;105:246–51. doi: 10.1073/pnas.0708097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton CL, Lukes Y, Nicholson D, Dinauer CA, Francis GL, Tuttle RM. The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J Clin Endocrinol Metab. 2000;85:1170–5. doi: 10.1210/jcem.85.3.6472. [DOI] [PubMed] [Google Scholar]
- Fugazzola L, Pilotti S, Pinchera A, Vorontsova TV, Mondellini P, Bongarzone I, et al. Oncogenic rearrangements of the RET proto-oncogene in papillary thyroid carcinomas from children exposed to the Chernobyl nuclear accident. Cancer Res. 1995;55:5617–20. [PubMed] [Google Scholar]
- Gacy AM, Goellner G, Juranic N, Macura S, McMurray CT. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell. 1995;81:533–40. doi: 10.1016/0092-8674(95)90074-8. [DOI] [PubMed] [Google Scholar]
- Gasparini P, Sozzi G, Pierotti MA. The role of chromosomal alterations in human cancer development. J Cell Biochem. 2007;102:320–31. doi: 10.1002/jcb.21481. [DOI] [PubMed] [Google Scholar]
- Glover TW. Common fragile sites. Cancer Lett. 2006;232:4–12. doi: 10.1016/j.canlet.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Glover TW, Stein CK. Induction of sister chromatid exchanges at common fragile sites. Am J Hum Genet. 1987;41:882–90. [PMC free article] [PubMed] [Google Scholar]
- Glover TW, Stein CK. Chromosome breakage and recombination at fragile sites. Am J Hum Genet. 1988;43:265–73. [PMC free article] [PubMed] [Google Scholar]
- Handt O, Baker E, Dayan S, Gartler SM, Woollatt E, Richards RI, et al. Analysis of replication timing at the FRA10B and FRA16B fragile site loci. Chromosome Res. 2000;8:677–88. doi: 10.1023/a:1026737203447. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Canfield TK, Fjeld AD, Mumm S, Laird CD, Gartler SM. A variable domain of delayed replication in FRAXA fragile X chromosomes: X inactivation-like spread of late replication. Proc Natl Acad Sci U S A. 1997;94:4587–92. doi: 10.1073/pnas.94.9.4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman A, Rahat A, Scherer SW, Darvasi A, Tsui LC, Kerem B. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol Cell Biol. 2000;20:4420–7. doi: 10.1128/mcb.20.12.4420-4427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman A, Zlotorynski E, Scherer SW, Cheung J, Vincent JB, Smith DI, et al. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 2002;1:89–97. doi: 10.1016/s1535-6108(02)00017-x. [DOI] [PubMed] [Google Scholar]
- Hewett DR, Handt O, Hobson L, Mangelsdorf M, Eyre HJ, Baker E, et al. FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site genesis. Mol Cell. 1998;1:773–81. doi: 10.1016/s1097-2765(00)80077-5. [DOI] [PubMed] [Google Scholar]
- Klugbauer S, Pfeiffer P, Gassenhuber H, Beimfohr C, Rabes HM. RET rearrangements in radiation-induced papillary thyroid carcinomas: high prevalence of topoisomerase I sites at breakpoints and microhomology-mediated end joining in ELE1 and RET chimeric genes. Genomics. 2001;73:149–60. doi: 10.1006/geno.2000.6434. [DOI] [PubMed] [Google Scholar]
- Kong Q, Maizels N. Breaksite batch mapping, a rapid method for assay and identification of DNA breaksites in mammalian cells. Nucleic Acids Res. 2001;29:E33. doi: 10.1093/nar/29.6.e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwano A, Kajii T. Synergistic effect of aphidicolin and ethanol on the induction of common fragile sites. Hum Genet. 1987;75:75–8. doi: 10.1007/BF00273845. [DOI] [PubMed] [Google Scholar]
- Mishmar D, Rahat A, Scherer SW, Nyakatura G, Hinzmann B, Kohwi Y, et al. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci U S A. 1998;95:8141–6. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F. 2008 [Google Scholar]
- Motomura T, Nikiforov YE, Namba H, Ashizawa K, Nagataki S, Yamashita S, et al. ret rearrangements in Japanese pediatric and adult papillary thyroid cancers. Thyroid. 1998;8:485–9. doi: 10.1089/thy.1998.8.485. [DOI] [PubMed] [Google Scholar]
- Murano I, Kuwano A, Kajii T. Fibroblast-specific common fragile sites induced by aphidicolin. Hum Genet. 1989;83:45–8. doi: 10.1007/BF00274145. [DOI] [PubMed] [Google Scholar]
- Musio A, Sbrana I. Aphidicolin-sensitive specific common fragile sites: a biomarker of exposure to pesticides. Environ Mol Mutagen. 1997;29:250–5. doi: 10.1002/(sici)1098-2280(1997)29:3<250::aid-em4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Nikiforov YE. Thyroid carcinoma: molecular pathways and therapeutic targets. Mod Pathol. 2008;21(Suppl 2):S37–43. doi: 10.1038/modpathol.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov YE, Koshoffer A, Nikiforova M, Stringer J, Fagin JA. Chromosomal breakpoint positions suggest a direct role for radiation in inducing illegitimate recombination between the ELE1 and RET genes in radiation-induced thyroid carcinomas. Oncogene. 1999;18:6330–4. doi: 10.1038/sj.onc.1203019. [DOI] [PubMed] [Google Scholar]
- Nikiforov YE, Rowland JM, Bove KE, Monforte-Munoz H, Fagin JA. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997;57:1690–4. [PubMed] [Google Scholar]
- Nikiforova MN, Nikiforov YE. Molecular genetics of thyroid cancer: implications for diagnosis, treatment and prognosis. Expert Rev Mol Diagn. 2008;8:83–95. doi: 10.1586/14737159.8.1.83. [DOI] [PubMed] [Google Scholar]
- Palakodeti A, Han Y, Jiang Y, Le Beau MM. The role of late/slow replication of the FRA16D in common fragile site induction. Genes Chromosomes Cancer. 2004;39:71–6. doi: 10.1002/gcc.10290. [DOI] [PubMed] [Google Scholar]
- Pelliccia F, Bosco N, Curatolo A, Rocchi A. Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenet Genome Res. 2008;121:196–200. doi: 10.1159/000138885. [DOI] [PubMed] [Google Scholar]
- Popescu NC. Genetic alterations in cancer as a result of breakage at fragile sites. Cancer Lett. 2003;192:1–17. doi: 10.1016/s0304-3835(02)00596-7. [DOI] [PubMed] [Google Scholar]
- Samadashwily GM, Raca G, Mirkin SM. Trinucleotide repeats affect DNA replication in vivo. Nat Genet. 1997;17:298–304. doi: 10.1038/ng1197-298. [DOI] [PubMed] [Google Scholar]
- Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur J Endocrinol. 2006;155:645–53. doi: 10.1530/eje.1.02289. [DOI] [PubMed] [Google Scholar]
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–47. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- Smanik PA, Furminger TL, Mazzaferri EL, Jhiang SM. Breakpoint characterization of the ret/PTC oncogene in human papillary thyroid carcinoma. Hum Mol Genet. 1995;4:2313–8. doi: 10.1093/hmg/4.12.2313. [DOI] [PubMed] [Google Scholar]
- Sutherland GR. Chromosomal fragile sites. Genet Anal Tech Appl. 1991;8:161–6. doi: 10.1016/1050-3862(91)90056-w. [DOI] [PubMed] [Google Scholar]
- Sutherland GR, Parslow MI, Baker E. New classes of common fragile sites induced by 5-azacytidine and bromodeoxyuridine. Hum Genet. 1985;69:233–7. doi: 10.1007/BF00293031. [DOI] [PubMed] [Google Scholar]
- Usdin K, Woodford KJ. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995;23:4202–9. doi: 10.1093/nar/23.20.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Darling J, Zhang JS, Huang H, Liu W, Smith DI. Allele-specific late replication and fragility of the most active common fragile site, FRA3B. Hum Mol Genet. 1999;8:431–7. doi: 10.1093/hmg/8.3.431. [DOI] [PubMed] [Google Scholar]
- Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res. 2008;18:114–24. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- Yunis JJ, Soreng AL. Constitutive fragile sites and cancer. Science. 1984;226:1199–204. doi: 10.1126/science.6239375. [DOI] [PubMed] [Google Scholar]
- Zhang H, Freudenreich CH. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cervisiae. Molecular Cell. 2007;27:367–379. doi: 10.1016/j.molcel.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotorynski E, Rahat A, Skaug J, Ben-Porat N, Ozeri E, Hershberg R, et al. Molecular basis for expression of common and rare fragile sites. Mol Cell Biol. 2003;23:7143–51. doi: 10.1128/MCB.23.20.7143-7151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.